





Abstract
5α-Reductase types 1 and 2, encoded by SRD5A1 and SRD5A2, are the two enzymes that can catalyze the conversion of testosterone to dihydrotestosterone, the most potent androgen receptor (AR) agonist in prostate cells. 5α-Reductase type 2 is the predominant isoform expressed in the normal prostate. However, its expression decreases during prostate cancer (PCa) progression, whereas SRD5A1 increases, and the mechanism underlying this transcriptional regulatory switch is still unknown. Interrogation of SRD5A messenger RNA expression in three publicly available data sets confirmed that SRD5A1 is increased in primary and metastatic PCa compared with nontumoral prostate tissues, whereas SRD5A2 is decreased. Activation of AR, a major oncogenic driver of PCa, induced the expression of SRD5A1 from twofold to fourfold in three androgen-responsive PCa cell lines. In contrast, AR repressed SRD5A2 expression in this context. Chromatin-immunoprecipitation studies established that AR is recruited to both SRD5A1 and SRD5A2 genes following androgen stimulation but initiates transcriptional activation only at SRD5A1 as monitored by recruitment of RNA polymerase II and the presence of the H3K27Ac histone mark. Furthermore, we showed that the antiandrogens bicalutamide and enzalutamide block the AR-mediated regulation of both SRD5A1 and SRD5A2, highlighting an additional mechanism explaining their beneficial effects in patients. In summary, we identified an AR-dependent transcriptional regulation that explains the differential expression of 5α-reductase types 1 and 2 during PCa progression. Our work thus defines a mechanism by which androgens control their own synthesis via differential regulatory control of the expression of SRD5A1 and SRD5A2.
Androgens are known to play a central role in prostate cancer (PCa) malignancy, from an initially localized tumor to a metastatic disease, where androgen deprivation therapy remains the standard of care (1). Progressive diseases, which were first thought to become “androgen independent,” are now clearly recognized as “castration resistant” rather than completely independent from the androgen axis. To explain this resistance to lower circulating androgens, several molecular mechanisms have been identified in aggressive PCa, including androgen receptor (AR) gene amplification, activating mutations of AR, but also increased expression of key genes involved in androgen synthesis locally in tumor cells (2–4).
Testosterone, secreted by the testis, and adrenal steroid precursors such as dehydroepiandrosterone, are among factors influencing androgen levels in the prostate and therefore AR activation status (5–7). Indeed, prostate cells contain a variety of steroidogenic enzymes required for synthesis of active androgens that originate from adrenal precursors circulating in the bloodstream (8–11). 5α-Reductase types 1 and 2, encoded by SRD5A1 and SRD5A2, are the 2 enzymes that can catalyze the conversion of testosterone to dihydrotestosterone (DHT), the most potent AR agonist in prostate cells. 5α-Reductase type 2 is the predominant isoform expressed in the prostate, whereas type 1 represents less than 10% of total 5α-reductase levels in normal prostate cells (12). However, although the expression of SRD5A2 decreases in PCa cells, SRD5A1 expression increases (3, 4, 12–14). Neither the mechanism underlying this transcriptional regulatory switch nor the consequences of such an imbalance between both 5α-reductases are known. It is clear, however, that these two enzymes are associated with not only PCa initiation but also its progression. Indeed, several polymorphisms in SRD5A1 and SRD5A2 have been associated with PCa recurrence (15). Moreover, PCa prevention trials demonstrated that 5α-reductase inhibitors influence the development of this cancer (16). The imbalance in SRD5A isoform expression in PCa illustrates the complex relation between 5α-reductases, DHT synthesis, and PCa progression.
In this study, we demonstrate that both SRD5A1 and SRD5A2 are direct AR target genes. However, their expression is inversely modulated by the androgen signaling pathway. Following activation, AR activates SRD5A1 transcription but represses SRD5A2, favoring a transcriptional isoform switch that correlates with SRD5A expression patterns in human tumors.
Materials and Methods
Cell culture
LNCaP, LAPC4, and 22rv1 cells were grown in phenol red–free RPMI supplemented with 10% fetal bovine serum, penicillin, streptomycin, and sodium pyruvate at 37°C and 5% CO2. After 48 hours of steroid deprivation using charcoal-stripped serum, cells were treated with 10 nM R1881 (metribolone; Steraloids) or vehicle. For small interfering RNA (siRNA) transfections, cells were trypsinized, seeded in steroid-depleted media, and subsequently transfected with either a nontargeted pool of siRNA (siControl) or a pool of siRNA targeting AR (SMARTpool; Dharmacon) with Hiperfect reagent per the manufacturer’s instructions (Qiagen).
RNA extraction and quantitative reverse transcription polymerase chain reaction
RNA was extracted with the RNeasy Mini Kit (Qiagen), and first-strand complementary DNA synthesis was performed with ProtoScript II reverse transcription (New England Biolabs). Complementary DNA expression was then quantified by SYBR green–based quantitative polymerase chain reaction (qPCR) techniques using the LightCycler 480 instrument (Roche). Relative expression was standardized to the expression of housekeeping genes, PUM1 in TBP (Supplemental Table 1).
Chromatin immunoprecipitation–qPCR
Chromatin immunoprecipitation (ChIP)–qPCR of AR (sc-816X; Santa Cruz Biotechnology), RNA polymerase II (05-623; Millipore), or H3K27Ac (ab4729; Abcam) was done as previously described (17) (Table 1). Briefly, cells were crosslinked after R1881 treatment or vehicle with 1% formaldehyde at room temperature, and nuclei were enriched by sequential centrifugations. After sonication, immunoprecipitations were performed overnight at 4°C with the corresponding antibody previously incubated with Dynabeads (Life Technologies). DNA was purified using the Qiagen PCR purification kit and analyzed by qPCR using a Roche LightCycler 480. Nontargeted rabbit immunoglobulin ChIP was used as a control of antibody nonspecific binding, and 2 negative regions were used for ChIP normalization between samples (Supplemental Table 2).
Antibody table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| AR (N-20) | NA | sc-816X | Santa Cruz Biotechnology | Rabbit; polyclonal | 3 μg per ChIP reaction | AB_1563391 |
| RNA polymerase II | NA | Anti-RNA polymerase II, clone CTD4H8 | Millipore, 05-623B | Mouse; monoclonal | 2 μg per ChIP reaction | AB_309852 |
| H3K27Ac | NA | Abcam, ab4729 | Rabbit; polyclonal | 2 μg per ChIP reaction | AB_2118291 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| AR (N-20) | NA | sc-816X | Santa Cruz Biotechnology | Rabbit; polyclonal | 3 μg per ChIP reaction | AB_1563391 |
| RNA polymerase II | NA | Anti-RNA polymerase II, clone CTD4H8 | Millipore, 05-623B | Mouse; monoclonal | 2 μg per ChIP reaction | AB_309852 |
| H3K27Ac | NA | Abcam, ab4729 | Rabbit; polyclonal | 2 μg per ChIP reaction | AB_2118291 |
Abbreviations: NA, not available; RRID, Research Resource Identifier.
Antibody table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| AR (N-20) | NA | sc-816X | Santa Cruz Biotechnology | Rabbit; polyclonal | 3 μg per ChIP reaction | AB_1563391 |
| RNA polymerase II | NA | Anti-RNA polymerase II, clone CTD4H8 | Millipore, 05-623B | Mouse; monoclonal | 2 μg per ChIP reaction | AB_309852 |
| H3K27Ac | NA | Abcam, ab4729 | Rabbit; polyclonal | 2 μg per ChIP reaction | AB_2118291 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| AR (N-20) | NA | sc-816X | Santa Cruz Biotechnology | Rabbit; polyclonal | 3 μg per ChIP reaction | AB_1563391 |
| RNA polymerase II | NA | Anti-RNA polymerase II, clone CTD4H8 | Millipore, 05-623B | Mouse; monoclonal | 2 μg per ChIP reaction | AB_309852 |
| H3K27Ac | NA | Abcam, ab4729 | Rabbit; polyclonal | 2 μg per ChIP reaction | AB_2118291 |
Abbreviations: NA, not available; RRID, Research Resource Identifier.
Clinical data and statistical analyses
SRD5A1 and SRD5A2 expression data from clinical specimens were retrieved and analyzed from publicly available microarrays on the Gene Expression Omnibus profile (GDS1439 and GDS2545) (18–20) and from the Cancer Genome Atlas cbioportal (21, 22) for the data by Taylor et al. (23). For quantitative reverse transcriptase–PCR and ChIP-qPCR data, Student t test was used to assess statistical significance.
Results
SRD5A1 and SRD5A2 expression during PCa progression
We first analyzed three publicly available clinical data sets to investigate the inverse relationship between SRD5A messenger RNA (mRNA) expression and PCa progression. The first data set, from Taylor et al. (23), showed an increase in SRD5A1 expression, whereas SRD5A2 was decreased in tumor samples [Fig. 1(a)]. We also had access to two data sets with primary and metastatic tumor samples. In both cohorts, SRD5A1 mRNA expression was significantly increased in localized PCa compared with nontumoral tissues [Fig. 1(b) and 1(c)]. Moreover, its expression was significantly higher in metastatic samples compared with nontumoral tissues in the two data sets [Fig. 1(b) and 1(c)]. On the other hand, SRD5A2 mRNA expression was significantly lower in localized and metastatic tumor samples compared with peritumoral prostate tissue in data from Chandran et al. (19) and Yu et al. (20) [Fig. 1(b)]. In the study by Varambally et al. (18), SRD5A2 expression in metastatic samples was significantly lower compared with both peritumoral and localized PCa tumor tissues [Fig. 1(c)]. These results thus validate the existence of a shift in SRD5A1 and SRD5A2 isoform expression favoring type 1 5α-reductase during PCa progression at the mRNA level, suggesting that the mechanism underlying their differential expression involves transcriptional regulation.
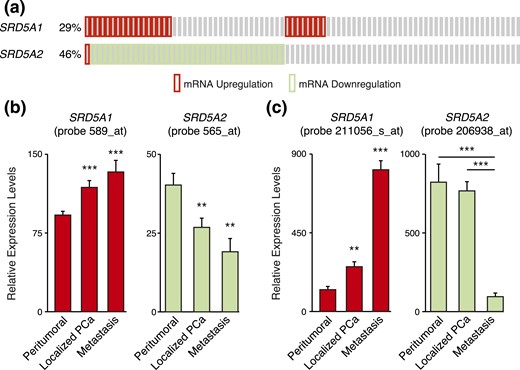
Differential expression of SRD5A1 and SRD5A2 during PCa progression. (a) Change in SRD5A1 and SRD5A2 expression levels in PCa tumors from data obtained from Taylor et al. (23). (b) Relative expression levels of SRD5A1 and SRD5A2 in normal adjacent prostate tissue, prostate tumor tissue, and metastatic PCa from data obtained by Chandran et al. (19). (c) Relative expression levels of SRD5A1 and SRD5A2 in benign prostate tissue, clinically localized PCa, and metastatic PCa from data obtained by Varambally et al. (18). *P < 0.05, **P < 0.01, and ***P < 0.001 by Student t test.
Androgens modulate the expression of both SRD5A1 and SRD5A2 in PCa cells
As androgen signaling is known to be central to PCa cell activity, we investigated the relationship between AR activation and 5α-reductase expression. As expected, the synthetic androgen R1881 induced the expression of KLK3 (encoding prostate-specific antigen) in the human PCa cell line LNCaP [Fig. 2(a), right panel]. Similarly, AR activation induced SRD5A1 expression by twofold [Fig. 2(a), left panel]. On the contrary, R1881 significantly repressed SRD5A2 expression by more than twofold [Fig. 2(a), left panel]. This transcriptional regulation was stable through time, with R1881 regulation of both 5α-reductases lasting at least up to 3 days [Fig. 2(b)].
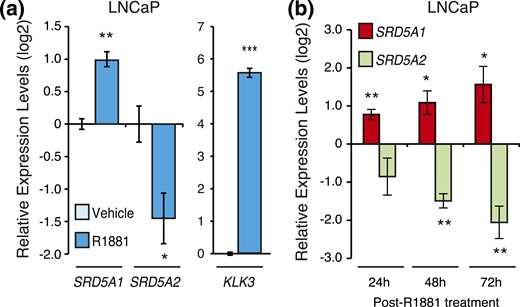
Androgens regulate SRD5A gene expression in LNCaP cells. (a) Relative mRNA expression levels of SRD5A1 and SRD5A2 genes in LNCaP cells following a 24-hour treatment with R1881 or vehicle. KLK3 is shown as a positive control. Control cells treated with vehicle were set at 0. Results are shown as the mean ± SEM of three independent experiments performed at least in duplicate. (b) Transcript levels of SRD5A1 and SRD5A2 following R1881 or vehicle treatment in LNCaP cells. *P < 0.05, **P < 0.01, and ***P < 0.001.
We then validated these results in two other human androgen-responsive PCa cell lines, LAPC4 and 22rv1. SRD5A2 expression was undetectable in LAPC4 cells. However, SRD5A1 expression was detectable and, as in LNCaP cells, its expression was significantly induced by androgens [Fig. 3(a)]. Similarly, SRD5A1 was significantly induced by R1881 treatment in 22rv1 cells, whereas SRD5A2, with expression levels 15 times lower than that of SRD5A1, did not vary following R1881 treatment [Fig. 3(b)]. Globally, these results show that SRD5A1 was positively modulated by androgens across several PCa cell lines and that SRD5A2 was repressed by androgens in LNCaP cells, the only cell line constitutively expressing SRD5A2.
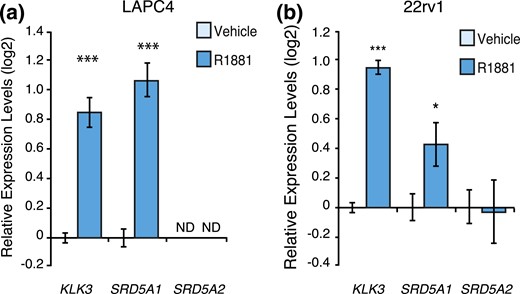
AR activation regulates SRD5A1 expression in 22rv1 and LAPC4 cells. Relative mRNA expression levels of KLK3 and SRD5A1 in LAPC4 cells (a) and KLK3, SRD5A1, and SRD5A2 in 22rv1 (b) cells following a 24-hour treatment with R1881 or vehicle. Control cells treated with vehicle were set at 0. Results are shown as the mean ± SEM of three independent experiments performed at least in duplicate. *P < 0.05 and ***P < 0.001. ND, expression not detected.
AR is recruited to both SRD5A1 and SRD5A2 in response to R1881
By reanalyzing a publicly available ChIP-sequencing peak list for AR binding to chromatin (24), we identified AR binding sites in both SRD5A genes in LNCaP cells. ChIP-qPCR experiments confirmed AR binding to the first intron of SRD5A1 following R1881 treatment in LNCaP, LAPC4, and 22rv1 cells [Fig. 4(a)], corroborating androgen-mediated positive regulation of SRD5A1 in these cells (Figs. 2 and 3). Similarly, we identified and validated AR recruitment at the SRD5A2 intron 1 in LNCaP and 22rv1 cells [Fig. 4(b)]. A second AR binding site was also identified in the last intron of SRD5A2, with recruitment of AR following R1881 treatment only detected in LNCaP cells [Fig. 4(b)]. No recruitment of AR was detected at SRD5A2 in LAPC4 cells.
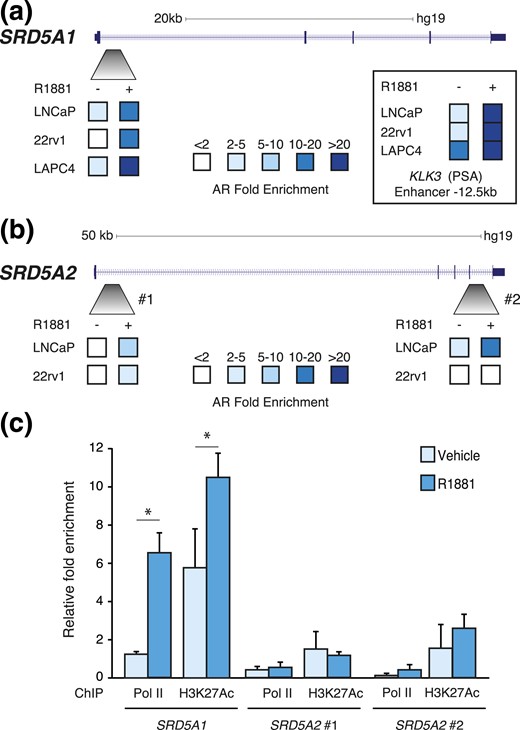
SRD5A1 and SRD5A2 are direct AR target genes in PCa cells. ChIP-qPCR analysis of AR binding following treatment with R1881 at one genomic region in SRD5A1 (a) and two genomic regions in SRD5A2 (b) in LNCaP, 22rv1, and LAPC4 cells (n = 3). Inset: AR recruitment at the −12.5-kb enhancer of KLK3. (c) Relative enrichment RNA polymerase II (pol II) and H3K27Ac at the AR binding sites identified in SRD5A1 and SRD5A2 in LNCaP cells. *P < 0.05.
Importantly, treatment with R1881 induced recruitment of RNA polymerase II (pol II) only at the AR binding site located in SRD5A1 [Fig. 4(c)], correlating with the increased expression of this gene under androgen stimulation (Figs. 2 and 3). The active promoter/enhancer mark H3K27ac was also increased under androgen stimulation, in a specific manner at SRD5A1. However, no recruitment of pol II or active enhancer mark enrichment was detectable at the 2 AR binding sites in SRD5A2 [Fig. 4(c)].
Antiandrogen treatment blocks AR-mediated transcriptional regulation of SRD5A1 and SRD5A2
To further evaluate the transcriptional regulation of SRD5A genes, we proceeded with genetic and pharmacological inactivation of AR in LNCaP cells. Knockdown of AR using RNAi completely blocked the induction of SRD5A1 by R1881 treatment [Fig. 5(a)]. In addition, treatment with the antiandrogen bicalutamide (Casodex) also blocked the modulation of SRD5A1 by androgens [Fig. 5(b)]. SRD5A2, strongly repressed by R1881 treatment, was significantly induced following transfection with a pool of siRNAs targeting AR [Fig. 6(a)]. Moreover, in the absence of functional AR, R1881 treatment did not repress SRD5A2 expression [Fig. 6(a)]. Surprisingly, bicalutamide treatment, like R1881 treatment, diminished SRD5A2 expression [Fig. 6(b)]. Bicalutamide is a first-generation AR inhibitor and does not block AR nuclear relocalization and its DNA binding, but it promotes blockade of PCa cell proliferation in vitro, similar to the absence of androgen stimulation (25, 26). It is known to block AR-mediated transcriptional activation of several target genes, such as KLK3, but its effect on AR-mediated transcriptional repression has been much less studied. To overcome this issue, we used the second-generation antiandrogen enzalutamide that blocks PCa cell proliferation by impairing AR nuclear translocation, as well as its DNA and cofactor binding, and by inducing AR degradation (26). In both the absence and presence of R1881, enzalutamide treatment induced SRD5A2 expression, therefore blocking androgen-mediated repression of this gene [Fig. 6(c)]. Taken together, our data show that AR is directly recruited to both SRD5A1 and SRD5A2 genomic loci and that AR activity is required for the androgen-mediated transcriptional regulation of these genes.
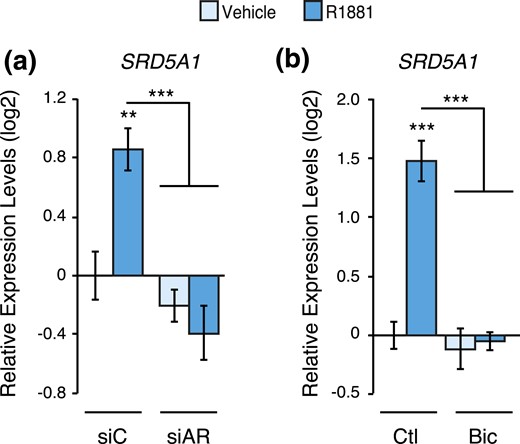
Genetic and pharmacological inhibition of AR blocks SRD5A1 gene modulation by androgens. Relative mRNA expression levels of SRD5A1 in LNCaP cells following a 48-hour treatment with R1881 or vehicle following AR knockdown by siRNAs (a) or inhibition by bicalutamide (b). Results are shown as the mean ± SEM of two independent experiments performed in triplicate. Control cells treated with vehicle were set at 0. **P < 0.01 and ***P < 0.001.
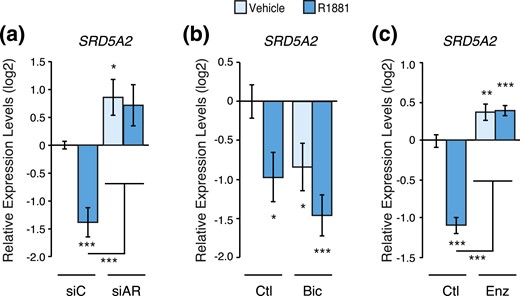
Genetic and pharmacological inhibition of AR blocks SRD5A2 gene modulation by androgens. Relative mRNA expression levels of SRD5A2 in LNCaP cells following a 48-hour treatment with R1881 or vehicle following AR knockdown by siRNAs (a) or inhibition by bicalutamide (b) or enzalutamide (c). Results are shown as the mean ± SEM of two independent experiments performed in triplicate. Control cells treated with vehicle were set at 0. *P < 0.05, **P < 0.01, and ***P < 0.001. Bic, bicalutamide; Ctl, control; Enz, enzalutamide; siAR, siRNA pool targeting AR; siC, control siRNA.
Discussion
The inverse correlation between SRD5A1 and SRD5A2 expression during PCa progression has been convincingly demonstrated in the past few years; however, this was done without identifying the underlying mechanism (3, 4, 12–14). In the current study, we identified a transcriptional switch that can explain why SRD5A1 expression is increased, whereas SRD5A2 expression is decreased during PCa progression. Indeed, SRD5A1 gene expression was induced in all PCa cell lines tested following androgen stimulation, correlating with the sustained AR signaling signature observed in PCa (27). On the contrary, we also noticed that in LNCaP cells, AR activation could repress SRD5A2 gene expression. Moreover, we showed that modulation of AR activity with siRNA or pharmacological treatments impairs this SRD5A1 and SRD5A2 gene regulation by the androgen signaling pathway.
In LNCaP cells, AR binds at two distinct loci at the SRD5A2 gene, and its expression is significantly decreased upon R1881 exposure. In cell lines with a very low level of SRD5A2 expression, LAPC4, and 22rv1 cells, the absence of recruitment of AR to the SRD5A2 gene indicates that the silencing of this genes is already set via other mechanisms. SRD5A2 expression was below our limit of detection in LAPC4 cells; although no major deletion at the SRD5A2 locus was reported in this cell line (28), we cannot rule out the existence of a small deletion impairing gene expression as reported in rare PCa cases (21, 22). It remains to be determined whether AR is directly involved in silencing SRD5A2 expression in actual tumors as it does in LNCaP cells. In addition, whether bicalutamide represses SRD5A2 expression in patients has yet to be shown. Further repression of SRD5A2 by bicalutamide, in addition to a blockade of the androgen-mediated induction of SRD5A1 expression, could potentially contribute to its beneficial effects for the management of PCa in patients by blocking the expression of the two enzymes that can synthesize DHT.
The fact that androgens and their receptor can induce SRD5A1 in prostate tumor cells identifies a positive feedback loop important for understanding the course of PCa (Fig. 7). These cancer cells are known to express the major enzymes required to produce androgens from adrenal precursors (3, 4). Now it appears that, once synthesized locally in tumor cells, androgens can trigger their own synthesis and therefore activate a feedforward positive loop to maintain chronic activation of AR in PCa cells. The use of 5α-reductase inhibitors, finasteride and dutasteride, in the clinic has been showed to decrease PCa incidence (29, 30). Although finasteride only inhibits type 2, dutasteride inhibits both types 1 and 2, but both compounds have a comparable effect in the setting of PCa prevention, at a stage where type 2 is the predominant isoform in the prostate. In advanced PCa cases, where 5α-reductase type 1 is the predominant isoform (3, 4, 12–14), combining antiandrogens or androgen deprivation therapy with a dual 5α-reductase inhibitor such as dutasteride may enhance clinical response, as shown by stronger DHT level suppression in tumor samples (31).
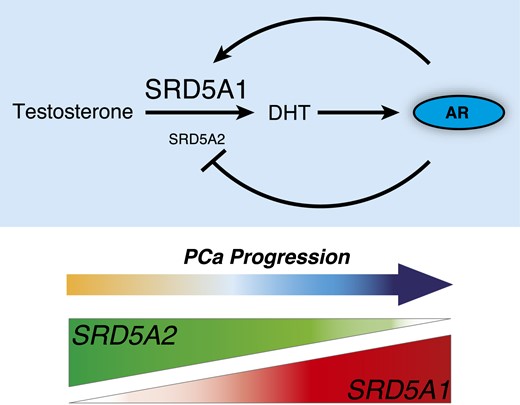
AR transcriptional control of SRD5A1 induces a positive feedback loop activating the androgen signaling pathway in prostate cancer. AR controls a transcriptional regulatory switch that promotes SRD5A1 over SRD5A2 in prostate cancer cells. In return, overexpression of SRD5A1 will promote DHT synthesis and, therefore, AR hyperactivation.
Four major UDP-glucuronosyltransferase (UGT) isoforms are primarily responsible for the inactivation of androgenic sex steroids—namely, UGT2B7, UGT2B15, UGT2B17, and UGT2B28, which have been linked with PCa progression (5, 32–35). Interestingly, AR is also known to target and suppress the expression of UGTs. With the results shown in the current study, it appears that AR activation leads to sustained DHT synthesis (SRD5A1 induction) in parallel with a decrease in DHT inactivation (repression of UGT enzymes).
The current study identified a mechanism involving an AR-mediated transcriptional switch between SRD5A1 and SRD5A2 in PCa cells that explains a clinical observation made several years ago. This study also highlights a potential mechanism of action of antiandrogens: in blocking AR function, 5α-reductase expression is impaired, further decreasing the activity of the intratumoral androgen signaling pathway. The results presented herein characterize, to our knowledge, a novel mechanism by which androgens can control their own synthesis and activity in PCa cells.
Abbreviations:
- AR
androgen receptor
- ChIP
chromatin immunoprecipitation
- DHT
dihydrotestosterone
- mRNA
messenger RNA
- PCa
prostate cancer
- qPCR
quantitative polymerase chain reaction
- siRNA
small interfering RNA
- UGT
UDP-glucuronosyltransferase.
Acknowledgments
The authors thank Chloé Laflamme and Catherine Rosa Dufour for critical reading of the manuscript.
This work was supported by a Terry Fox Research Institute Program Project Team Grant on Oncometabolism (116128, V.G.) and the Canadian Institutes of Health Research (CIHR) (MOP-111144, V.G.). E.A.-W. is recipient of a postdoctoral fellowship from CIHR, the Fonds de Recherche du Québec–Santé (FRQS), and supported by a McGill Integrated Cancer Research Training Program (MICRTP) scholarship. T.Y. was a recipient of summer MICRTP studentship. I.S.T. was supported a Micheal D’Avirro studentship.
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: Vincent Giguère, PhD, Goodman Cancer Research Centre, McGill University, 1060 Pine Avenue West, Montréal, Quebec H3A 1A3, Canada. E-mail: [email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}