





Abstract
Incretin and insulin responses to nutrient loads are suppressed in persons with diabetes, resulting in decreased glycemic control. Agents including sulfonylureas and dipeptidyl peptidase-4 inhibitors (DPP4i) partially reverse these effects and provide therapeutic benefit; however, their modes of action limit efficacy. Because somatostatin (SST) has been shown to suppress insulin and glucagonlike peptide-1 (GLP-1) secretion through the Gi-coupled SST receptor 5 (SSTR5) isoform in vitro, antagonism of SSTR5 may improve glycemic control via intervention in both pathways. Here, we show that a potent and selective SSTR5 antagonist reverses the blunting effects of SST on insulin secretion from isolated human islets, and demonstrate that SSTR5 antagonism affords increased levels of systemic GLP-1 in vivo. Knocking out Sstr5 in mice provided a similar increase in systemic GLP-1 levels, which were not increased further by treatment with the antagonist. Treatment of mice with the SSTR5 antagonist in combination with a DPP4i resulted in increases in systemic GLP-1 levels that were more than additive and resulted in greater glycemic control compared with either agent alone. In isolated human islets, the SSTR5 antagonist completely reversed the inhibitory effect of exogenous SST-14 on insulin secretion. Taken together, these data suggest that SSTR5 antagonism should increase circulating GLP-1 levels and stimulate insulin secretion (directly and via GLP-1) in humans, improving glycemic control in patients with diabetes.
Persons with type 2 diabetes have impaired insulin and incretin responses to nutrient loads, contributing to reduced glycemic control (1). Dipeptidyl peptidase-4 inhibitors (DPP4i), which decrease the rate of degradation of incretin hormones [i.e., gastric inhibitory polypeptide (GIP) and glucagonlike peptide-1 (GLP-1)], are efficacious in the treatment of such patients; however, the efficacy of DPP4i is limited because they only inhibit degradation of the incretins and do not stimulate their endogenous secretion (2). Insulin secretagogues (e.g., sulfonylureas and glinides) are used as antidiabetic therapies in patients who still have sufficient insulin secretory capacity, but their action is not glucose dependent, leading to hypoglycemic episodes (3, 4). Identification of therapeutics that can stimulate insulin secretion in a glucose-dependent manner, as well as agents that may stimulate incretin secretion and act synergistically with DPP4i, has therefore been the subject of research efforts in the last decade and led us to investigate further the role of somatostatin (SST) in the regulation of incretin and insulin levels.
SST is a peptide hormone produced in brain cells (25% of total expression), pancreatic islets (5%), and the gut (65%) (5). SST has two forms, SST-14 and SST-28, that agonize a family of five SST receptor (SSTR) subtypes that are class A Gαi-protein–coupled receptors functionally coupled to inhibition of adenylate cyclase (6–13). SST-14 is secreted primarily from hypothalamic neurons, δ cells in the gastric epithelium (specifically, the foregut), and the δ cells of the pancreatic islets (14), whereas SST-28 is secreted mainly from the δ cells in the intestinal epithelium (specifically, the mucosa of the ileum), and preferentially binds to SSTR5 (9, 15). In humans, SSTRs are expressed in brain, anterior pituitary, pancreatic islets, adrenal cortex and medulla, thyroid, ovary, testis, kidney, lung, myocardium, and skeletal muscle (16). Through its action on SSTRs in various tissues, SST has been shown to play a role in the regulation of growth and thyroid-stimulating hormones, gastrin and histamine release in the stomach, and gastrointestinal motility, bile flow, and intestinal fluid absorption in the gut (14),
In addition to these actions, SST suppresses the secretion of insulin and glucagon from pancreatic islets in humans and many other species (17–19), and has also been shown to suppress GLP-1 secretion in isolated, perfused porcine ileum (20), as well as from rat intestinal cultures and in fed sheep (21). Reversal of SST suppression of secretion via selective antagonism of the appropriate receptor subtype should result, therefore, in increased secretion of these key hormones and afford increased glycemic control and other benefits.
SSTR5 is highly expressed on the human β cell of the pancreatic islets (22), suggesting a key role for this receptor subtype in the regulation of insulin secretion in humans. In rodents, selective antagonism of SSTR5 and of Sstr5 gene knockout mice (Sstr5−/−) improved glycemic control (23–25), which has been attributed to increased insulin sensitivity (resulting in increased insulin secretion by reducing glucotoxicity and lipotoxicity), and increases in direct insulin secretion and/or other insulin-independent glucose disposal methods (24). Sstr5 is also highly expressed in mice and rats on the enteroendocrine cells of the gut responsible for GLP-1 and GIP secretion (26, 27), and it has been speculated that weight loss with chronic administration of SSTR5 antagonists may be the result of increased GLP-1 levels (24) due to SST-regulated incretin secretion. Hence, SSTR5 may be the key mediator of SST suppression of incretin and insulin secretion in humans.
In this study, we examined the effect of selective SSTR5 antagonism on insulin secretion from islets isolated from rats, mice, and humans, as well as on circulating levels of the incretin GLP-1 in normal and diabetic murine models. Our data indicate that, whereas there is not a robust insulin-secretion effect in isolated mouse or rat islets with selective SSTR5 antagonism, a potent and selective SSTR5 antagonist reverses the blunting effects of SST on insulin secretion from isolated human islets. Additionally, we demonstrate for that SSTR5 antagonism in rodents increases systemic levels of active GLP-1 in vivo, which is primarily responsible for the improvements in glucose tolerance in these animals. Therefore, in humans, selective antagonism of SSTR5 is hypothesized to blunt the inhibitory tone of SST on adenylate cyclase in pancreatic β cells and intestinal enteroendocrine cells, resulting in enhanced glucose-dependent insulin secretion directly from pancreatic β cells, as well as indirectly by enhanced GLP-1 secretion from the gut.
Materials and Methods
The SSTR5 antagonist (S5A1), 4-[8-[(2-cyclopropyl-5-ethoxy-4-methyl-phenyl)methyl]-2-oxo-1,3,8-triazaspiro[4.5]decan-3-yl] benzoic acid [Fig. 1(a)], was prepared as described in US patent application WO2016/205032 (Beauchamp, Coates, Martinez-Grau, Toledo, inventors; Eli Lilly and Company).
![(a) Chemical structure of S5A1. (b) S5A1 is a high-affinity ligand of SSTR5. Binding of [3H]-S5A1 to membranes expressing human SSTR5 was quantified using an SPA. [3H]-S5A1 binding to membranes was measured in the absence (total) or presence of an unlabeled competitor to determine nonspecific binding. S5A1 bound with a Kd of 260 pM, specific binding was >90%, and the maximum number if binding sites was 25 pmol/mg of protein. Data plotted are a representative experiment from three that were conducted.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/158/11/10.1210_en.2017-00639/1/m_en.2017-00639f1.jpeg?Expires=1750494559&Signature=FBFR8yVuo86-Z61H5f3AncS~j7aPhjZjbhhEkaQll5d~tT752D8GXUQRaENXQoted6oiujWsyvXInhIrri0Hr8l-BLnwqTqsMGl9XTe7efLwffEX2FbeG7IkO0cyWQOMX8KC3MKIf5kV2VTxx2bPg0PEctepPdOQAoAq0EVKNUrOaUO9WWlg9N6oCAp786fRf5UfTDAwFLA-wcis3TWu~R6LQRG-lsW75ts6a5s22U6UmhjTFruJXM5PWSMG0hkg3Ucd0RMiAS9OtrNcvrQiha~SGvcC9ANA4frD8uoZigUMgUJlQb02TteIK6~qmL7vXPxpJ4t9nNGBM~JJr3NqqQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(a) Chemical structure of S5A1. (b) S5A1 is a high-affinity ligand of SSTR5. Binding of [3H]-S5A1 to membranes expressing human SSTR5 was quantified using an SPA. [3H]-S5A1 binding to membranes was measured in the absence (total) or presence of an unlabeled competitor to determine nonspecific binding. S5A1 bound with a Kd of 260 pM, specific binding was >90%, and the maximum number if binding sites was 25 pmol/mg of protein. Data plotted are a representative experiment from three that were conducted.
Binding assays
Human, mouse, and rat SST receptor binding affinities were measured by competition binding, using a scintillation proximity assay (SPA). Profiling of SSTR binding affinity was measured in recombinant hSSTR1, SSTR2, SSTR3, and SSTR5 stably expressed Chinese hamster ovary (CHO) membranes (PerkinElmer, Waltham, MA) at a concentration of 0.4 to 1.0 µg of protein per well. The binding affinity for rodent receptors was assessed using HEK293 membranes transiently expressing mSstr1, -2, -3, -4, and -5 and rSstr5 using Lipofectamine 2000 (Life Technologies, Carlsbad, CA) at a concentration of 5 to 8 µg of protein per well. Membranes were homogenized and diluted in buffer [25 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, and 0.5% bovine serum albumin (BSA), pH 7.4]. [125I]-SST-14 (Vitrax, Placentia, CA) was added at a final concentration of 0.25 nM, and reactions were incubated for 1 hour at 25°C. Specific binding was determined by displacement with 1 μM unlabeled SST-14. Half-maximal inhibitory concentration (IC50) values were calculated using four-parameter nonlinear regression from concentration response curves (GraphPad Prism; GraphPad Software, San Diego, CA). Binding affinity (Ki) values were calculated from the IC50 by the equation of Cheng and Prusoff (28), in which Ki = IC50/(1 + [L]/Kd), where Kd is an equilibrium dissociation constant.
[3H]-S5A1 at 44 Ci/mmol was prepared by Quotient Bioresearch (Cardiff, United Kingdom). Quantification of binding to hSSTR5 membranes was performed analogously to [125I]-SST binding assays. Briefly, multiple concentrations of [3H]-S5A1 were incubated with 1.25 μg of hSSTR5 membranes and 0.5 mg of wheat germ agglutinin-coated SPA beads to generate a saturation binding isotherm. Nonspecific binding was determined by the addition of 10 μM unlabeled compound. After incubation for 1 hour at room temperature, bound ligand was quantified by SPA. Data were fit using a one-site model with total and nonspecific binding in GraphPad Prism (GraphPad Software).
hSSTR5 functional β-arrestin antagonist assay
Quantification of functional antagonism of hSSTR5 was performed by measuring hSST14-induced β-arrestin recruitment by hSSTR5 using a chemiluminescence-based enzyme-fragment complementation assay (29). Measurement of antagonism of SST-14–induced β-arrestin activity was assayed in 384-well format using PathHunter CHO-K1 hSSTR5β-arrestin cells (catalog no. 93-0402C2; DiscoveRx) according to the manufacturer’s instructions. Phosphate-buffered saline–washed cells were treated with increasing concentrations of S5A1 and equilibrated at 37°C for 30 minutes. Cells were then stimulated with an 80% maximal effective concentration of SST-14 (9 nM; catalog no. 1156; Tocris), after which chemiluminescence was quantified with an Envision plate reader (PerkinElmer). IC50 values were calculated from luminescence data, using four-parameter nonlinear regression over a concentration response of compound.
Functional G-protein activation guanosine triphosphate-γ-[35S] binding
Blockade of SSTR5-stimulated guanosine triphosphate (GTP)–γ-[35S] binding in CHO membranes stably expressing cloned human SSTR5 receptors was studied in an SPA format according to previously described protocols with minor modifications (30, 31). Reactions containing buffer (100 mM NaCl, 20 mM HEPES, 5 mM MgCl2, 1 mM EDTA, 0.4% BSA, 3 μM guanosine diphosphate), 0.5 nM GTPγ[35S], SPA beads (1 mg/well), and SSTR5 membranes (6 µg of protein per well) were stimulated using 0.8 nM SST-28 (Bachem). Plates were sealed and incubated for 2 hours at 25°C. Specific GTP-γ-[35S] binding was determined as the difference in counts per minute observed in the absence and presence of 10 µM unlabeled GTPγS. Concentration response curves were plotted as the percent of specific GTP-γ-[35S] inhibition and IC50 values were determined using a sigmoidal dose response curve with variable slope (GraphPad Prism; GraphPad). Antagonist affinity (Kb) was estimated according to DeLapp et al. (30), using a modification of the equation of Cheng and Prusoff (28) in which Kb = IC50/(1+[A]/EC50).
Cyclic adenosine monophosphate generation in mouse and human incretin-secreting cell lines
Mouse enteroendocrine GLUTag cells (D. Drucker, University of Toronto, Canada) were cultured in Dulbecco’s modified Eagle medium supplemented with 5.5 mM glucose, 10% fetal bovine serum, and 2 mM glutamine. Human colonic NCI-H716 cells (CCL-251; ATCC) were cultured in RPMI-1640 supplemented with 11 mM glucose and 10% fetal bovine serum. Before assay, cells were trypsinized, pelleted, and seeded in 96-well tissue culture assay plates at a density of 15,000 to 20,000 cells per well. Cells were allowed to attach and were incubated for 48 hours. On the assay day, cells were washed and incubated with test compounds in 50 µL balanced salt-solution buffer supplement with 0.1% BSA, 500 µM 3-isobutyl-1-methylxanthine, and 5 mM glucose. After compound treatment, cyclic adenosine monophosphate (cAMP) levels were determined using a Cisbio (Bedford, MA) Dynamic 2 cAMP HTRF kit. To each well, we added 25 µL of anti-cAMP cryptate and 25 µL of cAMP-d2, and plates were incubated for 1 hour at room temperature. The fluorescence signal was read at 620 nm and 665 nm. Fluorescence ratio values were converted to nanomolar cAMP per well using a cAMP standard curve. Data were analyzed with GraphPad Prism using a four-parameter nonlinear logistic algorithm.
Insulin secretion in rodent and human islets
Pancreatic islets were isolated from male C57BL6 mice (22 to 26 g; Harlan Laboratories, Indianapolis, IN) and male Sprague Dawley rats (225 to 260 g; Harlan Laboratories) by ductal collagenase injection, digestion, and density gradient separation (32). Human islets were acquired through the University of Miami and Prodo Laboratories. After isolation, islets were cultured for 48 to 72 hours to stabilize the viable β-cell mass, shipped in a preservation medium to Eli Lilly and Company, and used for insulin secretion assays (33). For all perifusion assays, 50 islets were scattered and immobilized in Bio-Gel P-4 Gel (BioRad, Hercules, CA) inside sample containers (chambers), and bathed with a HEPES-buffered solution (120 mM NaCl; 4.8 mM KCl; 2.5 mM CaCl2⋅2H2O; 1.2 mM MgCl2⋅6H; 10 mM HEPES; 24 mM NaHCO3) and 0.25% BSA. The perifusate was collected in a 96-well plate. Insulin in the perifusate was quantified by enzyme-linked immunosorbent assay [Meso Scale Discovery (MSD); Rockville, MD] following an internally optimized protocol (34). Data were normalized by the total DNA content of the islets inside the respective chamber to correct for small differences in islet mass among the chambers. Statistical differences were analyzed using the repeated-measures analysis of variance with donor as block and a compound symmetric covariance structure, followed by a Dunnett correction.
Perifusion insulin-secretion studies with mouse, rat, and human islets (five donors) were completed in which islets were perifused with a buffered solution containing either glucose (vehicle), forskolin (1000 nM for potentiation of cAMP-mediated glucose-stimulated insulin secretion); forskolin (1000 nM) and SST-14 (100 nM); or forskolin (1000 nM), SST-14 (100 nM) and either S5A1 or a selective SSTR3 antagonist [BN81674 (4n); 0.3 µM] (35). Experiments were deemed valid if an elevation in the concentration of glucose was followed by an increase in insulin secretion and this glucose-stimulated insulin secretion was potentiated by forskolin.
In vivo studies
All in vivo studies were conducted in accordance with accepted standards of humane animal care and approved by Eli Lilly and Company’s Institutional Animal Care and Use Committee. Animals were single-housed on a 12-hour light cycle from 06:00 to 18:00. Acute studies were performed in male C57BL/6 mice (Envigo Laboratories, Indianapolis, IN; diet: Teklad 2014) or male Zucker Diabetic Fatty (ZDF) rats (Charles River Laboratories; diet: LabDiet 5008). Diet-induced obese (DIO) male C57BL/6 mice (D12492 formula; Research Diets) were obtained from Jackson Laboratories (Bar Harbor, ME) at 20 to 22 weeks of age.
Determination of plasma GLP-1 concentrations
Immediately after collection, all blood samples for GLP-1 analysis were preserved with 5 µL of a mixture of DPP4i (catalog no. DPP-4; Bio Trend) and aprotinin (catalog no. BP250310; Fisher BioReagents), stored on wet ice, and then centrifuged (1328g, 4°C) to provide plasma, which was drawn off and plated. Samples were stored at −80°C until they could be analyzed using either the MSD Mouse/Rat GLP-1 (x-36) amide Assay Kit (catalog no. K150FAC-4), or the MSD Active GLP-1 (v2) Kit (catalog no. K150JWC-2). GLP-1 samples were analyzed at no more than a 1:1 dilution of assay buffer to diluent 13 (catalog no. R56BB-3; MSD).
Acute oral glucose tolerance tests
The day before the study, food was removed from the cages at 16:00. At 07:00, body weights were measured, and baseline blood samples were collected via tail vein for glucose (Aviva glucometer; Roche). Animals were dosed via oral gavage with vehicle [1% hydroxyethylcellulose (HEC), 0.25% Tween 80, and 0.05% Antifoam 1510-US; Dow Corning] or test agent(s) (n = 5 or 6 per group) 1.5 hours before glucose administration (10 mL/kg; catalog no. G7528; Sigma). Blood samples (30 µL) were collected for measurement of glucose, insulin, and/or GLP-1 (EDTA-coated Sarstedt Microvette® CB 300 K2E tubes) immediately before the glucose bolus and 15, 30, and 60 minutes after the dose. Mice were euthanized immediately after the final blood collection.
Fasted-state GLP-1 secretion in C57BL/6 mice
Food was removed from the cages at 18:00 on the day before the study. At 06:00 on the study day, body weights were measured, and vehicle (1% HEC, 0.25% Tween 80, and 0.05% Antifoam 1510-US) or test agent (n = 5 per group) was administered via oral gavage. Blood samples (130 µL) for measurement of active GLP-1 and plasma exposure were collected via tail vein at 0.25, 0.5, 1, 2, 4, 6, 9, 12 hours after administration (only two samples were collected from each mouse, and animals were euthanized immediately after the second blood collection).
Sstr5−/− mice
Male Sstr5−/− and wild-type (WT) littermates were obtained from Taconic Laboratories (line B6;129S5-Sstr5<tm1Lex>; Taconic Custom Breeding Solutions) at 7 to 15 weeks of age. These mice have a mixed 129/SvEv-C57Bl6 genetic background (Het × Het). Sstr5−/− and WT mice were evaluated for differences in body weight, glucose (Aviva glucometer; Roche), plasma insulin (insulin assay; Meso Scale Diagnostics), and GLP-1. Vehicle (1% HEC, 0.25% Tween 80, and 0.05% Antifoam 1510-US) alone or containing the Sstr5 antagonist was administered via oral gavage to overnight-fasted mice. Blood samples (130 µL) for measurement of active GLP-1 and plasma exposure were collected via tail vein at 2 and 4 hours after administration. Mice were euthanized immediately after the final blood collection.
Chronic in vivo evaluation
DIO mice were acclimated to the facility for 2 weeks before the study. Baseline body weights, as well as glucose and GLP-1 levels (60 µL of blood via tail vein blood sampling) were measured, and the mice were then randomized by body weight and glucose levels. Vehicle (1% HEC, 0.25% Tween 80, and 0.05% Antifoam 1510-US) or test agents were administered via oral gavage once daily, alone or in combination with a DPP4i for 15 days (n = 6 per group). On days 1, 8, and 15, blood samples were collected via tail vein for glucose and GLP-1 (x-36) amide levels before and 1 hour after dosing. On day 16, after an overnight fast, body weights were measured and test agents were administered 2 hours before an oral glucose tolerance test (OGTT; 2 g/kg; catalog no. G7528; Sigma). Blood samples were collected via tail vein for measurement of glucose and active GLP-1 (EDTA-coated Sarstedt Microvette® CB300 K2E tubes) immediately before the glucose bolus and 15, 30, and 60 minutes after the dose. Mice were euthanized immediately after the final blood collection.
Statistical analysis
Results are presented as mean values ± standard error of the mean, unless otherwise indicated. Data were analyzed with GraphPad Prism (GraphPad Software) using a four-parameter nonlinear logistic algorithm and analysis of variance.
Results
S5A1 is a potent and selective SSTR5 antagonist
S5A1 binds to the human, rat, and mouse isoforms of SSTR5 with high affinity, as demonstrated in competition binding experiments with [125I]-SST 14, using an SPA format (Table 1). S5A1 was highly selective toward other SSTRs (Ki for SSTR1-4 >1000 times compared with SSTR5 for human and mouse). To quantify direct binding of S5A1 to the hSSTR5 receptor we conducted saturation ligand binding studies using tritiated S5A1 [Fig. 1(b)]. S5A1 bound specifically and saturably to hSSTR5-expressing membranes (Kd, 280 ± 30 pM; standard deviation, 3). The data are concordant with [125I] competition binding (Ki, 500 pM) and demonstrate direct and high affinity interaction of S5A1 with hSSTR5.
SST Receptor Binding Affinity of S5A1
| Receptor Binding Assay | Ki, nM |
|---|---|
| Human SSTR5 | 0.54 (0.3; 5) |
| Human SSTR1 | >5190 (5) |
| Human SSTR2 | >10,000 1) |
| Human SSTR3 | >10,000 (1) |
| Mouse SSTR5 | 4.87 (0.8; 5) |
| Mouse SSTR1 | >6850 (5) |
| Mouse SSTR2 | >10,000 (1) |
| Mouse SSTR3 | >10,000 (1) |
| Mouse SSTR4 | >10,000 (1) |
| Rat SSTR5 | 3.77 (1) |
| Receptor Binding Assay | Ki, nM |
|---|---|
| Human SSTR5 | 0.54 (0.3; 5) |
| Human SSTR1 | >5190 (5) |
| Human SSTR2 | >10,000 1) |
| Human SSTR3 | >10,000 (1) |
| Mouse SSTR5 | 4.87 (0.8; 5) |
| Mouse SSTR1 | >6850 (5) |
| Mouse SSTR2 | >10,000 (1) |
| Mouse SSTR3 | >10,000 (1) |
| Mouse SSTR4 | >10,000 (1) |
| Rat SSTR5 | 3.77 (1) |
Data are geometric means, with standard deviation (in some entries) and number of replicates indicated in parentheses. A qualifier (>) indicates data did not reach 50% inhibition and the Ki was calculated using the highest concentration tested in the assay.
Abbreviation: SD, standard deviation.
SST Receptor Binding Affinity of S5A1
| Receptor Binding Assay | Ki, nM |
|---|---|
| Human SSTR5 | 0.54 (0.3; 5) |
| Human SSTR1 | >5190 (5) |
| Human SSTR2 | >10,000 1) |
| Human SSTR3 | >10,000 (1) |
| Mouse SSTR5 | 4.87 (0.8; 5) |
| Mouse SSTR1 | >6850 (5) |
| Mouse SSTR2 | >10,000 (1) |
| Mouse SSTR3 | >10,000 (1) |
| Mouse SSTR4 | >10,000 (1) |
| Rat SSTR5 | 3.77 (1) |
| Receptor Binding Assay | Ki, nM |
|---|---|
| Human SSTR5 | 0.54 (0.3; 5) |
| Human SSTR1 | >5190 (5) |
| Human SSTR2 | >10,000 1) |
| Human SSTR3 | >10,000 (1) |
| Mouse SSTR5 | 4.87 (0.8; 5) |
| Mouse SSTR1 | >6850 (5) |
| Mouse SSTR2 | >10,000 (1) |
| Mouse SSTR3 | >10,000 (1) |
| Mouse SSTR4 | >10,000 (1) |
| Rat SSTR5 | 3.77 (1) |
Data are geometric means, with standard deviation (in some entries) and number of replicates indicated in parentheses. A qualifier (>) indicates data did not reach 50% inhibition and the Ki was calculated using the highest concentration tested in the assay.
Abbreviation: SD, standard deviation.
In cell lines that express SSTR5, binding of S5A1 translates to functional efficacy. S5A1 induced cAMP-increases in both GLUTag and NCI-H716 cell lines (IC50, 35.9 and 1.05 nM, respectively). Additionally, S5A1 potently inhibited hSSTR5-mediated GTP-γ-[35S] binding with an antagonist affinity (Kb) of 1.99 nM.
SSTR5 antagonism reversed SST-14-mediated suppression of forskolin-induced insulin secretion in human islets but not in rodent islets
To determine whether SSTR5 and SSTR3 mediate SST suppression of insulin secretion, we studied the effect of S5A1 as well as the SSTR3-selective antagonist BN81674 on insulin secretion from perifused human, mouse, and rat islets. In isolated human islet perifusion studies, forskolin was used to stimulate cAMP-mediated insulin secretion. Adding exogenous SST-14 suppressed the forskolin-stimulated insulin secretion, whereas addition of S5A1 to the forskolin/SST-14 mixture reversed the inhibitory effects of exogenous SST-14 on first- and second-phase insulin secretion in human islets [Fig. 2(a)]. The insulin response with S5A1 was higher than the forskolin-stimulated insulin secretion, suggesting that S5A1 suppressed at least a portion of the paracrine tone of SST in the islets in addition to the exogenous SST added during the experiment. In the same system, BN81674 reversed the SST-14-mediated suppression, restoring insulin secretion back to the level seen in the forskolin treated islets [Fig. 2(b)].
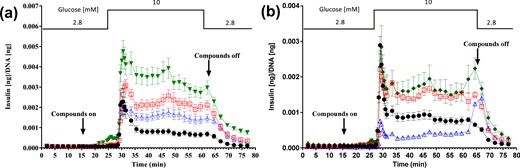
Reversal of SST-14–mediated suppression of forskolin-induced insulin secretion in perifused human islets. Perifusion insulin secretion with (a) SSTR5 antagonist (n = 3 donors combined) and (b) SSTR3 antagonist (n = 1 donor) from isolated human islets. Symbols represent islets treated with glucose control (●); glucose + 1000 nM forskolin (□); glucose + 1000 nM forskolin + 100 nM SST-14 (Δ); glucose + 1000 nM forskolin + 100 nM SST-14 + 0.3 µM S5A1 (▼); and glucose + 1000 nM forskolin + 100 nM SST-14 + 0.3 µM S3B1 (♦).
Following the same perifusion protocol used with isolated mouse islets, S5A1 added to the forskolin/SST-14 mixture did not reverse the SST-14-mediated suppression of first-phase insulin secretion, whereas BN81674 did. Addition of either S5A1 or BN81674 to the forskolin/SST-14 groups did not significantly alter second-phase insulin secretion [Fig. 3(a) and 3(c)]. In isolated rat islets, the addition of SST-14 to the forskolin treatment suppressed both phases of forskolin-stimulated insulin secretion, and neither phase of insulin secretion could be fully recovered by the addition of S5A1 or BN81674 [Fig. 3(b) and 3(d)].
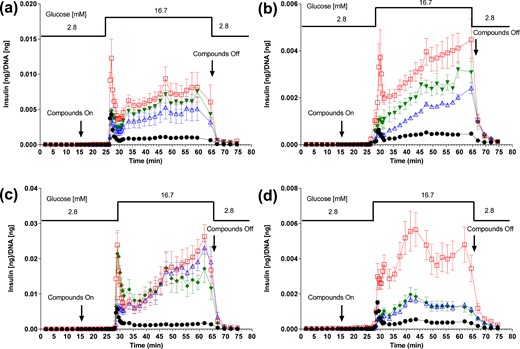
Reversal of SST-14–mediated suppression of forskolin-induced insulin secretion in perifused rodent islets. Perifusion insulin secretion with SSTR5 antagonist from isolated (a) C57BL/6 mice and (b)Sprague Dawley rats, and SSTR3 antagonist from (c) isolated C57BL/6 mice and (d) Sprague Dawley rats. Symbols represent islets treated with glucose control (●); glucose + 1000 nM forskolin (□); glucose + 1000 nM forskolin + 100 nM SST-14 (Δ); glucose + 1000 nM forskolin + 100 nM SST-14 + 0.3 µM S5A1 (▼); and glucose + 1000 nM forskolin + 100 nM SST-14 + 0.3 µM S3B1 (♦).
SSTR5 antagonism increased circulating GLP-1 levels and improved glycemic control in healthy mice
Given the expression of SSTR5 on the enteroendocrine cells of the gut, we wanted to see if SSTR5 antagonism plays an important role in regulating GLP-1secretion. To that end, S5A1 was dosed to normal C57BL/6 mice, resulting in a dose proportional increase in fasting state levels of GLP-1 (x-36), and these GLP-1 (x-36) levels were further increased following an OGTT [Fig. 4(a)]. GLP-1 area under the curve (AUC) increased 2 to 2.7 times [ED50, 0.38 mg/kg; Fig. 4(b)]. These increases in GLP-1 levels did not impact fasting glucose levels [Fig. 4(c)] but were associated with a dose-dependent improvement in glucose tolerance [AUC ED50, 0.18 mg/kg; Fig. 4(d)].
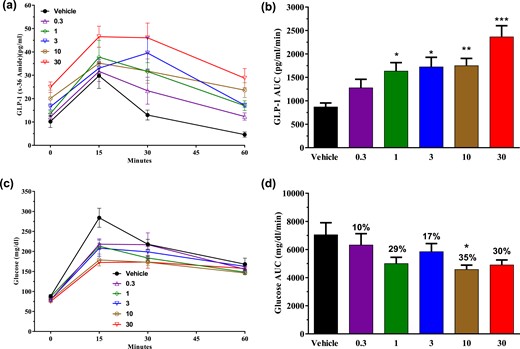
SSTR5 antagonism increased GLP-1 levels and improved glycemic control after an oral bolus of glucose in normal mice. Dose-response effects of S5A1 in normal C57BL/6 mice on (a, b) total GLP-1 (x-36) amide levels and (c, d) glucose tolerance after an oral bolus of glucose (3 g/kg). Data shown as mean ± standard error of the mean from five mice per group. *P < 0.05; **P < 0.01; ***P < 0.001.
To evaluate the direct, non-glucose–dependent effect of SSTR5 antagonism on the secretion of active GLP-1, S5A1 was dosed orally to fasted male C57BL/6 mice, and active GLP-1 levels were monitored for 12 h in the absence of food. Within minutes, active GLP-1 levels increased more than fourfold with 30 mg/kg of S5A1 (8.3 ± 2.0 pg/mL) compared with the vehicle control [2.0 ± 0.7 pg/mL; Fig. 5(a)]. The endogenous active GLP-1 remained elevated throughout the 12-hour study, resulting in a dose- and compound exposure-dependent increase of active GLP-1 AUC levels [ED50, 0.30; ED90, 5.54 mg/kg; Fig. 5(b)]. The unbound plasma exposure of S5A1 increased approximately linearly from 0.3 to 30 mg/kg, and its free plasma concentration exceeded the mSstr5 Ki (4.87 nM) from 0.5 to 4 hours after dosing at 0.3 mg/kg; it was greater than the binding affinity from 0.25 to 12 hours at 1, 3, 10, and 30 mg/kg, respectively [Fig. 5(c)]. Given the in vivo half-life of active GLP-1 of 1.5 minutes (36), the prolonged elevation of GLP-1 levels in the absence of a DPP4i indicates that GLP-1 secretion is increased due to SSTR5 antagonism.
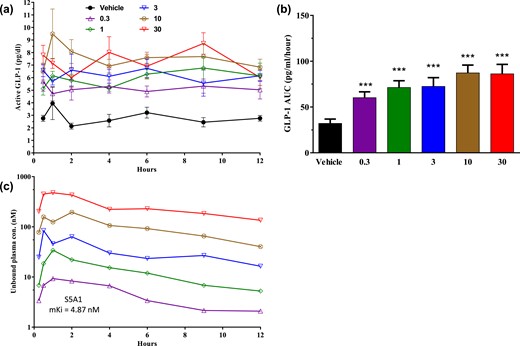
SSTR5 antagonism increases GLP-1 levels in a non-glucose-dependent manner in normal mice. (a, b) Effects of S5A1 on active GLP-1 levels over 12 hours in normal mice after an overnight fast. (c) Unbound plasma exposure of S5A1 over 12 hours. Data shown as mean ± standard error of the mean from five mice per group. ***P < 0.001. con, concentration.
The combination of SSTR5 antagonism and DPP4 inhibition resulted in further increases in circulating GLP-1 levels and greater glycemic control in healthy mice and ZDF rats
Agents that afford increased incretin secretion should provide even greater levels of circulating incretins and increased glycemic control in the presence of a DPP4i. To determine whether this was the case for SSTR5 antagonists, overnight-fasted, normal C57BL/6 mice were administered either S5A1 (0.3 and 30 mg/kg), a DPP4i (sitagliptin, 10 mg/kg), or a combination of the two before an OGTT. Treatment with the combination did not change plasma glucose levels in fasted mice as compared with vehicle control; however, GLP-1 (7-36) amide levels were increased 13-fold before the glucose bolus. In animals treated with the combination, active GLP-1 AUC over 60 minutes was threefold higher than in animals treated with the DPP4i alone and 20-fold higher than in vehicle-treated controls [Fig. 6(a) and 6(b)]. Whereas the glucose excursion was suppressed comparably in animals treated with S5A1 or the DPP4i alone, there was an even greater decrease in the glucose excursion in animals treated with the combination [Fig. 6(c) and 6(d)]. Plasma insulin levels were increased vs the control mainly at the 15-minute time point during the OGTT. The insulin levels of animals treated with both S5A1 and DPP4i were lower than those of animals treated with DPP4i alone and decreased relative to improved glycemic control [Fig. 6(e)].
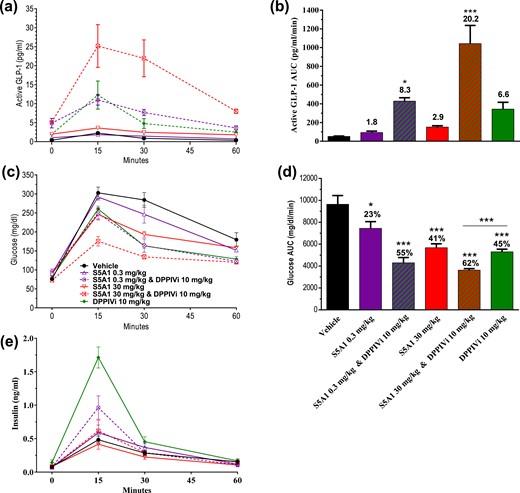
Combination of SSTR5 antagonism and DPP4 inhibition resulted in further increases in circulating GLP-1 levels and greater glycemic control in normal mice. Effects of S5A1 administered alone and in combination with a DPP4i on (a, b) active GLP-1 levels, (c, d) glucose tolerance, and (e) insulin after an oral bolus of glucose (3 g/kg). Data shown as mean ± standard error of the mean from six mice per group. *P < 0.05; **P < 0.01; ***P < 0.001.
To determine the effects of SSTR5 antagonism on glucose tolerance, active GLP-1 and insulin levels in insulin-resistant, type 2 diabetic animals [i.e., male ZDF rats (37) with postprandial glucose levels between 325 and 400 mg/dL] were fasted overnight, weighed, and then administered either S5A1 (3 and 30 mg/kg), a DPP4i (10 mg/kg), or a combination of the two. By 1.5 hours later, before receiving the bolus of glucose (2 g/kg), glucose levels were reduced and active GLP-1 levels were increased in animals treated with either the SSTR5 antagonist or DPP4i alone. The prebolus glucose levels of animals treated with the combination of both agents were 42% lower than in control animals [Fig. 7(b)], and the prebolus active GLP-1 levels in these animals were increased 18-fold compared with vehicle control [Fig. 7(a)]. Prebolus insulin levels were increased in animals treated with the DPP4i alone and more so in animals treated with the combination of both agents. The AUC of glucose-stimulated insulin secretion during the OGTT was not increased with S5A1 alone compared with the control [Fig. 7(e) and 7(f)].
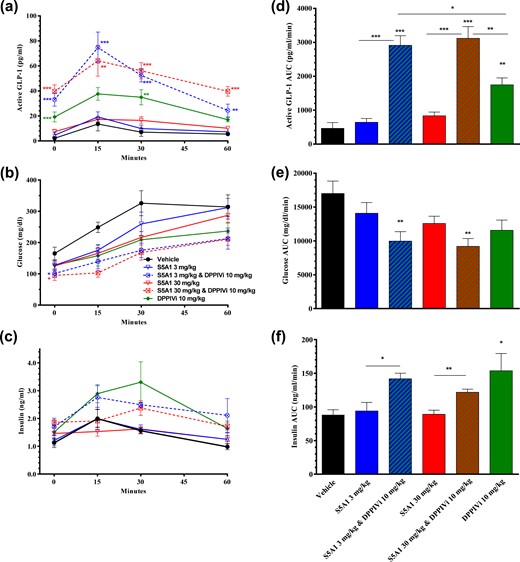
Combination of SSTR5 antagonism and DPP4 inhibition resulted in further increases in circulating GLP-1 levels and improved glycemic control in male ZDF rats. Effects of S5A1 administered alone and in combination with a DPP4i on (a, b) active GLP-1 levels, (c, d) glucose tolerance, and (e, f) insulin levels after an oral bolus of glucose (2 g/kg). Data are shown as mean ± standard error of the mean from five rats per group. *P < 0.05; **P < 0.01; ***P < 0.001.
Treatment of animals with either S5A1 or the DPP4i alone did not result in a statistically significant attenuation of the glucose excursion after the glucose challenge as measured by total glucose AUC, although trends were detected. In contrast, the glucose excursion of animals treated with the combination was suppressed significantly as compared with vehicle control [Fig. 7(c) and 7(d)]. It should be noted that glucose lowering, as measured by decreased AUC with all of the treatments groups, was primarily due to the initial decrease in fasting glucose (0 minutes). This combination treatment also resulted in profound increases in active GLP-1 levels that were significantly twice that of the DPP4i alone and sevenfold that of vehicle control as measured by AUC after the glucose challenge [Fig. 7(a) and 7(b)].
Global knockout of Sstr5 in mice increased plasma GLP-1 levels, decreased nonfasted glucose levels, and abolished S5A1-stimulated GLP-1 secretion
In light of the elevated circulating levels of GLP-1 noted in animals treated with S5A1, it seemed probable that the improved glycemic control reported in Sstr5−/− mice (23–25) could be due, at least in part, to elevated GLP-1 levels in these animals. Therefore, the nonfasted body weights, as well as the glucose, insulin, and GLP-1 (x-36) amide levels of 7-week-old homozygous Sstr5−/− mice (n = 16) and aged-matched WT littermates (n = 24), along with 15-week-old heterozygous Sstr5+/− mice (n = 5), were assessed. Sstr5−/− mice (24.7 ± 0.5 g) weighed significantly less than age-matched WT littermates (26.4 ± 0.5 g). Plasma GLP-1 levels were threefold higher (P < 0.001) in the Sstr5−/− mice [Fig. 8(a)] and remained elevated at up to 19 months of age [Fig. 8(d)]. The nonfasted plasma glucose levels in the Sstr5−/− mice were significantly lower [Fig. 8(b)] than in the WT littermates, and plasma insulin levels trended lower [Fig. 8(c)]. Despite the significantly greater body weight of the older Sstr5+/− mice (36.7 ± 0.7 g), the plasma total GLP-1 levels in these animals trended higher than in WT littermates, with no difference in plasma insulin levels.
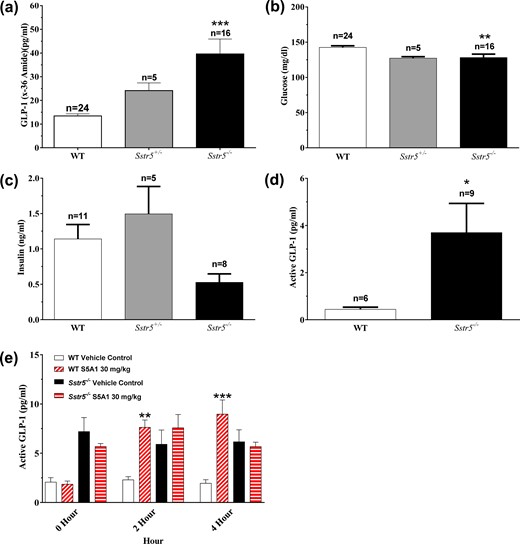
Sstr5−/− mice had increased circulating GLP-1 levels along with decreased nonfasted glucose and insulin levels, and lacked S5A1-stimulated GLP-1 secretion. Nonfasted (a) GLP-1, (b) glucose, and (c) insulin in Sstr5−/− mice compared with WT littermates at 7 to 15 weeks of age. (d) Nonfasted active GLP-1 levels in Sstr5−/− mice at 19 months of age compared with WT littermates. (e) Effects of S5A1 in Sstr5−/− mice and WT littermates on overnight-fasted active GLP-1 levels (n = 5 to 6 per group). Data shown as mean ± standard error of the mean. *P < 0.05; **P < 0.01; ***P < 0.001.
As further confirmation that the actions of S5A1 are mediated through SSTR5, the compound (30 mg/kg) was administered to overnight fasted Sstr5−/− mice and their WT littermates. The plasma active GLP-1 levels in S5A1-treated WT littermates were significantly increased at 2 and 4 hours after treatment vs those of vehicle-treated WT animals, whereas the levels of active GLP-1 in S5A1-treated Sstr5−/− mice were not increased vs those of vehicle-treated Sstr5−/− mice [Fig. 8(e)]. The free plasma concentration of S5A1 (83 to 129 nM) in both groups exceeded the mSstr5 binding affinity (Ki, 4.87 nM).
Treatment of DIO mice with S5A1 alone or in combination with a DPP4i increased plasma GLP-1 levels and improved postprandial glycemic control over 2 weeks of once-daily dosing
To probe whether compensatory mechanisms could reduce the SSTR5-mediated effects on prolonged treatment, S5A1 was administered once daily for 15 days both as a single agent (3 and 10 mg/kg) and in combination with a DPP4i (each agent at 10 mg/kg). The nonfasted total GLP-1 (x-36) amide levels in animals treated with S5A1 alone were elevated significantly at 1 hour after treatment on days 1, 8, and 15, as compared with the levels of vehicle-treated animals [Fig. 9(b)], and trended higher even at 24 hours after treatment, reaching significance for the 3 mg/kg dose on day 8 and for the 10 mg/kg dose on day 15 [Fig. 9(a)]. The nonfasted total GLP-1 (x-36) amide levels in animals treated with only the DPP4i were not different from those of vehicle-treated animals at the same time points, whereas elevations were seen in animals treated with both S5A1 and the DPP4i [Fig. 9(a) and 9(b)]. Nonfasted glucose levels were not different in any of the treated groups compared with vehicle control on days 8 and 15 (data not shown).
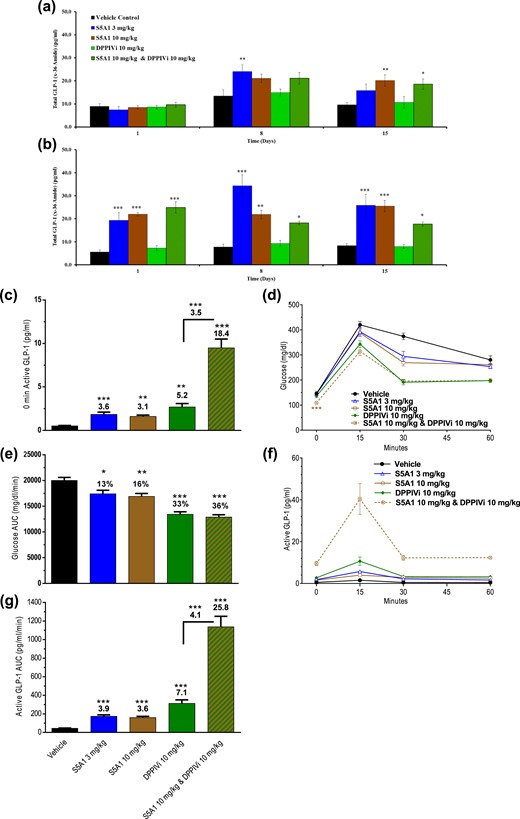
Once-daily treatment of DIO mice with S5A1 alone or in combination with DPP4 inhibition resulted in sustained increases of active GLP-1 levels and improved glycemic control over 2 weeks. Nonfasted, active GLP-1 levels (a) just before and (b) 1 hour after S5A1 administration over 15 days. Baseline (c) active GLP-1 levels, (d, e) glucose tolerance, and (f, g) active GLP-1 levels after an oral bolus of glucose on day 16 (1.5 g/kg). Data are shown as mean ± standard error of the mean from 6 mice per group. *P < 0.05; **P < 0.01; *** P < 0.001.
DIO mice were fasted overnight and subjected to an OGTT 2 hours after treatment on day 16. Baseline active GLP-1 levels were significantly increased (more than threefold) with S5A1 compared with vehicle control [Fig. 9(c)]. Whereas before dosing, glucose levels of overnight-fasted mice were not different from vehicle control for any of the treated groups (data not shown), fasted glucose levels in animals receiving the combination (but not the individual agents) were lowered significantly 2 hours after receiving the compound dose [Fig. 9(d)].
During the OGTT, S5A1 alone significantly improved glucose tolerance; however, the combination with the DPP4i produced an even significantly greater lowering of glucose AUC [Fig. 9(d) and 9(e)]. Active GLP-1 (AUC) was increased more than 3.5-fold with S5A1 compared with vehicle control. Additionally, S5A1 in combination with the DPP4i increased active GLP-1 levels fourfold higher than the DPP4i alone and 26-fold higher than vehicle control [Fig. 9(f) and 9(g)]. At the end of the OGTT, the unbound plasma concentration of S5A1 was greater than mSstr5 Ki at 3 and 10 mg/kg (37 and 83 nM, respectively). The unbound brain concentration was less than the mSstr5 binding affinity at both doses of S5A1. The unbound plasma and brain concentrations of S5A1 at 10 mg/kg were not altered with the coadministration of the DPP4i at 10 mg/kg.
Discussion
Previous authors have reported the glucose-lowering effects of SSTR5 antagonists in rodents (and Sstr5−/− mice) (23–25), and attributed it to either increased insulin sensitivity, increased insulin secretion, or other insulin-independent glucose-disposal pathways. To understand better the mechanisms by which SSTR5 antagonists lower glucose, we identified S5A1, a compound that potently and selectively binds to and antagonizes the human and mouse isoforms of SSTR5 (also potent against rSstr5 but selectivity was not determined). We demonstrated that S5A1 is a functional antagonist that increases cAMP levels in the GLP-1 secretory GLUTag and NCI-H716 cell lines by antagonizing endogenous SST, which both cell lines have been shown to secrete (38). We then used S5A1 to investigate more fully the effects of SSTR5 antagonism on insulin secretion in isolated human, mouse, and rat islets, and on the regulation of incretin levels in mice and rats.
It has been suggested that SST suppresses insulin secretion from islets through its action on SSTRs 2, 3, and/or 5 (18, 19, 39). The ability of selective SSTR5 and SSTR3 antagonists to reverse the suppression of insulin secretion by SST-14 in perifused human islets indicates a role for both receptors. Interestingly, whereas both S5A1 and BN81674 acutely reversed the inhibitory effect of exogenous SST-14 on forskolin/glucose-induced insulin secretion, the SSTR5 antagonist also blocked at least a portion of the paracrine SST tone. Furthermore, the relatively small enhancement in insulin secretion by addition of the SSTR5 antagonist at 2.8 mM glucose is consistent with previous reports that SST regulates or reduces insulin secretion from the β cell during times of high glucose concentration, but that its effects during periods of low glucose concentration are minimal (40, 41). Therefore, the risk of hypoglycemia due to SSTR5-mediated action on the islet is low. Of course, these studies do not address the impact of prolonged SSTR5 antagonism on the SST regulation of insulin secretion, nor do they define the potential role of SSTRs 1, 2, and 4 in the islet. Longer-term studies of SSTR5 antagonism in humans will be necessary to determine if other SSTRs begin to compensate for the suppression of this pathway, leading to a decrease in the action of SSTR5 antagonists on the islet over time.
In contrast to its effects on human islets, S5A1 was unable to reverse the inhibitory effect of exogenous SST-14 on the first phase of insulin secretion in mouse islets, whereas BN81674 fully restored the first phase insulin secretion in the mouse, indicating that mSstr3 is the more dominant regulator in mouse islets. Neither antagonist fully reversed the SST suppression of insulin secretion (first or second phase) in isolated rat islets. This diminished response in rodent islets to S5A1 is most likely due to the increased expression of other SSTRs, which presumably play a larger role than SSTR5 in the regulation of insulin secretion in these species. In fact, others have shown that Sstr1, Sstr2, and Sstr3 are highly expressed in rodent β cells, along with Sstr5 (42). The diminished effect of S5A1 on insulin secretion from rodent islets as compared with human islets implies that the improved glycemic control in rodents afforded by treatment with S5A1 (addressed later in the Discussion) must be due to other mechanisms.
Given our demonstration that treatment of normal and DIO mice, as well as male ZDF rats, with S5A1 resulted in increased active GLP-1 levels in both the fasted state and during an OGTT, and our observation of the marginal effects on insulin secretion from isolated rodent islets, we conclude that increased secretion of endogenous incretins caused by SSTR5 antagonism is a key driver of the glucose-lowering effects observed in earlier studies. As a result of the long duration of exposure with S5A1, GLP-1 levels were elevated the throughout the 12-hour monitoring period in healthy mice, and over 24 hours in the fed state in DIO mice. Furthermore, the durability of the effect in DIO mice over 2 weeks suggests there is no desensitization of the pathway. Increases in systemic GLP-1 have been shown to decrease the rate of gastric emptying (43) in rodent models, and this might also be the case in our studies.
The systemic levels of GLP-1 and insulin in Sstr5−/− mice are consistent with our conclusion that increased GLP-1 levels play a key role in SSTR5 antagonist-mediated glycemic control in rodents. Systemic GLP-1 levels in these mice were significantly higher than the levels seen in WT littermates, and they retained their elevated circulating GLP-1 levels up to 19 months of age, indicating that the endogenous secretory capability of the enteroendocrine cells in the gut can be maintained over a prolonged period of suppressed SSTR5 action. Systemic insulin levels in the Sstr5−/− mice were lower than in their WT littermates, presumably due to the decrease in systemic glucose levels. Finally, the lack of effect of S5A1 on GLP-1 levels in Sstr5−/− mice as compared with vehicle control supports our contention that the effects of S5A1 are, indeed, mediated through SSTR5.
Having demonstrated that SSTR5 antagonism in rodents increases GLP-1 secretion and affords increased glycemic control, we wanted to confirm that combining this mechanism with DPP4 inhibition would result in further increases in GLP-1 levels and greater glycemic control. As anticipated, codosing healthy mice with S5A1 and a DPP4i produced greater than additive increases in circulating GLP-1 levels than with either agent alone, resulting in even greater glucose tolerance, as measured by reduction in the glucose AUC in an OGTT. In male ZDF rats with an advanced disease state, treatment with S5A1 in combination with the DPP4i significantly increased GLP-1 levels and lowered overnight-fasted glucose levels, affording improved glycemic control in the OGTT. Greater than additive increases in circulating GLP-1 levels were also seen in DIO mice treated with the combination, although this did not translate into further reduction in glucose AUC in an OGTT, consistent with previous reports of GLP-1 effects in obese rodent models. Interestingly, in mice or male ZDF rats treated with a DPP4i, addition of S5A1 decreased insulin levels, presumably due to the further decrease in systemic glucose levels in animals treated with the combination and potentially a decreased rate of gastric emptying.
Our findings support species-dependent roles for SSTR5 in the SST-mediated regulation of insulin and GLP-1 secretion. In rodents, SSTR5 is involved in the regulation of GLP-1 secretion, and not in insulin secretion, whereas in humans, SSTR5 is clearly involved in the regulation of insulin secretion. Furthermore, the reported effects of SSTR agonists in humans suggest that SSTR5 is also a mediator of SST suppression of GLP-1 secretion. Whereas administration of the SST pan-agonist octreotide resulted in impaired glucose tolerance, elevated glucose, and decreased insulin secretion (44, 45), the prevalence of hyperglycemia and diabetes mellitus is even greater with pasireotide (46–48), which has a 39-fold higher binding affinity for SSTR5 than does octreotide (49, 50). After 7 days of administration, pasireotide significantly decreased plasma insulin, GLP-1, and GIP levels in healthy male volunteers, resulting in significantly impaired glucose tolerance (51). In longer phase 3 clinical trials (i.e., 6 and12 months), pasireotide treatment resulted in hyperglycemia and diabetes mellitus. Hemoglobin A1c levels in these patients were increased significantly, resulting in 46% of patients requiring a different glucose-lowering agent to control their diabetes (52). Taken together with our preclinical findings, these observations lead us to conclude that SSTR5 antagonism in patients with diabetes should reverse SST suppression of both insulin and GLP-1 secretion, resulting in improved glycemic control.
Abbreviations:
- AUC
area under the curve
- BSA
bovine serum albumin
- cAMP
cyclic adenosine monophosphate
- CHO
Chinese hamster ovary
- DIO
diet-induced obese
- DPP4i
dipeptidyl peptidase-4 inhibitor
- GIP
gastric inhibitory polypeptide
- GLP-1
glucagonlike peptide-1
- GTP
guanosine triphosphate
- IC50
half maximal inhibitory concentration
- HEC
hydroxyethylcellulose
- Kb
antagonist affinity
- Kd
equilibrium dissociation constant
- Ki
binding affinity
- MSD
Meso Scale Discovery
- OGTT
oral glucose tolerance test
- S5A1
somatostatin receptor 5 antagonist
- SPA
scintillation proximity assay
- SST
somatostatin
- SSTR
somatostatin receptor
- Sstr5−/−
knocked-out Sstr5 gene
- WT
wild-type
- ZDF rats
Zucker Diabetic Fatty rats.
Acknowledgments
Disclosure Summary: T.B.F., M.A., T.J.B., O.C., D.A.C., T.D.M., B.A.D., A.E., J.V. F., S.L.G., M.A.M.-G., V.M., G.R., M.A.S., T.M.S., S.K.S., M.A.T., F.S.W., X.Z., and D.G.B. are employees of Eli Lilly and Company and may hold company stock. K.B.B. was employed by and has equity interests in Eli Lilly and Company. T.J.B., D.A.C., M.A.M.-G., and M.A.T. are listed as inventors on US patent application 15/176,222.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}