





Abstract
Biased signaling describes the selective activation of signal transduction cascades by structurally related ligands downstream of shared G protein–coupled receptors (GPCRs). Although class I phosphoinositide 3-kinases (PI3Ks) are important components of GPCR-controlled transduction networks, little is known regarding the potential for biased regulation of class I PI3K-dependent signaling. The full complement of class I PI3K catalytic subunits (p110α, p110β, p110δ, and p110γ) first appears in bony fishes and, despite being associated with distinct cellular functions, all class I PI3Ks produce the lipid second-messenger phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3]. We have previously shown that 2 endogenous gonadotropin-releasing hormones (GnRH2 and GnRH3), which both signal through shared Gαq/11-coupled receptors, selectively activate different subsets of class I PI3K isoforms in their control of hormone release from goldfish (Carassius auratus) pituitary cells. Here, we tested the hypothesis that the biased activation of class I PI3K isoforms results in the selective recruitment of PtdIns(3,4,5)P3-sensitive effectors downstream of GnRH-stabilized GPCRs using pharmacological mapping. Our results reveal that distinct PtdIns(3,4,5)P3-sensitive effectors are involved in the differential control of GnRH2- and GnRH3-stimulated, as well as basal, hormone release and implicate the participation of noncanonical PtdIns(3,4,5)P3-sensitive transduction elements. Furthermore, observations using a selective inhibitor of the shared Gβγ-effector interaction surface indicate a role for Gβγ-dependent signaling in the integrated control of pituitary hormone exocytosis. These findings add to our understanding of functional selectivity in GPCR signal transduction networks, in general, and reveal the complexity of biased signaling downstream of class I PI3K catalytic activity.
Phosphoinositide 3-kinases (PI3Ks) are part of the intricate network of lipid-modifying enzymes that are universally important for the maintenance of phospholipid metabolism and membrane dynamics (1). In general, PI3K-dependent functions are mediated by the production of phosphoinositides, the low-abundance forms of phosphatidylinositol (PtdIns), phosphorylated at the 3-position hydroxyl group of the inositol ring (1). Eight isoforms of PI3K superfamily lipid kinases have been identified in vertebrates and are grouped into 3 functionally discrete classes on the basis of structural features and substrate preferences (2, 3). Among the PI3Ks, the class I PI3Ks are the best characterized and are also reliably activated downstream of cell-surface receptors, including receptor tyrosine kinases and G protein–coupled receptors (GPCRs) (4). Interestingly, GPCR-dependent activation of the class I PI3K catalytic isoforms p110β and p110γ occurs through direct interactions with Gβγ heterodimers (5, 6); therefore, in many systems, class I PI3K-dependent signaling forms an integral component of Gα-independent transduction pathways downstream of GPCR activation.
Growing evidence suggests that class I PI3K isoforms control distinct intracellular functions, although all 4 isoforms of class I PI3K produce the lipid second-messenger phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3] (1). Binding of PtdIns(3,4,5)P3 to specific lipid-binding domains facilitates membrane recruitment and, in some cases, activation of numerous intracellular effectors, including serine/threonine protein kinases, nonreceptor tyrosine kinases, adaptor proteins, guanine nucleotide exchange factors, guanosine triphosphatase–activating proteins, as well as direct interactions with plasma membrane transporters and ion channels (4). Studies in excitable cell types have identified phosphoinositide-dependent kinase 1 (PDK1), protein kinase B (Akt), and Bruton’s tyrosine kinase (BTK) as canonical intracellular effectors that contribute to the control of class I PI3K-dependent signaling through direct interactions between PtdIns(3,4,5)P3 and highly conserved pleckstrin homology (PH) lipid-binding domains. In general, PH domains are found in approximately 250 human proteins and directly bind phosphoinositides, although only a small fraction (∼10%) of PH domains actually exhibit high affinity and selectivity for a specific PtdIns species (7, 8). Interestingly, comprehensive analysis of PtdIns(3,4,5)P3-regulated proteins across species has shown that the number of PtdIns(3,4,5)P3-sensitive PH domain–containing proteins has increased from approximately 2 in Caenorhabditis elegans and 4 in Drosophila melanogaster to roughly 40 in vertebrates, including Danio rerio (zebrafish) (9). This coincides with the metazoan expansion of the class I PI3K catalytic subunits from a single ancestral PIK3CA-like ortholog within invertebrates to the modern complement of PIK3CA, PIK3CB, PIK3CD, and PIK3CG homologs found in true vertebrate lineages (10). Thus, it appears that the full complexity of class I PI3K-dependent signaling was first established in basal vertebrates and that PtdIns(3,4,5)P3-sensitive PH domains are evolutionarily conserved regulators of class I PI3K signaling functions.
Few studies have examined the involvement of PI3K-dependent signaling in the control of pituitary hormone release from untransformed cells by neuroendocrine factors, in general. Our previous work examined the role of PI3Ks during gonadotropin-releasing hormones (GnRH)–induced luteinizing hormone (LH) and growth hormone (GH) release using the goldfish (Carassius auratus) pituitary cell model (11–13). Briefly, in goldfish, 2 endogenous GnRH isoforms (GnRH2 and GnRH3) are expressed in the brain (14, 15), and both GnRHs directly stimulate the release of LH from gonadotropes and GH release from somatotropes (16, 17). Interestingly, studies using primary pituitary cells and membrane preparations isolated from the goldfish pituitary demonstrate that GnRH2 and GnRH3 bind and activate the same population of cell-surface receptors (18–20) that belong to the class A subfamily of GPCRs (21). However, although both of these GnRH isoforms share common GnRH receptors (GnRHRs), we recently demonstrated GnRH-selective utilization of the class I PI3K catalytic isoforms p110β, p110γ, and p110δ during the differential control of LH and GH release (13). These findings suggest that GnRH2 and GnRH3 binding to GnRHRs can bias the activation of class I PI3K-dependent signaling to mediate hormone release responses. The involvement of both class IA (p110β and p110δ) and IB (p110γ) PI3Ks implicates Gβγ subunits, as well as other known regulators of class I PI3Ks, as important components of GnRHR-mediated responses that could also influence GnRH-selective signaling in other cell types. In particular, the typical affinities measured for PtdIns(3,4,5)P3-PH domain interactions are in the nanomolar range, and PtdIns(3,4,5)P3 binding is a universal step required for the initiation of class I PI3K-dependent signal transduction (22). Thus, it is reasonable to hypothesize that ligand-selective activation of PtdIns(3,4,5)P3-dependent effectors contributes to the agonist-specific control of hormone release responses by GnRH2 and GnRH3. However, whether and how PtdIns(3,4,5)P3-sensitive downstream signaling components are required for GnRH actions on pituitary hormone release in goldfish, as well as in other animal study models, is not known.
In this study, we investigated whether PtdIns(3,4,5)P3-sensitive signaling effectors differentially participate in the control of GnRH2- and GnRH3-stimulated hormone release from primary cultures of dispersed goldfish pituitary cells. To do this, we performed pharmacological investigations of the possible roles played by PtdIns(3,4,5)P3-dependent signal transduction effectors in the control of basal and GnRH-induced LH and GH secretion in cell column perifusion experiments. Specifically, perturbation of signal transduction downstream of PtdIns(3,4,5)P3 production was achieved using a small molecule antagonist of PtdIns(3,4,5)P3-PH domain interactions in general [PITenin-1 (PIT-1)], as well as through the use of allosteric or adenosine triphosphate (ATP)–competitive inhibitors of the classical PtdIns(3,4,5)P3-dependent signaling effectors PDK1, Akt, and BTK [GSK2334470, Akt inhibitor VIII (Akti VIII), and CGI1746, respectively]. Unlike direct inhibition of class I PI3K catalytic activity, targeting PtdIns(3,4,5)P3-PH domain interactions represents a unique inhibition strategy that allows for the investigation of the noncatalytic roles played by PtdIns(3,4,5)P3-dependent signaling effectors (23, 24). Consequently, in addition to insights regarding the regulation of PDK1-, Akt-, and BTK-dependent signaling, results from this study also implicate PtdIns(3,4,5)P3-sensitive effectors in the control of hormone release responses independently of canonical class I PI3K transduction targets. Furthermore, although it has been speculated that GnRH actions involve Gβγ heterodimers (25), few studies have directly examined the involvement of Gβγ-dependent signal transduction downstream of GnRHR activation. To determine if Gβγ heterodimers, the upstream regulators of the class I PI3K catalytic isoforms p110β and p110γ, contributed to GnRH-dependent signal transduction, GnRH-stimulated hormone release was monitored in the presence of a selective inhibitor of Gβγ-effector interactions (NSC8668) (26–28).
Materials and Methods
Animals
Animal maintenance and handling protocols adhered to the Canadian Council for Animal Care guidelines and were approved by the Biosciences Animal Care and Use Committee of the Research Ethics Office at the University of Alberta. Common goldfish (Carassius auratus; postpubertal, 10–13 cm body length, 25–30 g body weight) were purchased from Aquatic Imports (Calgary, AB, Canada) and maintained at 18°C in flow-through aquaria (1800-L capacity; dechlorinated and charcoal-filtered municipal water). Within each aquarium, male and female goldfish were held together. Goldfish were cared for and fed to satiation with a commercial fish food during a scheduled morning feeding period by staff of the Faculty of Science Animal Support Services. All aquaria were housed under a simulated Edmonton, AB, Canada photoperiod (coordinates 53°32′N and 113°30′W) controlled by graded lighting and an astronomical clock set to the local sunrise and sunset. Goldfish were acclimated to these conditions for a minimum of 7 days prior to experimentation and were used within 1 month of purchase. Experiments were performed using mixed pituitary cell populations isolated from both male and female goldfish.
In temperate climates, goldfish are seasonal spawners with a tightly regulated reproduction cycle. Generally, spawning occurs between April and early June, after which the gonads undergo regression through the summer and fall. Gonadal recrudescence commences in midfall (around October/November), and a sexually matured (prespawning) stage is achieved by late March or April. Circulating LH levels are highest at the prespawning and spawning stages and lowest during late fall just prior to gonadal recrudescence (29, 30). On the other hand, serum GH levels are high in the early spring, coinciding with gonadal maturation, and remain elevated during the summer months when somatic growth is at its greatest. GH levels then decrease to low levels over the fall and early winter, when body growth slows, before increasing again late in the winter months as the fish approach mid to late recrudescence (31). During this period, GH plays an important role in potentiating the gonadal steroidogenic response to LH (32). However, despite these complex seasonal changes, GnRH stimulation of LH and GH release can be demonstrated at all times of the seasonal reproductive cycle, although the magnitude of the response may vary.
To minimize any variation due to seasonal reproductive changes, replicate experiments in most hormone release studies were performed within a short time period (8 to 14 days). However, different experiments were performed throughout the entire reproductive cycle, and testing of the involvement of a signaling pathway was carried out in more than 1 seasonal reproductive stage. Thus, the results from replicate experiments covering more than 1 gonadal maturational stage are generally consistent and were pooled for data presentation and analysis. In all instances, to facilitate future comparisons, the time of year and gonadal stage of the goldfish when individual experiments were performed are provided in the figure legends.
Preparation of dispersed pituitary cell cultures
Male and female goldfish were euthanized by cervical transection following deep anesthesia with 0.05% tricaine methanesulfonate (AquaLife; Syndel Laboratories, Vancouver, BC, Canada). Pituitaries were excised and collected in dispersion medium [Medium M199 with Hank’s salts (Gibco/Life Technologies, Grand Island, NY), supplemented with 26 mM NaHCO3, 25 mM HEPES, 100 mg/L streptomycin, 100,000 U/L penicillin, and 0.3% bovine serum albumin; pH adjusted to 7.2 with NaOH and sterilized by filtration through 0.22-μm filter units]. For preparing dispersed cells, pituitary fragments were processed using an established trypsin/DNAse dispersion protocol (33). Prior to plating, cell yield and viability were determined using trypan blue exclusion. Viability was routinely > 98%. Dispersed cells were resuspended and cultured overnight in plating medium [Medium M199 with Earles’ salts (Gibco/Life Technologies) containing 26 mM NaHCO3, 25 mM HEPES, 100 mg/L streptomycin, 100,000 U/L penicillin, and 1% horse serum; pH adjusted to 7.2 with NaOH and sterilized by filtration through 0.22-μm filter units] at 28°C, under 5% CO2 and saturated humidity. Dispersed pituitary cell cultures prepared in this manner lack residual hypothalamic neuronal terminals that have been shown to be present in pituitary fragments and thus allow for studies on the direct actions of neuroendocrine effectors on pituitary cell functions (33).
Drugs and reagents
GnRH isoforms
Synthetic GnRH2 [chicken GnRH-II; (His5, Trp7, Tyr8) GnRH] and GnRH3 [salmon GnRH; (Trp7, Leu8) GnRH] were purchased from Bachem (San Carlos, CA), and stock solutions were prepared in distilled, deionized water. Aliquots of stock solutions were kept frozen at –20°C until used. For all experiments, equimolar and maximally stimulatory concentrations of GnRH2 and GnRH3 (100 nM) (33) were used. Final concentrations used for experiments were obtained by diluting stock solutions in testing medium [Medium 199 with Hank’s salts (Gibco/Life Technologies) containing 26 mM NaHCO3, 25 mM HEPES, 100 mg/L streptomycin, 100,000 U/L penicillin, and 0.1% bovine serum albumin; pH adjusted to 7.2 with NaOH and sterilized by filtration through 0.22-μm filter units.
Small molecule inhibitors
In general, aliquots of each pharmacological modulator were prepared in dimethyl sulfoxide and stored at –20°C. Final drug concentrations used for experiments were achieved by diluting stock solutions (typically 1000-fold) in testing medium. When required, the concentrations of the organic solvent were always less than 0.1% and did not affect basal LH or GH release (34, 35).
Akti VIII [1,3-dihydro-1-(1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one] was purchased from Calbiochem (Billerica, MA). NSC8668 (Gβγ-binding inhibitor; also commonly called Gallein; 3′,4',5′,6'-tetrahydroxyspiro[isobenzofuran-1(3H),9'-(9H)xanthen]-3-one), and GSK2334470 [PDK1 inhibitor; (3S,6R)-1-[6-(3-amino-1H-indazol-6-yl)-2-(methylamino)-4-pyrimidinyl]-N-cyclohexyl-6-methyl-3-piperidinecarboxamide] were purchased from Tocris Biosciences (Bristol, United Kingdom). PIT-1 [PtdIns(3,4,5)P3-sensitive PH domain inhibitor; N-[[(3- chloro- 2- hydroxy- 5- nitrophenyl)amino]thioxomethyl]-benzamide] was purchased from Cayman Chemical (Ann Arbor, MI). CGI1746 [BTK inhibitor; 4-tert-butyl-N-(2-methyl-3-(4-methyl-6-(4-(morpholine-4-carbonyl)phenylamino)-5-oxo-4,5-dihydropyrazin-2-yl)phenyl)benzamide] was purchased from Axon Medchem (Reston, VA).
Rationale for the selection of compounds for pharmacological mapping
Detailed computational modeling and high-throughput screening identified a series of structurally related compounds that differentially modulate protein-protein interactions between Gβγ subunits and their effectors, demonstrating a unique approach for targeting GPCR signaling (26–28). In particular, the small molecule NSC8668 binds to a protein interaction “hot spot” on the top surface of the Gβ subunit that is normally contacted by the switch II region of the Gα subunit and, following dissociation, this hydrophobic surface facilitates binding of Gβγ with various downstream cellular effectors (36, 37). Thus, by directly interfering with crucial contacts between Gβγ subunits and effectors, NSC8668 can selectively inhibit signal transduction targets downstream of Gβγ dissociation from agonist-bound GPCRs. Importantly, sequence alignments show that the Gβγ interaction hot spot, consisting primarily of Lys57, Tyr59, Trp99, Val100, Met101, Leu117, Tyr145, Asp186, Met188, Asp228, Asn230, Asp246, and Trp332 within the Gβ1 subunit (28, 36, 37), are 100% conserved in basal vertebrates. Just as importantly, in silico modeling of the zebrafish Gβ1γ2 heterodimer shows that the predicted folding of the zebrafish Gβγ heterodimer should expose the hot spot in a manner that is identical to mammalian homologs (Supplemental Fig. 1).
Signal transduction responses downstream of class I PI3Ks involve the membrane translocation and activation of a diverse number of distinct PtdIns(3,4,5)P3-sensitive effectors. PIT-1 is a nonphosphoinositide antagonist of PtdIns(3,4,5)P3-PH domain interactions that was identified as part of a screen of ∼50,000 small molecules using a PtdIns(3,4,5)P3-Akt PH domain binding assay (38). PIT-1 was subsequently shown to selectively disrupt binding of PtdIns(3,4,5)P3 to the PH domains of Akt, PDK1, general receptor for phosphoinositides 1 (GRP1), and adenosine 5′-diphosphate–ribosylation factor (Arf) nucleotide-binding-site opener (ARNO) (38). Importantly, PITs failed to affect membrane translocation of PtdIns(4,5)P2- or PtdIns(3,4)P2-specific PH domain–containing effectors, including the PtdIns(4,5)P2-sensitive GPCR effector phospholipase C (PLC), and treatment with PIT-1 did not alter cellular levels of PtdIns(3,4,5)P3 (38). Taken together, these studies identify PIT-1 as a unique and powerful tool for examining the cellular actions of PtdIns(3,4,5)P3-selective PH domain–containing proteins. Furthermore, because of the high degree of conservation present in functionally important protein superfolds like PH domains, including the PtdIns(3,4,5)P3-coordinating residues identified in mammals and those found within the teleost homologs of GRP1, ARNO, PDK1, Akt, and BTK (Supplemental Fig. 2), selective targeting of protein-lipid interaction domains by PIT-1 is likely to be an especially useful tool for studies with nonclassical model organisms like the goldfish. On the other hand, PIT-1 does not inhibit binding of PtdIns(3,4,5)P3 to the PtdIns(3,4,5)P3-specific PH domain of BTK, suggesting that PIT-1 exhibits selectivity toward a subset of PtdIns(3,4,5)P3-selective PH domains (38).
The highly selective ATP-competitive inhibitor GSK2334470 was used to examine the role of PDK1-mediated signaling. This PDK1-selective compound has been shown to have little or no effect on the activity of 95 protein kinases and 15 lipid kinases assayed in cell-free systems at concentrations 100-fold greater than the reported half-maximal inhibitory concentration (IC50) (39). However, coordinate recruitment of Akt to PtdIns(3,4,5)P3 within the plasma membrane promotes intermolecular interactions between PDK1 and Akt on 2-dimensional membrane surfaces. Consequently, it is thought that only a small fraction of endogenous PDK1 is required for efficient and robust activation of Akt. As a result, Akt activation is often much less sensitive to inhibition using this PDK1 inhibitor when compared with the activation of other PDK1 substrates that lack a PtdIns(3,4,5)P3-binding PH domain, such as ribosomal protein s6 kinase (S6K), serine/threonine-protein kinase (SGK), or ribosomal s6 kinase (RSK) (39, 40). To characterize the direct involvement of Akt, the allosteric Akt inhibitor Akti VIII was used (41). Given that Akti VIII acts by binding to a unique PH domain–kinase domain interface, it is extremely specific for Akt isozymes over other AGC kinases and has been reported to show minimal inhibitory effects on the catalytic activity of a representative panel of 70 protein kinases (42, 43). Last, the involvement of BTK catalytic activity in the control of GnRH actions on hormone release was examined using the novel ATP-dependent inhibitor of BTK called CGI1746 (44). This compound has been reported to be extremely potent (IC50 = 1.9 nM), and highly selective for BTK, with more than 1000-fold selectivity over similar Tec and Src family tyrosine kinases, 205 other protein kinases, as well as negligible off-target activity against 82 nonkinase targets at 1 μM (44). Although the specificity of GSK2334470, Akti VIII, and CGI1746 in fish has not been directly examined, it is very likely that these drugs exert similar inhibitory activities and selectivity in teleosts as observed in mammals on the basis of the in silico structural information available for their respective targets. First, for the PDK1 inhibitor GSK2334470, its binding mode involves distinct pockets within the PDK1 G-loop, and a hydrogen bond network involving Lys111, Glu130, and Thr222 at the floor of the binding pocket, as well as with Ser160 and Ala162 in the hinge region (45). These coordinating residues, as well as important residues forming the bottom of the lipophilic pocket at the base of the G-loop (Met134, Val143, and Leu159) (46), are all conserved across vertebrate homologs of PDK1. Second, for Akti VIII, its specificity and inhibitory actions are dependent on its interaction with Trp80 within the kinase cleft (43, 46). Importantly, the Akti VIII–coordinating residue Trp80 and the Trp80 interacting residues within the c-terminal hydrophobic motif (Phe469 and Phe472) are conserved in all vertebrate homologs of Akt1, Akt2, and Akt3 (47). Last, CGI1746 is known to stabilize BTK in its inactive conformation by sequestering Tyr551 (44). Phosphorylated residues Tyr551 and Tyr223 are both conserved in zebrafish homologs of BTK, as are CGI1746-coordinating residues found within the H3 pocket of the SH3 domain, including Phe413, Asp521, Asn526, Leu542, and Val546 (44). CGI1746 also interacts with the hinge region of BTK that is located between the N- and C-lobes of the kinase domain (formed by Thr474, Glu475, and Met477), as well as through polar interactions with the catalytic lysine (Lys430) (44). These coordinating residues outside of the H3 pocket are also present in BTK sequences from Xenopus laevis and zebrafish.
As predicted from mammalian cell systems, acute treatment with PIT-1, GSK2334470, or Akti VIII attenuated basal phosphorylation of Akt (Ser473) within mixed goldfish pituitary cell extracts in preliminary studies (unpublished data).
Cell column perifusion for analysis of acute hormone release responses
Perfusion studies not only allow for quantification of the acute hormone release responses but also reveal the kinetics of the hormone release profile. This allows for the comparison of basal- and agonist-induced effects on hormone release across treatments, as well as in duplicate columns. In addition, with the continuous removal of perifusate from the column, potential autocrine and/or paracrine effects of pituitary secretions can be minimized. Column perifusion experiments were performed using an Acusyst-S System (Endotronics Inc., Minneapolis, MN; Forma Scientific), as previously described (33). Dispersed pituitary cells were cultured overnight with preswollen Cytodex beads (Cytodex I; Sigma-Aldrich, St. Louis, MO) in plating medium at 28°C, under 5% CO2 and saturated humidity. Cells attached to Cytodex beads were loaded into temperature-controlled columns (1.5 × 106 cells/column, 500-µL chamber volume, 18°C) and perifused with testing medium at a flow rate of 15 mL/h (time to clear total dead-space is approximately 5 to 6 minutes) for a minimum of 4 hours prior to the onset of experiments to stabilize basal hormone secretion. In general, individual perifusion experiments consisted of stimulator alone, inhibitor alone, and combinatorial treatments repeated in duplicate columns. Perifusion experiments always began with the collection of five 5-minute fractions of basal hormone secretion to determine the average pretreatment hormone release levels (application of testing medium without pharmacological agents). For combinatorial treatments, a 20-minute application of inhibitor alone, followed by a 5-minute pulse of stimulator in the presence of the inhibitor and a further 40-minute treatment with inhibitor, were applied. Control treatments with stimulator alone or inhibitor alone followed a timeline parallel to that established for the combination treatment. Fractions of perifusate were stored at –20°C until being assayed for LH and GH content by validated radioimmunoassays (Table 1; 48–50).
Antibody Table
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Carp gonadotropin II (cGTH-378; LH) | RAC cGTH-378 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:220,000 | |
| Carp growth hormone (GH) | KcGHA/S R#1 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:52,000 | |
| Rabbit gamma globulin | IgG-GARGG | IgG Corporation | Goat, polyclonal | 1:20 |
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Carp gonadotropin II (cGTH-378; LH) | RAC cGTH-378 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:220,000 | |
| Carp growth hormone (GH) | KcGHA/S R#1 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:52,000 | |
| Rabbit gamma globulin | IgG-GARGG | IgG Corporation | Goat, polyclonal | 1:20 |
Antibody Table
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Carp gonadotropin II (cGTH-378; LH) | RAC cGTH-378 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:220,000 | |
| Carp growth hormone (GH) | KcGHA/S R#1 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:52,000 | |
| Rabbit gamma globulin | IgG-GARGG | IgG Corporation | Goat, polyclonal | 1:20 |
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Carp gonadotropin II (cGTH-378; LH) | RAC cGTH-378 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:220,000 | |
| Carp growth hormone (GH) | KcGHA/S R#1 | Dr. R. E. Peter, University of Alberta (deceased) | Rabbit, polyclonal | 1:52,000 | |
| Rabbit gamma globulin | IgG-GARGG | IgG Corporation | Goat, polyclonal | 1:20 |
Results of LH and GH release from individual columns were expressed as a percentage of the corresponding pretreatment value (% pretreatment), which is calculated as the average hormone release (ng/mL) of the first 5 fractions of perifusate collected at the beginning of the experimental period prior to pharmacological manipulations. This conversion allowed for pooling of perifusion data from different columns without distorting the shape of the hormone release kinetic. The magnitude of hormone release responses to treatments with agonists were quantified from individual columns by calculating the net response (sum of the net change in hormone release in each of the collected fractions over the quantification period) of the LH or GH levels as the area under the response curve (baseline value subtracted, with baseline defined as the average % pretreatment value of the 3 fractions prior to the administration of agonist treatment). Termination of hormone release response to agonist application in a column was deemed to have occurred when hormone release values returned to within 1 standard error of the mean from the average baseline value. The net response for treatment columns exposed to small molecule inhibitors alone or combination treatments were quantified over the equivalent time frame as that used for agonist-alone treatments. Additionally, where indicated, the effect of selective inhibitors on basal release was evaluated by comparing the average of the % pretreatment hormone release values obtained over the duration of the inhibitor-alone treatment (normally 30 to 95 minutes of experiment, pooled across experiments) to the corresponding average values from 0 to 20 minutes of the treatment protocol in the same columns. All treatments were performed in duplicate columns in each experiment, and all perifusion experiments were repeated a minimum of 3 times using different cell preparations.
Bioinformatics
As an effort to ensure that pharmacological inhibitors that are designed for studies in mammalian systems are also suitable for use in comparative studies, where possible, in silico analysis using protein homology modeling and sequence alignments of basal vertebrate genomic sequence data from zebrafish, in particular (ZFIN zebrafish model organism database; https://zfin.org/) (51), were performed. These bioinformatics approaches were used to determine whether the drug-coordinating residues, as well as the 3-dimensional shape of the drug-binding pockets, are conserved within the teleost homologs of proteins targeted by the commonly used pharmacological modulators. Protein sequences were obtained and aligned using the integrated workflow at the Universal Protein Resource Knowledgebase (UniProtKB; http://www.uniprot.org) (52) and Clustal Omega Version 1.2.1 (http://www.clustal.org/omega). Searches used human sequences as the initial queries and identified homologs across species with a cut-off of the E value at ≤ 1.0e−10. Representative species for homology modeling were selected on the basis of sequence completeness. Advanced protein sequence alignments for structural predictions used the pairwise comparison of hidden Markov models (HMM-HMM comparison; HHpred; http://toolkit.tuebingen.mpg.de/hhpred) (53) and/or profile hidden Markov models (HMMER; http://www.hmmer.org) (54). Three-dimensional homology models were obtained by submitting query sequences or sequence alignments to the Protein Homology/AnalogY Recognition Engine, version 2.0 (Phyre2; http://www.sbg.bio.ic.ac.uk/phyre2) (55) Web portal. The quality of each computational model generated was evaluated using both the ProQ2 model assessment server (http://bioinfo.ifm.liu.se/ProQ2) and MolProbity (http://molprobity.biochem.duke.edu) to determine the Ramachandran statistics. Figures of structural models were prepared using the PyMOL Molecular Graphics System (version 1.7; Schrödinger, Cambridge, MA). Modular protein domains were identified and conserved structural features were integrated into homology models using the annotated National Center for Biotechnology Information Conserved Domain Database (http://www.ncbi.nlm.nih.gov/cdd/) (56). In general, as outlined in the "Rationale for the selection of compounds for pharmacological mapping" section and the Supplemental figures, the PtdIns(3,4,5)P3-PH domain interaction surfaces as well as the predicted drug binding sites of teleost PDK1, Akt, BTK, and the Gβ subunit are highly conserved relative to their mammalian counterparts.
Statistics
All values are expressed as mean ± standard error of the mean. Results were compared using analysis of variance. Where analysis of variance showed that differences existed between groups, further statistical analyses were performed using the Tukey’s honest significant difference multiple comparisons. The level of significance was set at P < 0.05.
Results
Basal and GnRH-stimulated hormone release involves Gβγ-dependent signal transduction
To characterize the role played by Gβγ heterodimers in the control of basal and GnRH-stimulated hormone release, we used NSC8668, which binds to Gβγ with submicromolar affinity and prevents Gβγ-effector interactions (26, 27) (Supplemental Fig. 1). Direct contact of Gβγ with a homologous dibasic interaction surface in the C2-helical loop of the class I PI3K p110γ (R552 and K553) and p110β (K532 and K533) subunits is required to activate catalytic activity (5, 6) (Supplemental Fig. 3). Importantly, NSC8668 can inhibit the interactions between Gβγ and holomeric p110γ, resulting in a loss of p110γ catalytic activity both in vitro and in vivo (27). Treatment with NSC8668 (10 μM) significantly reduced acute GnRH2- and GnRH3-stimulated LH and GH release responses measured in cell column perifusion (Figs. 1 and 2). In addition, inhibition of Gβγ-effector interactions significantly decreased the average basal LH and GH release from goldfish pituitary cells by approximately 17% and 15%, respectively (Fig. 3).
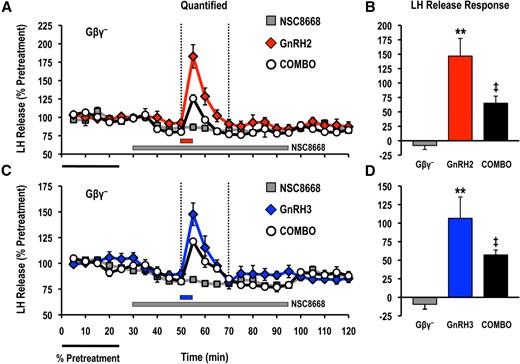
Effects of the Gβγ-binding inhibitor NSC8668 (Gβγ−; 10 μM) on LH release responses stimulated by (A, B) GnRH2 (100 nM) or (C, D) GnRH3 (100 nM). LH release profiles are shown in the left panel (A, C; gray solid square, inhibitor alone; red solid diamond, GnRH2 alone; blue solid diamond, GnRH3 alone; open circle, GnRH + inhibitor) and quantified net LH responses are shown on the right (B, D; mean ± standard error of the mean). Duration of LH release response quantification is indicated by the vertical dotted lines. LH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 4.08 ± 0.14 ng/mL). Pooled responses are shown from 4 experiments (n = 8) using individual cell preparations from goldfish with gonads that were at a sexually matured/prespawning stage (April). The solid gray horizontal bar indicates the duration of the inhibitor treatment, whereas the red (GnRH2) or blue (GnRH3) horizontal bar represents the 5-minute exposure to either GnRH. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).
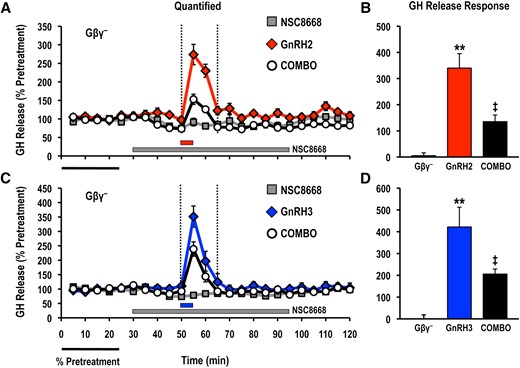
Effects of the Gβγ-binding inhibitor NSC8668 (Gβγ−; 10 μM) on GH release responses stimulated by (A, B) GnRH2 (100 nM) or (C, D) GnRH3 (100 nM). GH release profiles are shown in the left panel (A, C: gray solid square, inhibitor alone; red solid diamond, GnRH2 alone; blue solid diamond, GnRH3 alone; open circle, GnRH + inhibitor) and quantified net GH responses are shown on the right (B, D: mean ± standard error of the mean). Duration of GH release response quantification is indicated by the vertical dotted lines. GH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 9.18 ± 0.43 ng/mL). Pooled responses are shown from 4 experiments (n = 8) using individual cell preparations from goldfish with gonads that were at a sexually matured/prespawning stage (April). The solid gray horizontal bar indicates the duration of the inhibitor treatment, whereas the red (GnRH2) or blue (GnRH3) horizontal bar represents the 5-minute exposure to either GnRH. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).

Effects of the Gβγ-binding inhibitor NSC8668 (Gβγ−; 10 μM) on basal (A) LH and (B) GH release responses. Hormone release profiles are shown in the left panel and quantified responses are shown on the right (mean ± standard error of the mean). Basal release prior to inhibitor application (black bars) was quantified as the average percentage of the pretreatment values (average % pretreatment; black horizontal bar) of the first 4 perifusion fractions collected at the beginning of the experimental trial. The inhibitor-alone basal release (gold bars) was quantified as the average percentage of the pretreatment values over the entire duration of NSC8668 treatment (30 to 95 minutes; gray horizontal bar; n = 16, from 8 independent cell preparations completed in April, using fish with gonads that were at a sexually matured/prespawning stage). Treatments marked with asterisks (**) identify responses that were significantly different from the basal release prior to inhibitor application (paired Student t test; P < 0.05).
Pharmacological mapping of PtdIns(3,4,5)P3-dependent signaling
In general, we chose to monitor PtdIns(3,4,5)P3-dependent signaling by completing pharmacological mapping of both proximal and distal effectors of class I PI3Ks during basal- and GnRH-selective hormone release responses from both gonadotropes as well as somatotropes. The drug concentrations selected for use in these column perifusion studies are based on published studies of the efficacy and selectivity of these reagents in mammalian cell-based assays. However, to facilitate the understanding of the pharmacological manipulations used, the cellular specificity and mode of action used by each compound are outlined in the "Rationale for the selection of compounds for pharmacological mapping" section of the Materials and Methods. Although we could not test for drug selectivity directly using recombinant teleost proteins, 3 observations reported later strongly suggest that the inhibitors used do not nonspecifically alter cell functions: (1) divergence of hormone release responses between basal and agonist-stimulated release; (2) differential inhibition of basal LH compared with GH responses; and (3) occurrence of GnRH2- and GnRH3-selective effects.
PtdIns(3,4,5)P3-sensitive PH domain–containing proteins in general, but not PDK1, Akt, and BTK, are important regulators of GnRH-stimulated LH release
Treatment with PIT-1 (100 μM) significantly reduced acute GnRH2- and GnRH3-stimulated hormone release responses measured in cell column perifusion (Figs. 4 and 5). In contrast, inhibition of PDK1 (GSK2334470; 3 μM), Akt (Akti VIII; 10 μM), or BTK (CGI1746; 1 μM) did not alter the LH release responses stimulated by GnRH2 or GnRH3 (Figs. 4 and 5).
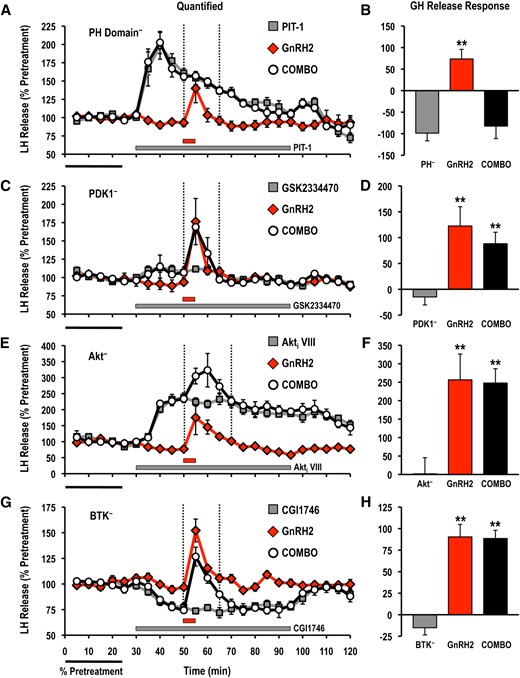
Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), (C,D) PDK1 (GSK2334470; PDK1−; 3 μM), (E, F) Akt (Akti VIII; Akt−; 10 μM), and (G, H) BTK (CGI1746; BTK−; 1 μM) on basal and GnRH2-stimulated LH release responses. For each panel, LH release profiles are shown on the left (A, C, E, G: gray solid square, inhibitor alone; red solid diamond, GnRH2 alone; open circle, GnRH2 + inhibitor), and quantified LH release responses are shown on the right (B, D, F, H). Duration of LH release response quantification is indicated by the vertical dotted lines. LH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 2.62 ± 0.39 ng/mL; n = 8, from 4 independent cell preparations). Pooled responses from experiments with pituitary cells prepared from goldfish in February to August (PIT-1, completed in February to March, times of gonadal recrudescence; GSK2334470, completed in February to April, time of gonadal recrudescence to sexually matured/prespawning; Akti VIII, completed in July to August, time of year with regressed gonads; CGI1746, completed in February, time of gonadal recrudescence) are shown. The gray horizontal bar indicates the duration of the inhibitor treatment, whereas the red horizontal bar represents the 5-minute GnRH2 (100 nM) exposure. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).
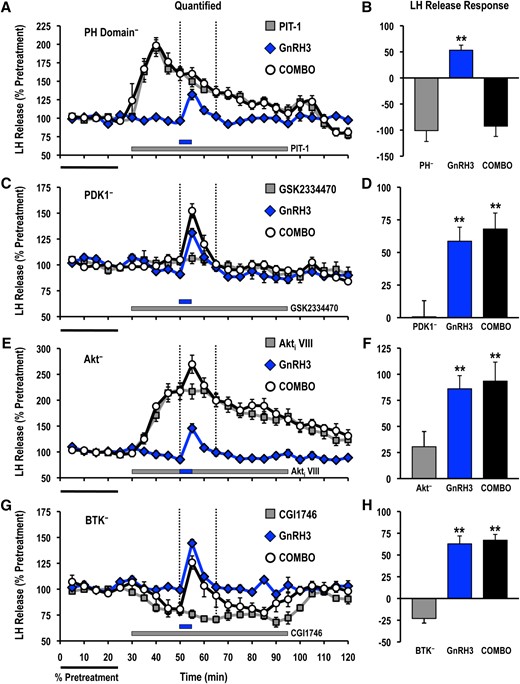
Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), (C, D) PDK1 (GSK2334470; PDK1−; 3 μM), (E, F) Akt (Akti VIII; Akt−; 10 μM), and (G, H) BTK (CGI1746; BTK−; 1 μM) on basal and GnRH3-stimulated LH release responses. For each panel, LH release profiles are shown on the left (A, C, E, G: gray solid square, inhibitor alone; blue solid diamond, GnRH3 alone; open circle, GnRH3 + inhibitor), and quantified LH release responses are shown on the right (B, D, F, H). Duration of LH release response quantification is indicated by the vertical dotted lines. LH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 2.54 ± 0.45 ng/mL; n = 8, from 4 independent cell preparations). Pooled responses from experiments with pituitary cells prepared from goldfish in February to August (PIT-1, completed in February to March, time of gonadal recrudescence; GSK2334470, completed in February to April, time of gonadal recrudescence to sexually matured/prespawning; Akti VIII, completed in July to August, time of year with regressed gonads; CGI1746, completed in February, time of gonadal recrudescence) are shown. The gray horizontal bar indicates the duration of the inhibitor treatment, whereas the blue horizontal bar represents the 5-minute GnRH3 (100 nM) exposure. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).
PtdIns(3,4,5)P3-sensitive PH domain–containing proteins are important regulators of GnRH-stimulated GH release
Pretreatment with PIT-1 abolished GH release responses induced by both GnRH2 and GnRH3 (Figs. 6 and 7), whereas application of the BTK-selective inhibitor CGI1746 also reduced GnRH2-elicited GH secretion (Fig. 6). However, the GnRH2-stimulated GH release was unaffected by both GSK2334470 and Akti VIII (Fig. 6). GnRH3-induced GH secretion was not altered by GSK2334470 or CGI1746, but was enhanced in the presence of Akti VIII (Fig. 7).
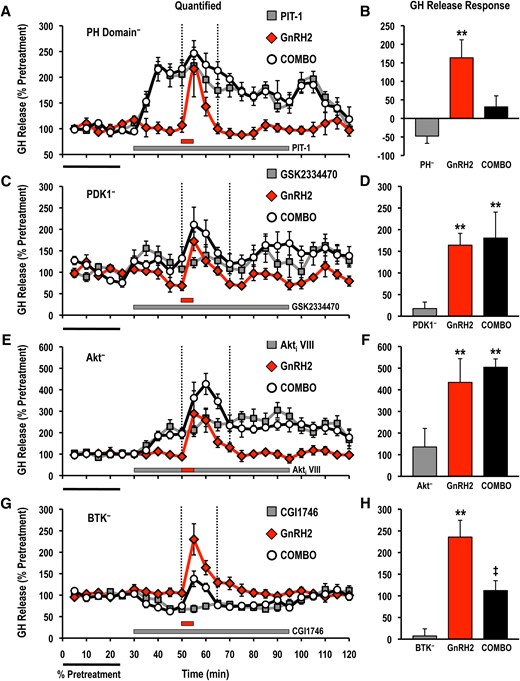
Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), (C, D) PDK1 (GSK2334470; PDK1−; 3 μM), (E, F) Akt (Akti VIII; Akt−; 10 μM), and (G, H) BTK (CGI1746; BTK−; 1 μM) on basal and GnRH2-stimulated GH release responses. For each panel, GH release profiles are shown on the left (A, C, E, G: gray solid square, inhibitor alone; red solid diamond, GnRH2 alone; open circle, GnRH2 + inhibitor), and quantified GH release responses are shown on the right (B, D, F, H). Duration of GH release response quantification is indicated by the vertical dotted lines. GH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 5.79 ± 1.39 ng/mL; n = 8, from 4 independent cell preparations). Pooled responses from experiments with pituitary cells prepared from goldfish in February to August (PIT-1, completed in February to March, time of gonadal recrudescence; GSK2334470, completed in February to April, time of gonadal recrudesce to sexually matured/prespawning; Akti VIII, completed in July to August, time of year with regressed gonads; CGI1746, completed in February, time of gonadal recrudescence) are shown. The gray horizontal bar indicates the duration of the inhibitor treatment, whereas the red horizontal bar represents the 5-minute GnRH2 (100 nM) exposure. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).
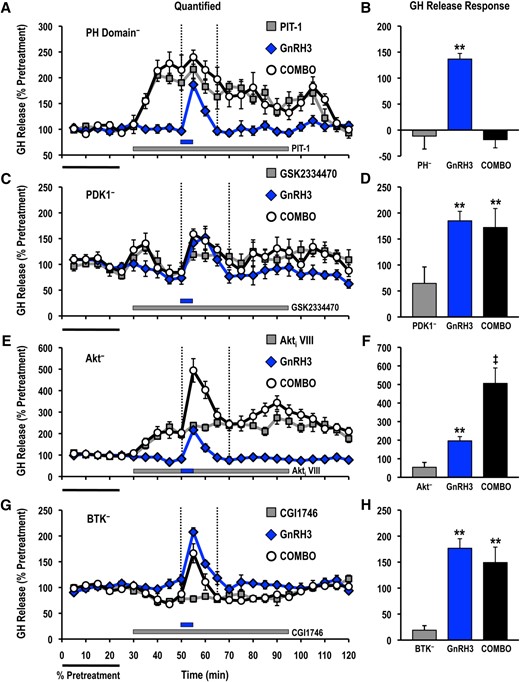
Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), (C, D) PDK1 (GSK2334470; PDK1−; 3 μM), (E, F) Akt (Akti VIII; Akt−; 10 μM), and (G, H) BTK (CGI1746; BTK−; 1 μM) on basal and GnRH3-stimulated GH release responses. For each panel, GH release profiles are shown on the left (A, C, E, G; gray solid square, inhibitor alone; blue solid diamond, GnRH3 alone; open circle, GnRH3 + inhibitor), and quantified GH release responses are shown on the right (B, D, F, H). Duration of GH release response quantification is indicated by the vertical dotted lines. GH release responses were expressed as a percentage of the pretreatment values (% pretreatment, average of the first 5 perifusion fractions collected; black horizontal bar; 4.69 ± 0.77 ng/mL; n = 8, from 4 independent cell preparations). Pooled responses from experiments with pituitary cells prepared from goldfish in February to August (PIT-1, completed in February to March, time of gonadal recrudescence; GSK2334470, completed in February to April, time of gonadal recrudescence to sexually matured/prespawning; Akti VIII, completed in July to August, time of year with regressed gonads; CGI1746, completed in February, time of gonadal recrudescence) are shown. The gray horizontal bar indicates the duration of the inhibitor treatment, whereas the blue horizontal bar represents the 5-minute GnRH3 (100 nM) exposure. Treatments that were significantly different from one another are identified by different symbols (** vs ‡; analysis of variance followed by Tukey’s honest significant difference multiple comparisons; P < 0.05).
Differential involvement of PtdIns(3,4,5)P3-dependent signal transduction effectors in the control of basal LH and GH release
Data from perifusion studies using PIT-1, GSK2334470, Akti VIII, and CGI1746 treatments alone were pooled, and the average hormone release observed during the inhibitor exposure was compared with those prior to inhibitor application. Inhibition of Akt catalytic activity or antagonism of PtdIns(3,4,5)P3-PH domain interactions significantly increased basal LH by approximately 87% and 39%, respectively (Fig. 8). Similar effects were observed for pituitary somatotropes, whereby Akti VIII (223% pretreatment) and PIT-1 (175% pretreatment) significantly increased basal GH release (Fig. 9). Treatment with the selective PDK1 inhibitor GSK2334470 selectively increased basal GH release (Fig. 9). Inhibition of BTK using CGI1746 significantly reduced basal LH and GH release by approximately 20% (Figs. 8 and 9). These observations suggest that basal LH and GH release were selectively altered by distinct PtdIns(3,4,5)P3-sensitive effectors.
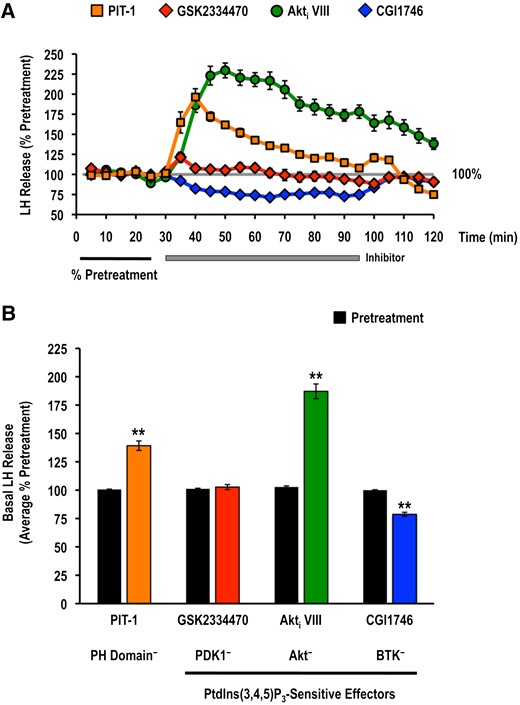
Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), PDK1 (GSK2334470; PDK1−; 3 μM), Akt (Akti VIII; Akt−; 10 μM), and BTK (CGI1746; BTK−; 1 μM) on basal LH release. LH release profiles are shown in the top panel (A: orange solid square, PIT-1 alone; red solid diamond, GSK2334470 alone; green solid circle, Akti VIII alone; blue solid diamond, CGI1746 alone) and quantified responses are shown on the bottom (B: mean ± standard error of the mean). Basal release prior to inhibitor application (black bars) was quantified as the average percentage of the pretreatment values (average % pretreatment; black horizontal bar) of the first 4 perifusion fractions collected at the beginning of the experimental trial. The inhibitor-alone basal release (colored bars: orange, PIT-1 alone; red, GSK2334470 alone; green, Akti VIII alone; blue, CGI1746 alone) was quantified as the average percentage of the pretreatment values over the entire duration of the inhibitor treatment (30 to 95 minutes; gray horizontal bar; n = 16, from 8 independent cell preparations; results from inhibitor-alone treatment columns in Figs. 4 and 5). Treatments marked with asterisks (**) identify responses that were significantly different from the basal release prior to inhibitor application (paired Student t test; P < 0.05).

Effects of selective small molecule inhibitors of PtdIns(3,4,5)P3-sensitive (A, B) PH domain interactions (PIT-1; PH−; 100 μM), PDK1 (GSK2334470; PDK1−; 3 μM), Akt (Akti VIII; Akt−; 10 μM), and BTK (CGI1746; BTK−; 1 μM) on basal GH release. GH release profiles are shown in the top panel (A: orange solid square, PIT-1 alone; red solid diamond, GSK2334470 alone; green solid circle, Akti VIII alone; blue solid diamond, CGI1746 alone) and quantified responses are shown on the bottom (B: mean ± standard error of the mean). Basal release prior to inhibitor application (black bars) was quantified as the average percentage of the pretreatment values (average % pretreatment; black horizontal bar) of the first 4 perifusion fractions collected at the beginning of the experimental trial. The inhibitor-alone basal release (colored bars; orange, PIT-1 alone; red, GSK2334470 alone; green, Akti VIII alone; blue, CGI1746 alone) was quantified as the average % pretreatment value over the entire duration of the inhibitor treatment (30 to 95 minutes; gray horizontal bar; n = 16, from 8 independent cell preparations; results from inhibitor-alone columns in Figs. 6 and 7). Treatments marked with asterisks (**) identify responses that are significantly different from the basal release prior to inhibitor application (paired Student t test; P < 0.05).
Discussion
This study examined the role of PtdIns(3,4,5)P3-sensitive intracellular signaling proteins downstream of PI3K activation in the regulation of pituitary hormone secretion. Consistent with the known involvement of class I PI3Ks, the present results indicate that recruitment of PtdIns(3,4,5)P3-sensitive PH domain–containing proteins, in general, is important for mediating GnRH effects on LH and GH release in goldfish. Although the candidate PtdIns(3,4,5)P3-sensitive signaling protein(s) that participates in GnRH stimulation of LH release has not been identified, Akt and BTK differentially modulate acute GH release responses in a GnRH-selective manner. Unique pituitary cell type–specific differences in the involvement of PtdIns(3,4,5)P3-dependent signaling in the control of basal hormone release responses are also revealed. Just as importantly, results using NSC8668 provide direct physiological evidence for Gβγ-dependent signaling downstream of GnRHR activation in the control of hormone exocytosis; in addition, these findings support the hypothesis that Gβγ-directed bias represents a unique mechanism for controlling ligand-selective signaling downstream of shared GPCRs.
Gβγ-dependent signal transduction regulates basal and GnRH-stimulated hormone release
In addition to its role during the assembly and trafficking of GPCR signaling complexes, Gβγ subunits can modulate the activity of numerous cellular effectors via direct interactions (57). However, in contrast to the influence of Gα subunits, the importance of Gβγ heterodimers in GnRHR-mediated signaling has received relatively little attention (25). Results from this study strongly suggest that both GnRH2 and GnRH3 activate Gβγ-dependent intracellular signaling to acutely control hormone exocytosis in both gonadotropes and somatotropes. Interestingly, previous results suggest that GnRHR activation leads to activation of the Gβγ-sensitive p110β and p110γ isoforms of class I PI3K in a GnRH- and cell type–specific manner (13). Direct contact of Gβγ with highly conserved dibasic motifs within the C2–helical domain linker of both the p110β (532KK) and p110γ (552RK) catalytic subunits is required for membrane recruitment and enzymatic activity (5, 6). The involvement of Gβγ-sensitive signaling and the presence of the conserved Gβγ interaction surfaces in teleost homologs of p110β and p110γ (Supplemental Fig. 3) strongly suggest that activity of these 2 class I PI3Ks is dependent upon Gβγ-effector interactions downstream of GnRHR activation in goldfish pituitary cells. In addition, Gβγ subunits can also modulate the signaling activity of numerous cellular effectors via direct interactions with the same highly conserved binding surface(s) (28, 36, 37). These effectors include isoforms of PLCs, GPCR kinases (GRKs), voltage-sensitive calcium channels (VSCCs), inwardly rectifying K+ channels, adenylate cyclases, and mitogen-activated protein kinases (MAPKs) (57, 58). In this regard, GnRH stimulation of LH and GH secretion in goldfish requires the activation of PLC, VSCC, and the extracellular signal–regulated kinase (ERK) 1/2 family of MAPKs; in addition, GnRH-stimulated LH subunit and GH messenger RNA expression also involves ERK1/2 (16, 59, 60). Interestingly, regulation of the Raf–MAPK kinase (MEK)–ERK transduction cascade can also be Gβγ dependent (61, 62). Whether and how Gβγ participates in mediating PLC-, VSCC-, and/or ERK1/2-dependent signaling following GnRHR activation in goldfish pituitary cells requires further investigation.
Results with NSC8668 indicate that Gβγ also participates in the control of basal LH and GH release. Interestingly, the magnitude of NSC8668 effects on basal LH release mirrored those observed using selective inhibitors of BTK (in this study) and p110β, p110γ, and MEK1/2 catalytic activity (13, 60). However for somatotropes, although NSC8668 mirrored the responses observed for the class I PI3K and BTK inhibitors, basal GH release was only sensitive to inhibition of p110γ, but not p110β, and the kinetics of MEK1/2-dependent basal GH release were dissimilar to those seen with inhibition of Gβγ-effector interactions (13, 60). It is possible that p110β and p110γ may be redundant Gβγ-sensitive signals during the constitutive control of basal LH release and that their downstream signaling may include the activation of BTK and/or MEK1/2. Conversely, Gβγ heterodimers may selectively target p110γ to control basal GH release.
The pattern of involvement observed for Gβγ-dependent effectors during basal hormone release is distinct from GnRH-stimulated responses; although GnRH2 selectively activates p110β, and GnRH3 utilizes p110γ, to control LH release, both GnRHs are sensitive to inhibition of p110β and p110γ in terms of GH secretion (13). Thus, we hypothesize that the repertoire of heterodimeric Gβγ subunits involved in the control of basal and GnRH-stimulated actions might be different. Interestingly, the heterotrimeric Gαβγ complex is assembled from a pool of 16 Gα subunits, 5 Gβ subunits, and 12 Gγ subunits (57). A single GPCR can discriminate between different Gαβγ complexes, and different Gα subunits display preferences for the identity of the Gγ subunits within the heterotrimer (63). Furthermore, variation among the basic and hydrophobic residues present in different Gγ subunits contributes to differences in Gβγ-membrane dissociation rates and translocation kinetics in response to GPCR activation (64). Therefore, heterogeneity in the molecular identity of the Gαβγ complex, and, in particular, the Gβγ heterodimer, likely contributes to signal-specific downstream responses that are dependent upon the agonist-stabilized conformation of the GPCR. This would also allow for the biased regulation of signaling outside of, or in concert with, Gαβγ-independent mechanisms such as the recruitment of arrestins. In support of biased signaling mediated by Gβγ subunits, a novel biased ligand has been identified that selectively inhibits Gβγ-dependent signaling without influencing transduction responses downstream of Gα subunits (65). Although these findings need to be reconciled with structural models of Gαβγ nucleotide exchange, they suggest that GPCR-Gβγ interactions may contribute to the activation of Gβγ-dependent signaling (66). Interestingly, a recent report has demonstrated that ligand-dependent conformational differences in receptor-bound Gαβγ complexes directly contribute to ligand efficacy and intracellular signaling kinetics by influencing Gαβγ receptor residency (67). Thus, although ligand bias depends on changes to the conformational landscape of the receptor, ligand binding also significantly influences the receptor recruitment and structural dynamics of intracellular effectors, including Gαβγ. Undoubtedly, understanding the physiological significance of Gβγ-mediated signaling bias, as well as outlining the structural basis for Gβγ-selective signaling, will be an exciting avenue for future studies of functional selectivity downstream of activated GPCRs.
PtdIns(3,4,5)P3-sensitive PH domain–containing proteins are important regulators of GnRH-stimulated hormone release
The present findings using PIT-1 strongly suggest that signaling effectors containing PtdIns(3,4,5)P3-sensitive PH domains contribute to signal transduction responses stimulated by agonist-stabilized GnRHRs, as well as support the hypothesis that class I PI3K activation and downstream PtdIns(3,4,5)P3-PH domain interactions mediate GnRH stimulation of acute LH and GH release in goldfish (11–13). Interestingly, results with inhibitors of PDK1, Akt, and BTK suggest that these canonical downstream PtdIns(3,4,5)P3-PH domain-sensitive signaling elements are not universal mediators of GnRH-induced hormone release from goldfish gonadotropes and somatotropes.
Differences in the GnRH-selective involvement of PtdIns(3,4,5)P3-dependent signaling within pituitary gonadotropes and somatotropes
PDK1, Akt, and BTK are common intracellular signaling elements downstream of PtdIns(3,4,5)P3. Akt activation is tightly regulated by phosphorylation events at both Thr308 by PDK1 and Ser473 by mechanistic target of rapamycin complex 2 (68). Results from studies focusing exclusively on the regulation of Akt phosphorylation at Ser473 suggest that Akt-dependent signaling is modulated by mammalian GnRH (GnRH1) or its analogs in immortalized and oncogenic cell types (69, 70); however, the direct involvement of PDK1, Akt, and BTK in the control of pituitary hormone release have not been examined. Present results demonstrate unique differences in the roles of PDK1, Akt, and BTK in GnRHR-mediated signaling in goldfish pituitary gonadotropes and somatotropes. The observations that GnRH2-stimulated GH release was significantly reduced by treatment with the BTK inhibitor CGI1746, whereas stimulation of GH release by GnRH3 was potentiated by an allosteric inhibitor of Akt, reveal ligand-dependent bias in the involvement of Akt- and BTK-dependent signaling downstream of a GPCR. Furthermore, these results suggest that the biased activation of class I PI3K isoforms observed previously (13) leads to selective involvement of downstream PtdIns(3,4,5)P3-sensitive PH domain–containing effectors in goldfish pituitary somatotropes. In contrast, the inability of GSK2334470, Akti VIII, and CGI1746 to alter GnRH-elicited LH release suggests that neither bias in the recruitment of PtdIns(3,4,5)P3-sensitive effectors nor the participation of canonical PtdIns(3,4,5)P3-sensitive elements (PDK1, Akt, and BTK) occurs in goldfish gonadotropes. However, such a conclusion is premature, given that other PtdIns(3,4,5)P3-sensitive mechanisms have not been examined. In addition, because of the unique role of PIT-1 in antagonizing the membrane translocation of PtdIns(3,4,5)P3-sensitive effectors, we cannot exclude the possibility that kinase-independent functions of PDK1, Akt, and BTK may also contribute to basal or GnRH-stimulated signaling in both goldfish pituitary gonadotropes and somatotropes. Nevertheless, results reinforce the idea that cell type–specific differences exist in the roles of Akt- and BTK-dependent signaling in GnRH actions. Whether PtdIns(3,4,5)P3-sensitive effectors directly interact with GnRHRs is also unknown, but there are established roles for class I PI3Ks in the recruitment of GRKs and arrestins to activated GPCRs (71–75). Similar roles for PtdIns(3,4,5)P3-dependent effectors in gonadotropes and somatotropes could contribute to the GnRHR signaling responsible for differential control of hormone release responses.
Integration of BTK-dependent signaling with known components of GnRH2-mediated signal transduction in goldfish pituitary somatotropes
The present results using CGI1746 suggest that BTK-dependent signaling is selectively activated by GnRH2 to stimulate GH secretion. In isolated goldfish somatotropes, PI3Ks are upstream effectors of GnRH2-induced Ca2+-dependent signaling (11). In mammalian immune cells, BTK plays a central role in the control of PLCγ-mediated increase in [Ca2+]i by phosphorylating tyrosine residues that are important for regulating PLCγ catalytic activity (76–78). In addition, the PLCγ PH domain directly binds to PtdIns(3,4,5)P3 (79). Consequently, generation of PtdIns(3,4,5)P3 by receptor-associated class I PI3Ks causes acute increases in Ins(1,4,5)P3 production and intracellular Ca2+ release through membrane recruitment and activation of PLCγ (80). Interestingly, GnRH2 actions on GH release are dependent on PLC and protein kinase C (PKC), but GnRH2-stimulated GH release does not use Ins(1,4,5)P3-sensitive intracellular Ca2+ stores (81). Thus, BTK-dependent activation of PLCγ may participate in the activation of diacylglycerol-sensitive isoforms of PKC following GnRHR activation by GnRH2 in goldfish somatotropes. Interestingly, Ca2+-dependent and Ca2+-independent PKC isoforms also directly interact with the PH domain of BTK and prevent BTK membrane translocation and activation (82, 83). Thus, PKC activation may also modulate the strength and duration of BTK-dependent signaling (83). Adding to this complexity, BTK can directly associate with PtdIns(4)P 5-kinases and transport them to the plasma membrane to stimulate PtdIns(4,5)P2 synthesis (84). This allows BTK to regulate local phosphoinositide turnover by stimulating the production of the common substrate required for both class I PI3Ks and PLCs (84, 85). Understanding how the intricate relationship between PLC, PKC, and BTK contributes to biased signaling downstream of activated Gαq/11-coupled receptors will be an important area of future research.
The involvement of BTK in GnRH2 actions on goldfish GH release also provides a conduit for crosstalk between GPCR- and tyrosine kinase–dependent signaling in pituitary cells. In particular, phosphorylation of Tyr551 and subsequent autoactivation of membrane-associated BTK is controlled by Src family kinases (SFKs), including c-Src (86–88). SFK-dependent signaling controls GnRHR-mediated actions in a number of mammalian cell types (89). Furthermore, GnRH2-stimulated GH release involves p110γ, and full activation of BTK downstream of agonist-stimulated GPCRs is dependent upon the catalytic activity of both p110γ and c-Src (90). Whether and how SFKs contribute to ligand-selective signaling by GnRHRs remains to be examined. Interestingly, membrane recruitment and activation of BTK can also be controlled through direct interactions with the Gαq class of G proteins (91) or Gβγ heterodimers (92, 93). Previous studies have demonstrated that GnRHRs couple predominantly to Gαq/11, and results from this study indicate that GnRH actions on hormone release are also dependent upon signaling responses stimulated by Gβγ subunits. Therefore, it is possible that activated Gαβγ complexes could also directly contribute to BTK-dependent signaling downstream of GnRH2-stabilized GnRHRs in goldfish somatotropes.
Integration of Akt-dependent signaling with known components of GnRH3-mediated signal transduction in goldfish pituitary somatotropes
Results from this study reveal that Akt is a selective negative regulator of GnRH3-stimulated, but not GnRH-2-induced, GH secretion. How signal selectivity is achieved is not known, but a possible mechanism involving an interaction between Akt- and Ca2+-dependent signaling can be postulated. In somatotropes, GnRH3-dependent GH release selectively utilizes Ins(1,4,5)P3-sensitive intracellular Ca2+ stores (81). Akt phosphorylates IP3R subtypes and reduces their Ca2+ release activity in many cell types (94–96). Inhibition of Akt might relieve inhibitory IP3R phosphorylation to selectively potentiate GnRH3-stimulated GH release. Interestingly, PKCζ is expressed in goldfish pituitary cells (97), and PKCζ can negatively regulate Akt phosphorylation and activity (98). The possible involvement of PKCζ in GnRH signaling in goldfish pituitary cells is also quite intriguing considering that this isoform is directly activated by PtdIns(3,4,5)P3 (99). However, GnRH3 actions on LH release also involve Ins(1,4,5)P3-sensitive intracellular Ca2+ stores, but the effects of Akt inhibition are selective for GnRH3 actions on GH and not LH release. The basis for this cell-type difference is at present unknown but may be a result of selective uncoupling of Akt from class I PI3K-dependent signaling.
In contrast to the apparent role of Akt in regulating GnRH3 actions on GH secretion discussed earlier, previous results reveal that PI3K-dependent signaling contributes to GnRH3-dependent signal transduction at steps that are independent or downstream of increases in [Ca2+]i (11). Paradoxically, PI3K- and Akt-dependent signaling can be uncoupled in several models of GPCR-mediated signaling. In particular, signaling downstream of GPCRs coupled to Gαq can both positively and negatively regulate Akt activity. Stimulation of class I PI3Ks by Gβγ heterodimers increases phosphorylation of Akt, whereas Gαq inhibits Akt activation downstream of Gβγ-effector interactions (100). Gαq-coupled receptors also control Akt-dependent signaling using G protein–independent mechanisms (74, 75). In particular, β-arrestins can form signaling complexes that recruit Akt and the protein phosphatases PP2A (protein phosphatase 2A) (101–103) or PHLPP (PH domain leucine-rich repeat-containing protein phosphatase) (104); dephosphorylation of Akt reduces Akt catalytic activity. Akt can also physically interact with GRK2 (G protein–coupled receptor kinase 2), and this interaction inhibits Akt activity (105). Furthermore, nonvisual arrestins bind PtdIns(3,4,5)P3 with high affinity, and treatment with PtdIns(3,4,5)P3 can enhance arrestin interactions with GPCRs (106). As a result, increases in the local effective PtdIns(3,4,5)P3 concentration around the receptor by class I PI3Ks could stabilize GPCR-arrestin complexes that inactivate Akt. It is established that phosphorylation of GPCRs by GRKs facilitates the formation of arrestin signaling scaffolds, resulting in the uncoupling of receptors from G proteins (107). Whether negative regulation of Akt activity contributes to GnRH3-selective signaling through interactions with GRKs or arrestins will be an important area for future research. Regardless of what these future studies may reveal, it is clear that Akt serves a unique role as a convergence point for G protein–dependent and G protein–independent signaling downstream of GPCRs.
Noncanonical PtdIns(3,4,5)P3-dependent signaling downstream of GnRHR activation
Differences between the abilities of PIT-1, and those of GSK2334470, Akti VIII, or GCI1746 to affect GnRH-induced hormone release responses suggest novel PtdIns(3,4,5)P3-dependent regulators participate in the control of LH and GH release. Potential candidates include the cytohesin family of Arf guanine nucleotide exchange factors GRP1 and ARNO (108, 109) (Fig. 10). PIT-1 can inhibit the membrane translocation and signaling initiated by GRP1 and ARNO (110). GRP1 and ARNO regulate actin remodeling and membrane dynamics through activation of the Ras superfamily small guanosine triphosphatases ARF1 and ARF6 (111–113). In particular, ARF6 is an important component of Ca2+-regulated exocytosis in a number of model endocrine cell types (114). In pancreatic β-cells, GPCR activation of p110γ-dependent actin depolymerization enhances targeting of secretory granules to the plasma membrane (115, 116). It is possible that the GRP1 and ARNO regulate Arf-dependent cytoskeletal dynamics to facilitate basal and/or agonist-stimulated hormone release. Whether these mechanisms are involved in hormone secretion in pituitary gonadotropes and somatotropes should be explored.
![Summary model depicting the diversity of noncanonical class I PI3K signal transduction elements and the proposed integration of PtdIns(3,4,5)P3-dependent signal transduction in the GnRH-selective stimulation of acute hormone release from goldfish gonadotropes and somatotropes. In the majority of excitable cell models tested, production of PtdIns(3,4,5)P3 by activated class I PI3Ks results in the recruitment of the canonical effectors PDK1, Akt, and BTK. More specifically, results in goldfish have demonstrated that GnRH-stimulated LH and GH release responses involve the activation of PLC, PKCs, and Raf-MEK-ERK cascades as well as Ca2+-dependent signaling mechanisms. PDK1 directly phosphorylates the T-loop activation segment of conventional (cPKC), novel (nPKC), and atypical (aPKC) isozymes of PKC. PKC activation is known to activate the Raf-MEK-ERK cascade through either direct phosphorylation of Raf, or indirectly through the activation of the Ras superfamily guanine nucleotide exchange factor Ras guanyl nucleotide-releasing protein (RasGRP). BTK activity also directly regulates the signaling activity of several families of MAPKs: including ERK1/2, p38 MAPK, and JNK. In addition to these classical targets, PtdIns(3,4,5)P3-sensitive PH domains are present in a wide variety of intracellular signaling proteins. Examples of important noncanonical class I PI3K signaling effectors are shown. The involvement of the Arf guanine nucleotide exchange factors (GEFs) GRP1 and ARNO in the control of basal and GnRH-stimulated hormone release are implicated. Alternatively, PtdIns(3,4,5)P3 is also known to recruit the Rac-GEF PtdIns(3,4,5)P3-dependent Rac exchanger protein 1 (P-Rex1) to control, in concert with Arfs, actin remodeling and activate the Raf-MEK-ERK subfamily of MAPKs. Acute stimulation of PtdIns(3,4,5)P3 production has also been implicated in the rapid trafficking of l-type voltage-sensitive Ca2+ channels and the direct gating of some isoforms of transient receptor potential channels to enhance extracellular Ca2+ entry. Last, isoforms of PLCγ directly bind and are activated by PtdIns(3,4,5)P3 at the plasma membrane, although catalytic activity is also tightly regulated by Src family tyrosine kinases (such as c-Src) and BTK. PLCγ activation results in the activation of diacylglycerol (DAG)-sensitive isoforms of PKC, as well as stimulates intracellular Ca2+ release from inositol 1,4,5-trisphosphate [Ins(1,4,5)P3]-gated (IP3R-containing) Ca2+ stores. Overall, these noncanonical mechanisms for the control of PtdIns(3,4,5)P3-dependent signaling could result in the coordinate control of actin cytoskeleton dynamics and alter intracellular Ca2+ concentrations ([Ca2+]i) to facilitate basal and agonist-stimulated hormone release responses. In fact, the recruitment of noncanonical effectors may help displace constitutively active PtdIns(3,4,5)P3-sensitive effectors such as PDK1 or Akt to alleviate negative regulatory actions on exocytic vesicle docking or fusion events. In particular, Akt activation has been shown to phosphorylate and inactivate specific IP3R subtypes to decrease [Ca2+]i and, in many cell systems, Akt also directly attenuates Raf-dependent activation of MEK-ERK signaling. Relieving the inhibitory actions of Akt could significantly modulate basal, as well as agonist-stimulated, hormone release responses. PtdIns(3,4,5)P3-dependent signaling has the potential to interact with many important transduction elements that are known to be activated downstream of GnRHRs in goldfish gonadotropes and somatotropes.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/158/2/10.1210_en.2016-1552/2/m_en.2016-1552f10.jpeg?Expires=1750240199&Signature=Zfs29Yvl48NPQ8U0GJJqKzClrTBAl7KOoovp8QKPWlV-CQL14a8V48qiPC3hjfoB0Vv8wRcUuU3Ry8V7dUa82nGoRI50bqJA6owq0N2iSVKQPHtgG9sTdru~LxX79JKy3fdRYo9ypyUFzlj6hci07x1WKA3KEVzI6Wd9NXk6muunwSMsFKMv4jLSAlM-UNnqIbWobwXWdaAEG~fYG-6ktvZnTXa3QvqPT6ckJtZluH7sQ7cif0C6~zvrqhELZ-16A55gRCSQOhvYnMl5YAZUqYAG0kguDBeh7HORDcXOLVEDd0jEhUsEYq-Qb7kh0EmVs2kLTT2xMyeaTwAewZkTVA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Summary model depicting the diversity of noncanonical class I PI3K signal transduction elements and the proposed integration of PtdIns(3,4,5)P3-dependent signal transduction in the GnRH-selective stimulation of acute hormone release from goldfish gonadotropes and somatotropes. In the majority of excitable cell models tested, production of PtdIns(3,4,5)P3 by activated class I PI3Ks results in the recruitment of the canonical effectors PDK1, Akt, and BTK. More specifically, results in goldfish have demonstrated that GnRH-stimulated LH and GH release responses involve the activation of PLC, PKCs, and Raf-MEK-ERK cascades as well as Ca2+-dependent signaling mechanisms. PDK1 directly phosphorylates the T-loop activation segment of conventional (cPKC), novel (nPKC), and atypical (aPKC) isozymes of PKC. PKC activation is known to activate the Raf-MEK-ERK cascade through either direct phosphorylation of Raf, or indirectly through the activation of the Ras superfamily guanine nucleotide exchange factor Ras guanyl nucleotide-releasing protein (RasGRP). BTK activity also directly regulates the signaling activity of several families of MAPKs: including ERK1/2, p38 MAPK, and JNK. In addition to these classical targets, PtdIns(3,4,5)P3-sensitive PH domains are present in a wide variety of intracellular signaling proteins. Examples of important noncanonical class I PI3K signaling effectors are shown. The involvement of the Arf guanine nucleotide exchange factors (GEFs) GRP1 and ARNO in the control of basal and GnRH-stimulated hormone release are implicated. Alternatively, PtdIns(3,4,5)P3 is also known to recruit the Rac-GEF PtdIns(3,4,5)P3-dependent Rac exchanger protein 1 (P-Rex1) to control, in concert with Arfs, actin remodeling and activate the Raf-MEK-ERK subfamily of MAPKs. Acute stimulation of PtdIns(3,4,5)P3 production has also been implicated in the rapid trafficking of l-type voltage-sensitive Ca2+ channels and the direct gating of some isoforms of transient receptor potential channels to enhance extracellular Ca2+ entry. Last, isoforms of PLCγ directly bind and are activated by PtdIns(3,4,5)P3 at the plasma membrane, although catalytic activity is also tightly regulated by Src family tyrosine kinases (such as c-Src) and BTK. PLCγ activation results in the activation of diacylglycerol (DAG)-sensitive isoforms of PKC, as well as stimulates intracellular Ca2+ release from inositol 1,4,5-trisphosphate [Ins(1,4,5)P3]-gated (IP3R-containing) Ca2+ stores. Overall, these noncanonical mechanisms for the control of PtdIns(3,4,5)P3-dependent signaling could result in the coordinate control of actin cytoskeleton dynamics and alter intracellular Ca2+ concentrations ([Ca2+]i) to facilitate basal and agonist-stimulated hormone release responses. In fact, the recruitment of noncanonical effectors may help displace constitutively active PtdIns(3,4,5)P3-sensitive effectors such as PDK1 or Akt to alleviate negative regulatory actions on exocytic vesicle docking or fusion events. In particular, Akt activation has been shown to phosphorylate and inactivate specific IP3R subtypes to decrease [Ca2+]i and, in many cell systems, Akt also directly attenuates Raf-dependent activation of MEK-ERK signaling. Relieving the inhibitory actions of Akt could significantly modulate basal, as well as agonist-stimulated, hormone release responses. PtdIns(3,4,5)P3-dependent signaling has the potential to interact with many important transduction elements that are known to be activated downstream of GnRHRs in goldfish gonadotropes and somatotropes.
Class I PI3Ks and PtdIns(3,4,5)P3-dependent signal transduction in the control of basal LH and GH release
Novel roles for PDK1-, Akt-, and BTK-dependent signal transduction in the control of basal LH and GH release from goldfish pituitary cells are also revealed. In this study, inhibition of PDK1 increased basal GH, but not basal LH release responses. Because GSK2334470 is known to spare Akt activation in vivo (39, 40), PDK1 catalytic activity may contribute to basal GH release, at least in part, in an Akt-independent manner. The divergent effects of Akti VIII and GSK2334470 on basal LH release are also consistent with this hypothesis. This possibility is also supported by recent studies identifying a prominent role for Akt-independent PDK1 signaling downstream of oncogenic mutations in p110α (117). In these systems, cells retain robust PDK1 membrane localization and exhibit a dependency on the PDK1 substrate SGK3, whereas Akt activation is suppressed (117–119). Thus, the Akt-independent actions of PDK1 on basal GH release could involve SGK-mediated signaling. However, the involvement of additional PDK1-regulated AGC family kinases or kinase-independent roles of PDK1 cannot be excluded.
On the other hand, results with Akti VIII suggest that Akt activation negatively regulates basal LH and GH secretion. Likewise, inhibition of Akt enhances newcomer granule fusions and the motility of intracellular insulin granules in insulin-secreting β-cells (120). Interestingly, acute treatment with the p110α-selective inhibitor PIK-75 significantly increases the exocytic responses originating from newcomer granules during glucose-stimulated insulin secretion, whereas fusion events arising from previously docked granules are decreased by a mechanism independent of [Ca2+]i dynamics (120). Taken together, these findings identify a role for class IA PI3K- and Akt-dependent signaling in the control of vesicular trafficking and fusion events governing basal secretion in representative endocrine cell types.
In goldfish, basal LH and GH release require PKA- and cyclic adenosine monophosphate (cAMP)–dependent signaling (121). The C-terminal of PKA can directly interact with the kinase domain of PDK1 (122), and phosphorylation of PKA at Thr197 by PDK1 activates the catalytic subunit of PKA (123, 124). Therefore, Akt and PKA are common substrates of PDK1 that both physically interact with PDK1 at the membrane. Interestingly, although our results demonstrate that Akt inhibition increases both basal LH and GH release, PKA inhibitors decrease basal LH and GH secretion (121). These results suggest opposing roles for cAMP/PKA- and Akt-dependent signaling in the regulation of basal LH and GH release. In mammalian systems, cAMP-dependent signaling also inhibits Akt activation by reducing PtdIns(3,4,5)P3 production and translocation of PDK1 to the plasma membrane (125). Conversely, Akt can phosphorylate and activate phosphodiesterase (PDE) 3B, reducing intracellular cAMP concentrations (126). As a result, cAMP/PKA-induced inhibition of Akt-dependent signaling may also feed forward by inactivating PDE-mediated hydrolysis of cAMP. In support of this, the broad-spectrum PI3K inhibitor wortmannin increases insulin secretion by inhibiting PDE activity to increase cAMP content (127). Whether the negative influence of Akt-dependent signaling can be relieved through tonic activation of PKA in goldfish gonadotropes and somatotropes remains to be examined, but it is likely that the catalytic activities of these effectors are tightly regulated in the control of basal hormone release responses.
Unlike manipulations of PDK1 and Akt, inhibition of BTK catalytic activity reduced both basal LH and GH release, suggesting a positive role for BTK-dependent signaling in regulating the basal secretion of these hormones. Interestingly, the magnitude of changes in unstimulated LH and GH release following treatment with CGI1746 are similar to those observed using ATP-dependent inhibitors of class I PI3Ks (13), as well as an antagonist of Gβγ-dependent signaling in the current study. These observations suggest that BTK-dependent signaling participates in the basal control of LH and GH release, at least in part, in a PtdIns(3,4,5)P3-sensitive manner that is independent of PDK1 and Akt. It is also likely that BTK actions are dependent upon Gβγ-mediated activation of p110β and p110γ catalytic activity, given that inhibition of these 2 class IA PI3Ks similarly reduces basal LH and GH release (13). As mentioned previously, BTK can also be directly activated by Gβγ heterodimers, although membrane translocation of BTK would still be expected to be dependent on the production of PtdIns(3,4,5)P3.
The divergent effects of PIT-1 and those of the selective inhibitors of PDK1-, Akt-, and BTK-dependent signaling on basal LH and GH secretion indicate that PtdIns(3,4,5)P3-sensitive effectors have both stimulatory and inhibitory influences. Previous results have revealed that although LY294002 and wortmannin acutely increase basal LH and GH release, isoform-selective inhibitors of individual class I PI3K catalytic subunits generally decrease basal hormone secretion (11, 13). Likewise, in pancreatic β-cells, broad-spectrum inhibitors of PI3K enhance insulin secretion (128–130), whereas isoform-selective inhibition of p110γ or class II PI3Ks results in the reduction of insulin secretion (131–133). Differential regulation of the PtdIns(3,4,5)P3-sensitive signaling effectors described in this study not only provides the basis for these paradoxical observations using pan-PI3K inhibitors, but also highlights the need for additional studies using isoform-selective inhibitors of the class II and class III PI3Ks.
PtdIns(3,4,5)P3 signaling may also affect basal LH and GH release via mechanisms independent of PH domain interactions. In addition to direct interactions with PH domains, PtdIns(3,4,5)P3 and PtdIns(4,5)P2 can regulate plasma membrane availability of signaling, cytoskeletal, and transport proteins through ionic protein-lipid interactions (134). PtdIns(4,5)P2 clusters also act as molecular scaffolds for vesicle docking (135) and facilitate the formation of membrane microdomains independent of cholesterol or lipid phases (136). Maintenance of the PtdIns(4,5)P2 to PtdIns(3,4,5)P3 ratio by interconverting enzymes is also important for the regulation of intracellular Ca2+ signaling (137). Thus, it is likely that treatment with pan-PI3K inhibitors causes both an overall reduction in PtdIns(3,4,5)P3 availability, as well as a transient accumulation of the class I PI3K substrate PtdIns(4,5)P2, which together contribute to changes in basal hormone release. Additionally, comparisons of the cellular responses to inhibitors of PtdIns(3,4,5)P3-sensitive PH domains, in general, with those elicited by ATP-dependent inhibitors of the class I PI3Ks or downstream kinases, suggest additional noncatalytic roles for PtdIns(3,4,5)P3-PH domain interactions, such as during the formation of scaffolds important for GnRHR-stimulated signaling or hormone-vesicle trafficking. Elucidating whether and how these PH domain–independent mechanisms contribute to the control of PtdIns(3,4,5)P3-sensitive hormone release responses will be important in future studies.
Summary
Class I PI3Ks are complex regulators of intracellular signal transduction. Overall, the current study adds to the general understanding of functional selectivity in GPCR signal transduction networks by demonstrating the complexity of ligand-biased signaling downstream of class I PI3K catalytic activity. Results demonstrate that PtdIns(3,4,5)P3-sensitive PH domain–containing signal transduction components differentially participate in GnRH-elicited LH and GH secretion in a GnRH- and cell type–selective manner. In addition, important mechanistic differences in the control of basal and agonist-stimulated hormone release by PtdIns(3,4,5)P3-dependent effectors are demonstrated. Overall, these data suggest that biased activation of class I PI3Ks by GnRH variants could result in divergent PtdIns(3,4,5)P3 signaling responses that facilitate the selective coupling of PH domain–containing components in response to activated GnRHRs. Given that the control of PtdIns(3,4,5)P3 production is both dynamic and subject to modulation by complex regulatory networks, class I PI3K signaling is an attractive intracellular platform for integrating the multifactorial neuroendocrine regulation of gonadotrope and somatotrope activity. Taking into account the participation of the known Gβγ-binding effectors p110β and p110γ in GnRH actions on hormone release, results with NSC8668 also provide physiological evidence for the functional involvement of Gβγ-dependent transduction downstream of GnRHR activation. Furthermore, the presence of a clear structure-activity relationship between GnRH variants and GnRHRs in goldfish (19, 138–141), coupled with the GnRH-selective regulation of Gβγ-sensitive effectors, strongly implies that agonist binding could result in the stabilization of receptor conformations that promote biased signaling outcomes dependent upon Gβγ heterodimers, and not only Gα subunits.
Abbreviations:
- Akt
protein kinase B
- Arf
adenosine 5′-diphosphate ribosylation factor
- ARNO
adenosine 5′-diphosphate ribosylation factor nucleotide-binding-site opener
- ATP
adenosine triphosphate
- BTK
Bruton’s tyrosine kinase
- cAMP
cyclic adenosine monophosphate
- ERK
extracellular signal–regulated kinase
- GH
growth hormone
- GnRH
gonadotropin-releasing hormone
- GnRHR
gonadotropin-releasing hormone receptor
- GPCR
G protein–coupled receptor
- GRK
G protein–coupled receptor kinase
- GRP1
general receptor for phosphoinositides 1
- LH
luteinizing hormone
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- PDE
phosphodiesterase
- PDK1
phosphoinositide-dependent kinase 1
- PH
pleckstrin homology
- PI3K
phosphoinositide 3-kinase
- PIT-1
PITenin-1
- PKC
protein kinase C
- PLC
phospholipase C
- PtdIns(3,4,5)P3
phosphatidylinositol 3,4,5-trisphosphate
- SFK
Src family kinase
- SGK
serine/threonine-protein kinase
- VSCC
voltage-sensitive calcium channel
Acknowledgments
This work was supported by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) to J.P.C. Support for J.G.P. is provided by NSERC (Postgraduate Scholarship D), the Killam Trusts (Izaak Walter Killam Memorial Scholarship), Alberta Innovates Health Solutions (Graduate Studentship), the Province of Alberta Advanced Education and Technology Graduate Scholarship, and the University of Alberta (Department of Biological Sciences Graduate Teaching Assistantship, Dr. Richard E. Peter Memorial Scholarship, President’s Doctoral Prize of Distinction, and Dissertation Fellowship).
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: John P. Chang, PhD, CW405 Biology Science Building, Department of Biological Sciences, University of Alberta, Edmonton, Alberta, Canada T6G 2E9. E-mail: [email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}