





The cortisol-inactivating enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) that catalyzes the intracellular inactivation of glucocorticoids plays a pivotal role in human pregnant maintenance and normal fetal development. Given the fact that the main components of Hedgehog (HH) signaling pathway are predominantly expressed in syncytial layer of human placental villi where 11β-HSD2 is robustly expressed, in the present study, we have investigated the potential roles and underlying mechanisms of HH signaling in 11β-HSD2 expression. Activation of HH signaling by a variety of approaches robustly induced 11β-HSD2 expression as well as the 11β-HSD2 activity, whereas suppression of HH signaling significantly attenuated 11β-HSD2 expression as well as the 11β-HSD2 activity in both human primary cytotrophoblasts and trophoblast-like BeWo cells. Moreover, among glioma-associated oncogene (GLI) family transcriptional factors in HH signaling, knockdown of GLI2 but not GLI1 and GLI3 significantly attenuated HH-induced 11β-HSD2 expression and activity, and overexpression of GLI2 activator alone was sufficient to induce 11β-HSD2 expression and activity. Finally, GLI2 not only directly bound to the promoter region of gene hsd11b2 to transactivate hsd11b2 but also formed a heterodimer with RNA polymerase II, an enzyme that catalyzes the transcription of DNA to synthesize mRNAs, resulting in up-regulation of hsd11b2 gene transcription. Taken together, the present study has uncovered a hitherto uncharacterized role of HH/GLI2 signaling in 11β-HSD2 regulation, implicating that HH signaling through GLI2 could be required for the human pregnant maintenance and fetal development.
Hedgehog (HH) proteins comprise a family of secreted signaling molecules essential for many fundamental processes in embryonic development within metazoan (1, 2). In vertebrates, 3 HH subtypes have been found, including the Sonic HH (SHH), India HH, and Desert HH. Absence of HH ligand permits the 12 transmembrane-spanning receptor on the surface of HH-responsive cells Patched1 (PTC1) to interact with and inhibit its coreceptor smoothened (SMO), a 7 transmembrane-spanning protein. Interaction of HH ligand with PTC1 prevents PTC1 from inhibiting the activity of SMO. Activated SMO in turn initiates a series of intracellular events that culminate in activation and nuclear localization of glioblastoma (glioma-associated oncogene [GLI]) family transcription factors, which further promotes transcription of HH-responsive genes, such as Bcl2, myc, and cyclin D as well as Ptc1 and Gli1, 2 components of the HH signaling pathway itself (3–5).
As a transitory endocrine organ, human placenta is responsible for the synthesis of a number of steroid hormones via their metabolizing enzymes (6, 7). 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) (encoded by hsd11b2), a high-affinity dehydrogenase that inactivates biologically active cortisol to inactive cortisone, is one such enzyme that is highly expressed in human placental trophoblasts. The distribution of 11β-HSD2 in the human placenta is well positioned to serve as the barrier for maternal glucocorticoids entering fetal circulation (8). It is well known that overexposure to glucocorticoids can not only retard fetal growth but also program the development of chronic diseases such as hypertension, diabetes, and obesity in later life (9, 10). Thus, high expression of 11β-HSD2 in placental trophoblasts can ensure the maintenance of pregnancy and the normal development of the fetus in the presence of approximately 10-fold higher maternal cortisol (11). Moreover, genetic evidence from mice also indicates the crucial role of placental 11β-HSD2 in fetal development. Either down-regulation of 11β-HSD2 expression or genetic disruption of placental 11β-HSD2 results in increased exposure of the fetus to maternal glucocorticoid, leading to significant reductions in fetal growth and birth weight (12). Although 7 cellular signals have been demonstrated to act as regulators of 11β-HSD2 activity (13, 14), whether HH signaling affects 11β-HSD2 expression and activity in human placental trophoblasts remains unexplored.
Recently, we demonstrated the expression patterns of HH ligands and activity of the HH signaling pathway in both human and murine placentas, and further confirmed the versatile roles of HH signaling in placental development and functions including steroidogenesis, epithelial-mesenchymal transition and syncytialization (2, 15–17). In the present study, we have revealed that activation of HH pathway promotes the conversion of active cortical to inactive cortisone through GLI2-mediated induction of 11β-HSD2 expression in human tropoblasts. The findings, linking HH pathway activity with the expression and activity of enzyme 11β-HSD2, demonstrate a novel mechanism governing the conversion of active glucocorticoids to inactive glucocorticoids, providing with the potential therapeutic target for impaired placental glucocorticoid barrier and abnormal fetal development.
Materials and Methods
Cell culture
All the cell lines used in the experiments were obtained from ATCC. The human choriocarcinoma cell line BeWo was maintained in Ham's F-12K (Kaighn's) (Gibco BRL)/DMEM supplemented with 10% fetal bovine serum, 100-U/mL penicillin, and 100-mg/mL streptomycin in 5% CO2/95% air at 37°C as described previously (18). HEK293 EcR Shh cells (SHH-expressing cells) and control HEK293 cells were used for the production of biologically active murine SHH conditional medium (SM) and control medium (CM), respectively, in the presence of ecdysone. 293FT packaging cells (Life Technologies) for generating the lentiviruses were cultured as described previously (2).
Isolation and culture of cytotrophoblasts
Human placentas were obtained from uncomplicated normal term (38–40 W) pregnancy after elective cesarean section without labor, following a protocol approved by the Ethics Committee of School of Medicine of Zhejiang University. The primary cytotrophoblasts were isolated and purified as described previously (11). Briefly, tissue aliquots were removed from the maternal side of the placenta and digested with 0.125% trypsin (Sigma) and 0.02% Deoxyribonuclease I (Sigma) 4 times in DMEM (Life Technologies). The primary cytotrophoblasts were purified by using a 5%–65% Percoll (Sigma) gradient at step increments of 5% and cultured at 37°C with 5% CO2/95% air in DMEM containing 10% newborn calf serum (Life Technologies).
RNA isolation, RT-PCR, and quantitative real-time PCR
Total RNA was isolated from BeWo cells and cytotrophoblasts by using a TRIzol reagent (Takara Biotechnology Co, Ltd) according to the manufacturer's instructions. Five-microgram total RNA in a volume of 20 μL was reversely transcribed by using SuperScript III reagent (Life Technologies) and the oligo-(deoxythymidine) primer with incubation at 42°C for 1 hour. After the termination of cDNA synthesis, each reaction mixture was diluted with 80-μL Tris-EDTA buffer. mRNA levels of target genes were determined by RT-PCR and quantitative RT-PCR as described previously (2). The relative amounts of the mRNA levels of the target genes were normalized to the β-actin, glyceraldehyde-3-phosphate dehydrogenase, or α-tubulin levels, and the relative difference in mRNA levels was calculated by Ct (threshold cycle), the number of cycles at which the fluorescence exceeds the threshold. The primers used are shown in Supplemental Table 1.
Western blotting and immunoprecipitation
Western blotting and immunoprecipitations were done as previously described (19). For Western blotting, cells were washed 3 times with cold PBS and total protein extracts were prepared in whole-cell lysis buffer (50mM HEPES, 150mM NaCl, 1mM EGTA, 10mM sodium pyrophos-phate, 1.5mM MgCl2, 100mM sodium fluoride, 10% glycerol, and 1% Triton X-100) containing an inhibitor mixture (1mM phenylmethylsulfonyl fluoride, 10-μg/mL aprotinin, and 1mM sodium orthovanadate). Protein concentrations were determined by using a standard Bradford assay, and 50 μg of total protein were subjected to SDS-PAGE followed by a transfer onto Polyvinylidene Fluoride membranes (Millipore). Membranes were incubated overnight at 4°C with primary antibodies against GLI1 (sc-20687; Santa Cruz Biotechnology, Inc), GLI2 (ab26056; Abcam Ltd), GLI3 (ab69838; Abcam Ltd), Flag (14793; Cell Signaling Technology), His (sc-803; Santa Cruz Biotechnology, Inc), 11β-HSD2 (sc-20176; Santa Cruz Biotechnology, Inc), POL-II (RNA polymerase II sc-900; Santa Cruz Biotechnology, Inc), and β-actin (sc-69879; Santa Cruz Biotechnology, Inc) followed by incubation with secondary antibodies. For immunoprecipitation, cells were washed 3 times with cold PBS, and total protein extracts were prepared in proper whole-cell lysis buffer. The obtained lysates were immunoprecipitated with various antibodies. The immunoprecipitates were resolved by SDS-PAGE and subjected to immunoblotting. Signals were developed using the Enhanced Chemiluminescence System. National Institutes of Health Image software (ImageJ; http://rsb.info.nih.gov/ij/) was used to quantify the immunoreactive bands, and the normalized antigen signals were calculated from target protein-derived and β-actin-derived signals. The mean density of bands from the control cells was set to 1.
11β-HSD2 activity assay
11β-HSD2 activity was determined by measuring the conversion of cortisol to cortisone in the BeWo cells and cytotrophoblasts treated with the indicated conditions in serum-free DMEM. After washing, the cells were incubated with cortisol (1M) for 3 hours, and then the culture medium was collected. Cortisol and cortisone levels were then measured with enzyme immunoassay kits for cortisol (R&D Systems) and cortisone (Innovative Research).
Immunofluorescence
Immunocytochemistry was performed on chamber slides (Nalge Nunc International). BeWo cells or cytotrophoblasts were rinsed in PBS, fixed in ice-cold methanol, and permeabilized with 0.1% Triton X-100 in PBS. After incubation with blocking buffer (1% BSA) for 30 minutes, the cells were incubated with primary antibody against 11β-HSD2 (sc-20176; Santa Cruz Biotechnology, Inc), POL-II (sc-900; Santa Cruz Biotechnology, Inc), or His (sc-803; Santa Cruz Biotechnology, Inc) overnight at 4°C. After washing with 0.1% Triton X-100 in PBS, the cells were incubated with Alexa Fluor 488-conjugated secondary antibody (Invitrogen) or Alexa Fluor 555-conjugated secondary antibody (Invitrogen). The nuclei were stained with 6′-diamidino-2-phenylindole. Imagination was performed by a laser scanning microscope (Zeiss).
Generation of lentiviruses expressing GLI1-, GLI2-, or GLI3-shRNA
Generation of lentivirus expressing short hairpin RNA (shRNA) was described previously (2). For construction of lentiviral shRNA-expressing vectors, the hairpin shRNA templates of complementary oligonucleotides containing and overhangs were digested. The synthesized complementary oligonucleotides were annealed and inserted into the sites XbaI and NotI of a lentiviral shRNA expression vector, Pll3.7. The integrity of the shRNA-expressing construct was amplified by a DNA sequencer. The sequences of oligonucleotides were listed in the Supplemental Table 2. 293FT packaging cells were transfected with 6 μg of each construct by Lipofectamine 2000 reagents, 72 hours after transfection, lentivirus-containing supernatants were harvested, and the titers more than 1 × 106 colony forming U/mL were used for infection in BeWo cells or cytotrophoblasts for 24 hours in the presence of 8-μg/mL polybrene (Sigma).
Reporter construction, transient transfection, and reporter assays
Complex human 11β-HSD2 (hsd 11b2: nt −1683/+77, nt −1113/+77, nt −804/+77, and nt −261/+77) gene promoter regions were amplified by PCR (primers listed in the Supplemental Table 3) in the presence of genomic DNA from BeWo cells as described previously (20). The PCR products were cloned into pGL3-Basic vector (Promega) to generate the luciferase reporter constructs of 11β-HSD2. All of these constructs were verified by a DNA sequencer. Transient transfection was performed by using Lipofectamine 2000 as per the manufacturer's instructions. Briefly, BeWo cells were seeded in 24-well culture plates overnight to reach 50%–70% before transfection. The cells were transfected for 8 hours in the absence of serum. Each well contained 2-μL Lipofectamine 2000 reagents, 2-μg luciferase reporter plasmids, and 0.02-μg pRL-null-expressing Renilla luciferase (Promega) with or without 2 μg of constitutively active form of SMO (SMO*), GLI1, GLI2, N-terminally truncated GLI2 (ΔN-GLI2), or GLI3. The cells were then either cultured in the normal growth medium for 48 hours before harvest or incubated in either CM or SM for 48 hours. After the cells were harvested, the cellular lysates were prepared in 100-μL reporter lysis buffer (Promega), and the 20 μL of supernatants were used for dual-luciferase assay according to the manufacturer's instructions (Promega). The firefly luciferase levels were normalized to Renilla luciferase levels. The first bar was defined as 1.
Chromatin immunoprecipitation (ChIP) assays
ChIP was conducted using a commercial kit (17–295; Millipore) and a method modified from the manufacturer's protocol. Briefly, the cells were fixed with 1% formaldehyde to cross-link the transcription factors to chromatin DNA. After washing with PBS, the cells were resuspended with lysis buffer supplemented with protease inhibitor cocktail. The shearing of chromatin DNA was performed by sonication to produce an approximate 500 bp of input DNA and was subjected to immunoprecipitation with GLI2 antibody, POL-II antibody, or control IgG. After the immunoprecipitates were incubated with protein A agarose/salmon sperm DNA, the antibody-protein-DNA-agarose complex was washed and harvested for subsequent reverse cross-linking. The sheared DNA fragments from reverse cross-linking was extracted with a DNA extraction kit for further PCR amplification by using the primers listed in the Supplemental Table 4.
Statistical analysis
All the numerous data were expressed as mean ± SD and were analyzed by one-way ANOVA and Tukey-Kramer multiple comparison test (SPSS 13.0J software; SPSS, Inc). Statistical significance was assessed at P < .05 and P < .01. Experiments were independently triplicated, and results were qualitatively identical. Representative experiments are shown.
Results
Induction of 11β-HSD2 levels by HH in human placental trophoblasts
Human trophoblast-like BeWo cells and primary cytotrophoblasts (CTBs) capable of mimicking the biological behavior of placental villous trophoblasts are valuable models for the study of 11β-HSD2 expression (18). To investigate the effects of HH on 11β-HSD2 expression, we treated BeWo cells with either recombinant human SHH protein (SHH-N) ranging from 0 to 100 ng/mL or SM. SHH-N dose dependently induced the 11β-HSD2 mRNA levels, and SHH-N at 50 and 100 ng/mL increased the 11β-HSD2 mRNA levels by 0.8- and 2.4-fold, respectively (Figure 1A). Consistently, SM treatments for 24 and 48 hours led to the increases in 11β-HSD2 mRNA levels by 1.0- and 2.1-fold, respectively, as compared with the CM treatments (Figure 1B). Similar results were obtained from quantitative real-time PCR (qRT-PCR) assays in which other 2 housekeeping genes were used to normalize the 11β-HSD2 mRNA levels (Supplemental Figure 1, A–D). Likewise, the conversion of cortisol to cortisone was also significantly induced after SM treatment for 24 and 48 hours, and at 48 hours, SM treatment potentiated the conversion rate of cortisol to cortisone by 33.2% as compared with the CM treatment (Figure 1C and Supplemental Figure 1E). To further confirm the universality of HH in inducing 11β-HSD2 levels and activity, we treated CTBs with SHH-N and SM. Similarly, SHH-N at 50 and 100 ng/mL increased the 11β-HSD2 mRNA levels by 2.5- and 3.5-fold, respectively (Figure 1D), and increased the 11β-HSD2 protein levels by 1.8- and 3.2-fold, respectively (Figure 1G). SM at 24 and 48 hours increased the 11β-HSD2 mRNA levels by 1.3- and 3.2-fold, respectively (Figure 1E), and SM at 48 hours increased the 11β-HSD2 protein levels by 2.7-fold (Figure 1H). Likewise, 11β-HSD2 activity converting cortisol to cortisone was also significantly induced in CTBs followed by SM treatment for 24 and 48 hours, and at 48 hours, SM treatment potentiated the conversion rate of cortisol to cortisone by 17.3% as compared with the CM treatment (Figure 1F and Supplemental Figure 1F). However, pretreatment with 5E1 SHH neutralizing antibody almost completely abolished SM-induced 11β-HSD2 mRNA levels and protein levels (Figure 1I; data not shown). Taken together, HH induces the 11β-HSD2 expression levels not only in human trophoblast-like BeWo cells but also in human primary CTBs.
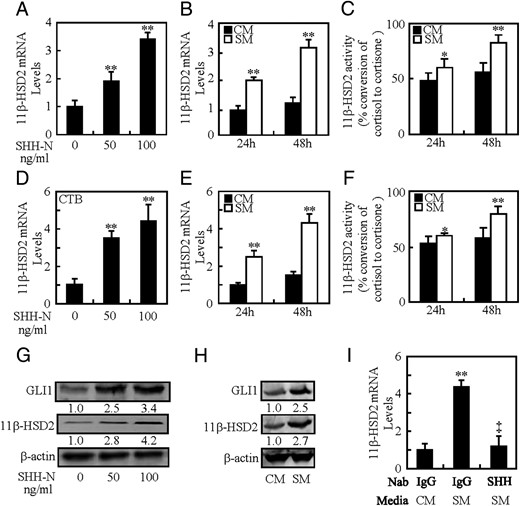
Induction of 11β-HSD2 expression by SHH. A and B, 11β-HSD2 mRNA levels in BeWo cells after culture with N-SHH or SM. C, 11β-HSD2 activity in BeWo cells after culture with SM. D and E, 11β-HSD2 mRNA levels in CTBs after culture with N-SHH or SM. F, 11β-HSD2 activity in CTBs after culture with SM. G and H, 11β-HSD2 and GLI1 protein levels in BeWo cells after culture with N-SHH or SM. I, 11β-HSD2 mRNA levels in BeWo cells after culture with 5E1 anti-SHH neutralizing antibody. mRNA and protein abundance was normalized to β-actin, respectively; *, P < .05; **, P < .01 vs CM or vehicle; ‡, P < .01 vs SM with control antibody; n = 6; error bar, SD.
Involvement of SMO in HH-induced 11β-HSD2 expression
To determine whether HH-induced 11β-HSD2 is SMO dependent or not, we activated HH signaling by a SMO agonist, purmorphamine, or overexpression of a constitutively active form of SMO (SMO*), and suppressed HH signaling by a SMO inhibitor, cyclopamine. Activation of SMO by purmorphamine increased 11β-HSD2 mRNA and protein levels in dose-dependent manners. Purmorphamine at 2μM induced 11β-HSD2 mRNA and protein levels by 3.3- and 2.5-fold, respectively (Figure 2, A and B), whereas it increased the conversion of cortisol to cortisone by 31% (Figure 2C and Supplemental Figure 2A). Likewise, overexpression of SMO* increased 11β-HSD2 mRNA and protein levels by 3.4- and 1.7-fold, respectively (Figure 2, D and E), and it increased the conversion of cortisol to cortisone by 37% (Figure 2F and Supplemental Figure 2B). Conversely, pretreatment of cells with cyclopamine significantly reversed the effect caused by SM. Cyclopamine dose dependently decreased SM-induced not only the expression of 11β-HSD2 at mRNA and protein levels but also the activity of 11β-HSD2. Cyclopamine at 5μM decreased the SM-induced 11β-HSD2 mRNA and protein levels by 67% and 70%, respectively (Figure 2, G and H) and suppressed SM-induced conversion of cortisol to cortisone by 20.2% (Figure 2I and Supplemental Figure 2C). Thus, HH-induced 11β-HSD2 expression is SMO dependent, and activation of SMO alone is sufficient to induce 11β-HSD2 expression.
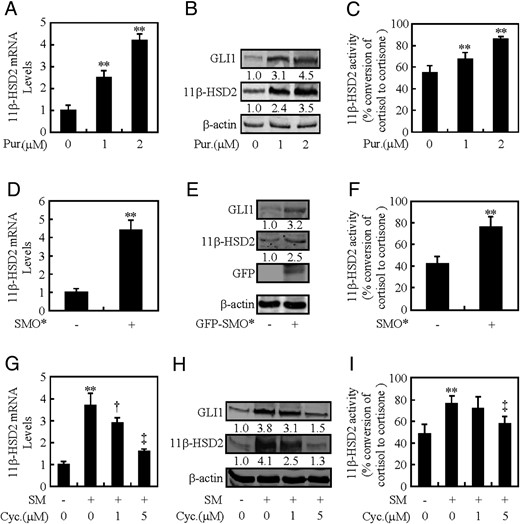
SMO is involved in HH-induced 11β-HSD2 expression. A–C, 11β-HSD2 mRNA levels, protein levels of 11β-HSD2 and GLI1, and 11β-HSD2 activity after treatment with Pur for 48 hours in BeWo cells. D–F, 11β-HSD2 mRNA levels, protein levels of 11β-HSD2 and GLI1, and 11β-HSD2 activity after overexpression of SMO* in BeWo cells. G–I, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity after treatment with Cyc for 48 hours in BeWo cells. RNA and protein abundance was normalized to β-actin, respectively. †, P < .05; **, ‡, P < .01 vs vehicle, empty vector, CM, or SM with vehicle; n = 6; error bar, SD.
Requirement of GLI2 but not GLI1 and GLI3 in HH-induced 11β-HSD2 expression
In order to assess the potential involvement of GLI transcriptional factors in HH-induced 11β-HSD2 expression, we generated lentiviruses expressing GLI1-, GLI2-, or GLI3-shRNA which knocked down the expression of GLI1, GLI2, and GLI3 by as much as 50%–70% at either mRNA or protein levels (Supplemental Figure 3, A–C). Knockdown of GLI2 attenuated SM-induced 11β-HSD2 expression at mRNA and protein levels by 52% and 50%, respectively, and as almost completely abolished SM-induced conversion of corticol to cortisone (Figure 3, D–F, and Supplemental Figure 3, F and G), whereas knockdown of GLI1 or GLI3 affected SM-induced neither the 11β-HSD2 mRNA and protein levels nor the conversion of corticol to cortisone (Figure 3, A–C and G–I, and Supplemental Figure 3G). On the other hand, overexpression of GLI2 alone induced 11β-HSD2 mRNA and protein levels by 2.6- and 1.2-fold, respectively, and increased the conversion of corticol to cortisone by 28% (Figure 3, J–L, and Supplemental Figure 3, H and I). Likewise, overexpression of constitutively active form of GLI2 (ΔN-GLI2, N-terminally truncated GLI2) induced 11β-HSD2 mRNA and protein levels by 5.9- and 2.7-fold, respectively, and increased the conversion of corticol to cortisone by 82% (Figure 3, J–L, and Supplemental Figure 3, H and I). However, overexpression of neither GLI1 nor GLI3 significantly affected the 11β-HSD2 levels and activities (Supplemental Figure 3, D and E; data not shown). Taken together, these data demonstrate GLI2 but not GLI1 and GLI3 is required for HH-induced 11β-HSD2 expression, and overexpression of GLI2 alone is sufficient to induce 11β-HSD2 expression.
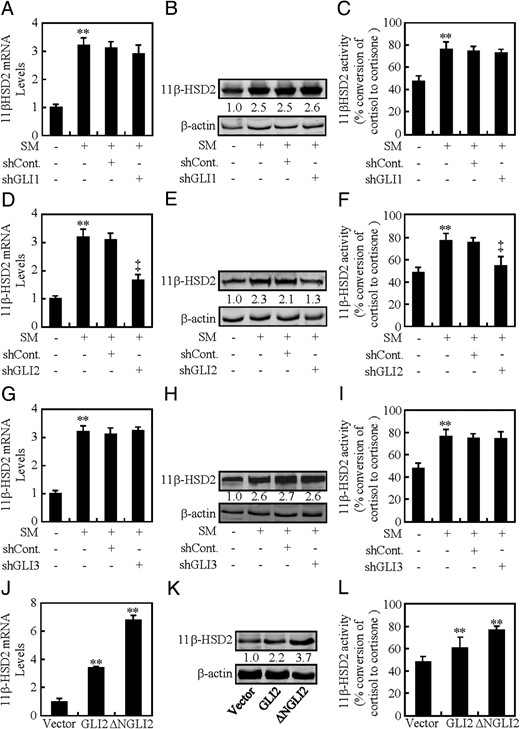
GLI2 is essential for HH-induced 11β-HSD2 expression. A–C, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity after GLI1 was knocked down in BeWo cells in the presence of CM or SM. D–F, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity after GLI2 was knocked down in BeWo cells in the presence of CM or SM. G–I, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity after GLI3 was knocked down in BeWo cells in the presence of CM or SM. J–L, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity in BeWo cells transfected with the indicated plasmids. RNA and protein abundance was normalized to β-actin, respectively. ** and ‡, P < .01 vs CM with control-shRNA, SM with control-shRNA, or empty vector; n = 6; error bar, SD.
HH/GLI2-induced 11β-HSD2 expression is independent of syncytialization
As previously reported, 11β-HSD2 expression is up-regulated during the process of placental trophoblasts fusion, namely syncytialization, and HH/GLI2 is involved in directing syncytialization (15, 21). To exclude the possibility that HH/GLI2-induced 11β-HSD2 expression is resulted from the trophoblasts fusion, we knocked down GLI2 in human trophoblast-like choriocarcinoma JAR cells, which express 11β-HSD2 but show a weak ability to fuse (22, 23), and checked the 11β-HSD2 mRNA and protein levels. As a result, knockdown of GLI2 attenuated SM-induced 11β-HSD2 mRNA and protein levels by 70% and 60%, respectively, as well as the 11β-HSD2 activity (Figure 4, A, B, and D, and Supplemental Figure 4A). Similarly, overexpression of GLI2 in trophoblast-like JAR cells increased 11β-HSD2 protein levels by 2.1-fold (Figure 4C). Immunostaining of a single cell also showed that treatment with SM increased the 11β-HSD2 expression significantly as compare with that of CM (Figure 4E). Previous study identified that placental trophoblasts fusion is dependent on the transcomplex formation of GLI2 and Glial Cell Missing-a (GCMa), and loss of either GLI2 or GCMa leads to the failure of trophoblasts fusion (15). To further confirm whether HH/GLI2-induced 11β-HSD2 expression is independent of trophoblasts fusion, we knocked down either GLI2 or GCMa in combination with overexpression of GCMa and GLI2. As a result, although overexpression of GLI2 in combination with knockdown of GCMa inhibited trophoblasts fusion, 11β-HSD2 expression level was still comparable with that of overexpression of GLI2 alone (Figure 4, F and G). Taken together, HH/GLI2-induced 11β-HSD2 expression is independent of trophoblasts syncytialization.
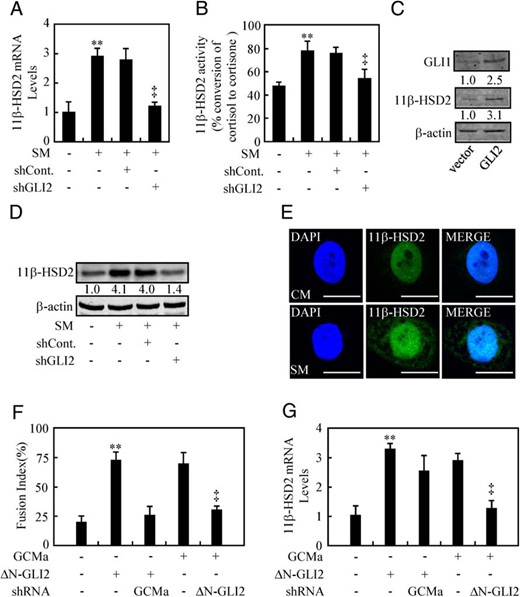
HH-induced 11β-HSD2 expression is independent of syncytialization. A, B, and D, 11β-HSD2 mRNA and protein levels, and 11β-HSD2 activity after GLI2 was knocked down in JAR cells in the presence of CM or SM. C, 11β-HSD2 and GLI1 protein levels in BeWo cells transfected with the indicated plasmids. E, Immunofluorestaining of 11β-HSD2 in BeWo cells after 48 hours of culture in either SM or CM. F and G, Fusion index and 11β-HSD2 mRNA levels after BeWo cells were transient transfected and infected with the indicated plasmids and lentiviruses. RNA and protein abundance was normalized to β-actin, respectively; ** and ‡, P < .01 vs CM, empty vector with control-shRNA, ΔN-GLI2 with control-shRNA, or GCMa with control-shRNA; n = 6; error bar, SD. Scale bar, 80 μm.
GLI2 directly binds to the promoter region of hsd11b2
To further confirm how GLI2 regulates 11β-HSD2 expression, we cloned 11β-HSD2 promoter region (hsd11b2, nt −1683 to nt +77) containing 2 potential GLI response elements and generated luciferase reporter construct and its deletion mutants lacking 1 or 2 responsive elements (Figure 5A). In human BeWo cells, after treatment with SM or CM for 48 hours, the full-length construct (nt −1683 to nt +77) exhibited a robust response to SM over CM (12.1-fold), whereas all deletion mutants led to significant diminishments in luciferase expression (Figure 5B). Likewise, inhibition of HH signaling by cyclopamine ranging from 0μM to 5μM attenuated SM-induced luciferase activity in a dose-dependent manner, and knockdown of GLI2 attenuated SM-induced luciferase activity by 71%, whereas overexpression of ΔN-GLI2 almost completely reversed GLI2-shRNA-negated luciferase activities (Figure 6, C–E). Conversely, activation of HH signaling by either purmorphamine at 2μM or overexpression of SMO* significantly stimulated luciferase activities by 7.7- and 6.2-fold, respectively, and overexpression of ΔN-GLI2 alone induced the luciferase activity by approximately 16.5-fold (Figure 6, F–H), whereas overexpression neither GLI1 nor GLI3 affected the luciferase activity (Supplemental Figure 5, A and B). To examine the physical interaction between GLI2 and the promoter region of hsd11b2 gene, we performed ChIP-PCR assays in CTBs by using the specific antibody against GLI2. The PCR primers were designed to amplify specific gDNA fragments of nt −1200/−850 and nt −800/−450, and the ChIP assays were normalized to their corresponding input. As expected, GLI2 robustly bound to the fragment of nt −1200/−850 but not nt −800/−450 (Figure 6I). Thus, we identified that 11β-HSD2 is a direct target of HH/GLI2 signaling.
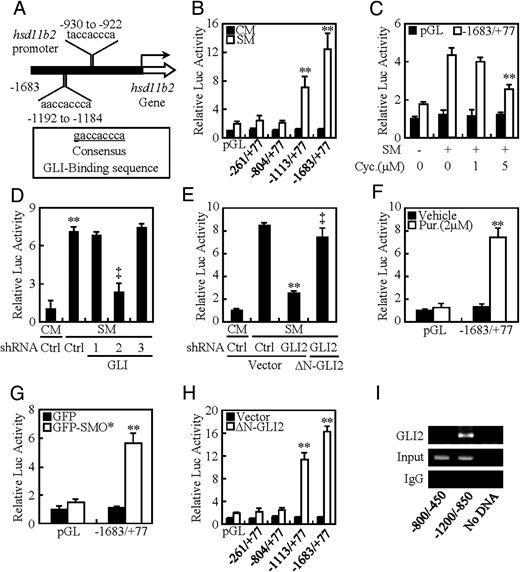
hsd11b2 is a direct downstream target of HH signaling. A, Schematic presentations of promoter regions with potential GLI consensus and nonconsensus binding sites of hsd11b2 gene encoding 11β-HSD2. B–H, Dual-luciferase assays in BeWo cells treated with the indicated conditions. I, ChIP followed by PCR (ChIP-PCR) in CTBs by using antibody against GLI2. The abundance was normalized to the corresponding input. ** and ‡, P < .01 vs empty vector with control-shRNA, ΔN-GLI2 with control-shRNA, CM, SM, SM with vehicle, empty vector, CM with control-shRNA, SM with control-shRNA, or SM with GLI2-shRNA; n = 6; error bar, SD.
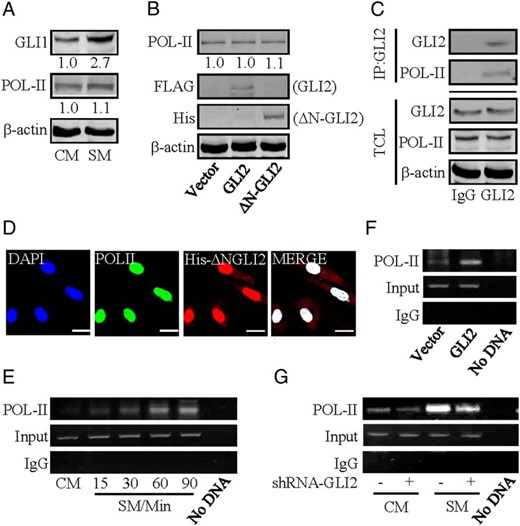
Complex formation of GLI2 and RNA polymerase II. A and B, Protein levels of RNA polymerase II in BeWo cells after treatment with the indicated conditions. Protein abundance was normalized to β-actin, respectively. C, Coimmunoprecipitation of endogenous GLI2 and RNA polymerase II in BeWo cells. IP: GLI2; WB: RNA polymerase II. Protein abundance was normalized to β-actin. D, Immunofluorescent staining of His-tagged ΔN-GLI2 and RNA polymerase II in HEK293 cells. E–G, ChIP followed by PCR (ChIP-PCR) in CTBs by using antibody against RNA polymerase II. The abundance was normalized to the corresponding input; n = 6. Scale bar, 20 μm.
Stimulation of RNA polymerase II binding to the promoter of hsd11b2 by activation of HH signaling
As an enzyme that catalyzes the transcription of DNA to synthesize mRNAs, RNA polymerase II plays a vital role in the binding of a wide range of transcription factors to upstream gene promoters (24, 25). Western blotting data showed that either SM treatment or overexpression of GLI2 failed to cause an increase in RNA polymerase II protein levels (Figure 6, A and B). In coimmunoprecipitation assays, protein complexes precipitated with IgG contained no GLI2 and RNA polymerase II, whereas the protein complexes precipitated with a GLI2 antibody contained abundantly endogenous RNA polymerase II, which is consistent with the data showing the colocalization of ΔN-GLI2 and RNA polymerase II in HEK293 cells (Figure 6, C and D). To further confirm whether GLI2 affects the binding of RNA polymerase II to hsd11b2 promoter, CHIP-PCR was performed by using chromatins isolated from CTBs at different time intervals after HH stimulation. Enhanced RNA polymerase II binding to the hsd11b2 promoter was detected 60 minutes after HH stimulation (Figure 6E). Similarly, overexpression of GLI2 alone is sufficient to enhance RNA polymerase II binding to the hsd11b2 promoter (Figure 6F). Conversely, knockdown of GLI2 attenuated SM-induced RNA polymerase II binding to the hsd11b2 promoter by 55% (Figure 6G), whereas knockdown of either GLI1 or GLI3 did not affect the binding (data not shown). Collectively, activation of HH enhances RNA polymerase II binding to the promoter of hsd11b2 gene by activated GLI2, which further promotes hsd11b2 transcription.
Discussion
The present study, to our best knowledge for the first time, has revealed that GLI2 not only directly binds to the promoter region of gene hsd11b2 to transactivate hsd11b2 but also forms a heterodimer with RNA polymerase II to promote hsd11b2 gene transcription in response to HH (Figure 7). Thus, our study has uncovered a hitherto uncharacterized role of HH/GLI2 signaling in human placental 11β-HSD2 expression.
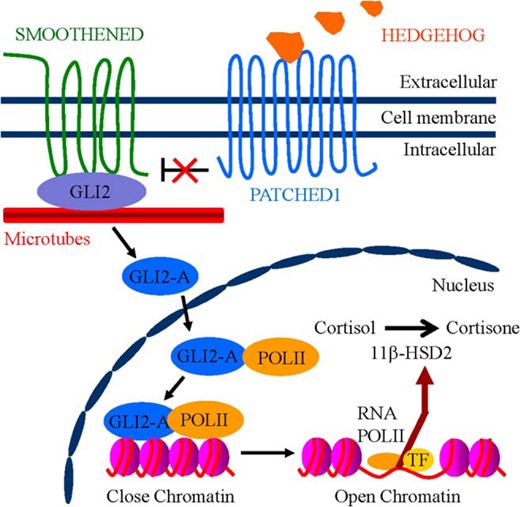
Schematic graph showing the proposed model for HH/GLI2-induced 11β-HSD2 expression in human trophoblasts. In the presence of HH ligands, binding of HH to PTCH1 relieves the inhibition of SMO, resulting in activation of GLI2-A (active form of GLI2). GLI2-A enters the nucleus, which thereby forms a transcriptional complex with RNA polymerase II that transactivates downstream target hsd11b2 converting cortisol to cortisone.
There is robust evidence that the expression of 11β-HSD2 is highly tissue specific in human body (26–28). High expression of 11β-HSD2 is observed in mineralocorticoid target organs where the mineralocorticoid receptor is shielded against cortisol. Although the placenta is not considered as a typical mineralocorticoid target organ, 11β-HSD2 is abundantly expressed in the syncytial layer of the villi where it forms the protective barrier for the fetus against the 10-fold higher maternal cortisol (14). Abnormal placental 11β-HSD2 expression has been shown to be associated with intrauterine fetal growth retardation. Thus, maintenance of appropriate 11β-HSD2 expression in the placenta is crucial for the normal development of the fetus. Despite the previous knowledge of the differential expression pattern of 11β-HSD2 in the trophoblasts, the underlying molecular mechanisms remain obscure.
HH ligands secreted by HH-producing cells act on the HH-responding cells to fulfill signaling transduction and regulate cell behaviors (1). Given the fact that PTC1 and SMO are robustly and predominantly expressed in the syncytial layers of human placental villi, we assume that HH proteins may signal through paracrine mechanism to regulate 11β-HSD2 of CTBs. This hypothesis has been confirmed by the following solid evidence in the present study. 1) In both BeWo cells and primary human placental CTBs culture models, activation of HH signaling induces the 11β-HSD2 expression. 2) Inhibition of HH signaling by multiple approaches results in attenuation of 11β-HSD2 expression. 3) Knockdown of GLI2 or overexpression of active form of GLI2 significantly affects 11β-HSD2 expression. And 4) GLI2 forms a complex with POL-II to transactivate gene hsd11b2. Thus, our findings not only uncover a hitherto uncharacterized role of HH signaling but also expand the list of tissues and cells influenced by HH signaling. These findings correspond to our previous results showing that knockout of SHH (SHH−/−) in murine placentas leads to defects in the development of placental labyrinth, and that placenta-specific knockdown of GLI2 by lentiviral GLI2-shRNA in SHH−/+ murine placentas phenocopies labyrinthine defects of SHH−/− placentas (17). However, whether GLI2 indeed promotes 11β-HSD2 expression, genetic evidence from trophoblast-specific knockout of GLI2 in murine placenta is worthy of further investigation.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Amino acids 781–1080 of GLI-1 of human origin | GLI1 | sc-20687, Santa Cruz Biotechnology, Inc | Rabbit; polyclonal | 1:200 | |
| Peptide corresponding to human Gli2 | RNDVHLRTPLLKENGDSEAGTEPGGPESTEASSTSQAVEDCLHVRAIKTE | GLI2 | ab26056, Abcam Ltd | Rabbit; polyclonal | 1:2000 |
| Synthetic peptide conjugated to KLH derived from within residues 1–100 of human Gli3 | GLI3 | ab69838, Abcam Ltd | Rabbit; polyclonal | 1:2000 | |
| DYKDDDDK tag (D6W5B) | FLAG | 14793, Cell Signaling Technology | Rabbit; monoclonal | 1:2000 | |
| His-probe antibody (H-15) | HIS | sc-803, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Amino acids 261–405 mapping at the C terminus of 11β-HSD2 of human origin | 11β-HSD2 | sc-20176, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Epitope mapping within the tandem repeat domain of the large subunit of RNA polymerase II (RPB1) of mouse origin | POL-II | sc-900, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| A slightly modified synthetic peptide corresponding to β-cytoplasmic actin | β-Actin | sc-69879, Biotechnology, Inc | Mouse; monoclonal | 1:1000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Amino acids 781–1080 of GLI-1 of human origin | GLI1 | sc-20687, Santa Cruz Biotechnology, Inc | Rabbit; polyclonal | 1:200 | |
| Peptide corresponding to human Gli2 | RNDVHLRTPLLKENGDSEAGTEPGGPESTEASSTSQAVEDCLHVRAIKTE | GLI2 | ab26056, Abcam Ltd | Rabbit; polyclonal | 1:2000 |
| Synthetic peptide conjugated to KLH derived from within residues 1–100 of human Gli3 | GLI3 | ab69838, Abcam Ltd | Rabbit; polyclonal | 1:2000 | |
| DYKDDDDK tag (D6W5B) | FLAG | 14793, Cell Signaling Technology | Rabbit; monoclonal | 1:2000 | |
| His-probe antibody (H-15) | HIS | sc-803, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Amino acids 261–405 mapping at the C terminus of 11β-HSD2 of human origin | 11β-HSD2 | sc-20176, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Epitope mapping within the tandem repeat domain of the large subunit of RNA polymerase II (RPB1) of mouse origin | POL-II | sc-900, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| A slightly modified synthetic peptide corresponding to β-cytoplasmic actin | β-Actin | sc-69879, Biotechnology, Inc | Mouse; monoclonal | 1:1000 |
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Amino acids 781–1080 of GLI-1 of human origin | GLI1 | sc-20687, Santa Cruz Biotechnology, Inc | Rabbit; polyclonal | 1:200 | |
| Peptide corresponding to human Gli2 | RNDVHLRTPLLKENGDSEAGTEPGGPESTEASSTSQAVEDCLHVRAIKTE | GLI2 | ab26056, Abcam Ltd | Rabbit; polyclonal | 1:2000 |
| Synthetic peptide conjugated to KLH derived from within residues 1–100 of human Gli3 | GLI3 | ab69838, Abcam Ltd | Rabbit; polyclonal | 1:2000 | |
| DYKDDDDK tag (D6W5B) | FLAG | 14793, Cell Signaling Technology | Rabbit; monoclonal | 1:2000 | |
| His-probe antibody (H-15) | HIS | sc-803, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Amino acids 261–405 mapping at the C terminus of 11β-HSD2 of human origin | 11β-HSD2 | sc-20176, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Epitope mapping within the tandem repeat domain of the large subunit of RNA polymerase II (RPB1) of mouse origin | POL-II | sc-900, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| A slightly modified synthetic peptide corresponding to β-cytoplasmic actin | β-Actin | sc-69879, Biotechnology, Inc | Mouse; monoclonal | 1:1000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Amino acids 781–1080 of GLI-1 of human origin | GLI1 | sc-20687, Santa Cruz Biotechnology, Inc | Rabbit; polyclonal | 1:200 | |
| Peptide corresponding to human Gli2 | RNDVHLRTPLLKENGDSEAGTEPGGPESTEASSTSQAVEDCLHVRAIKTE | GLI2 | ab26056, Abcam Ltd | Rabbit; polyclonal | 1:2000 |
| Synthetic peptide conjugated to KLH derived from within residues 1–100 of human Gli3 | GLI3 | ab69838, Abcam Ltd | Rabbit; polyclonal | 1:2000 | |
| DYKDDDDK tag (D6W5B) | FLAG | 14793, Cell Signaling Technology | Rabbit; monoclonal | 1:2000 | |
| His-probe antibody (H-15) | HIS | sc-803, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Amino acids 261–405 mapping at the C terminus of 11β-HSD2 of human origin | 11β-HSD2 | sc-20176, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| Epitope mapping within the tandem repeat domain of the large subunit of RNA polymerase II (RPB1) of mouse origin | POL-II | sc-900, Biotechnology, Inc | Rabbit; polyclonal | 1:1000 | |
| A slightly modified synthetic peptide corresponding to β-cytoplasmic actin | β-Actin | sc-69879, Biotechnology, Inc | Mouse; monoclonal | 1:1000 |
Most importantly, the present study has identified that GLI2 not only directly binds with the promoter region of gene hsd11b2 but also interacts with POL-II and promotes POL-II-controlled transcription of gene hsd11b2. The interaction between GLI2 and POL-II is attested by the following evidence. 1) Immunoprecipitation experiments indicate that both GLI2 activator and POL-II exist in 1 immunocomplex. 2) Immunofluorescent staining indicates both GLI2 activator and POL-II are colocalized in the nuclei of trophoblasts. And 3) ChIP-PCR experiments demonstrate that GLI2 physically interacts with POL-II in the promoter region of gene hsd11b2. However, whether the interaction between GLI2 and POL-II is direct or not and which exact domains links GLI2 and POL-II are needed to be further determined.
HH family proteins have been recognized as one of the major intercellular signals that play vital roles in cell growth, cell survival, and decision of cell fate (29–31). 11β-HSD2 is expressed not only in the human placenta, but also several other organs, including distal nephron and colon (28, 32). Although the present study has revealed the mechanisms by which HH regulates 11β-HSD2 in human placenta, whether HH regulates 11β-HSD2 in other tissues and whether that regulation is through the same way need to be further addressed. On the other hand, given the fact that the placenta not only expresses 11β-HSD2 but also the isoenzyme 11β-HSD1 that converts cortisone into cortisol, and that 11β-HSD1 expressions are down-regulated by HH stimulation (our preliminary data, data not shown), the interaction between HH and 11β-HSD1 is worthy of further investigation.
In conclusion, we have demonstrated that activation of HH signaling by paracrine or autocrine is crucial for the up-regulation and maintenance of 11β-HSD2 expression in human placenta. Given the essential role of placental 11β-HSD2 in the human pregnancy maintenance and fetal development, our uncovered molecular mechanism underlying the basal and induced expression of 11β-HSD2 by HH/GLI2 signal may lay a foundation for developing novel therapeutic and preventive strategies for the pregnancy failure and diseases such as intrauterine growth retardation.
Acknowledgments
We thank Dr Sasaki for his generous donation of plasmid.
Author contributions: H.Z. performed all the experiments; C.Z. performed the Western blotting for 11β-HSD2 of Supplemental data; X.F. performed the qRT-PCR for 11β-HSD2 of Supplemental data; W.X. performed the Western blotting for GLI1, GLI2, and GLI3; L.T. performed the qRT-PCR for GLI1, GLI2, and GLI3; X.W. and C.T. designed the study; and C.T. wrote the manuscript. All authors read and approved the final manuscript.
This work was supported by the 973 Program Grant 2011CB944403; National Natural Science Foundation of China Grants 31271561, 81170016, 81170787, 81370713, and 31571493; and the Natural Science Foundation of Zhejiang Province Grant LY13H150002.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- ChIP
chromatin immunoprecipitation
- CM
control medium
- CTB
cytotrophoblast
- GCMa
Glial Cell Missing-a
- GLI
glioma-associated oncogene
- HH
Hedgehog
- 11β-HSD2
11β-hydroxysteroid dehydrogenase type 2
- POL-II
RNA polymerase II
- PTC1
Patched1
- qRT-PCR
quantitative real-time PCR
- SHH
Sonic HH
- shRNA
short hairpin RNA
- SM
SHH conditional medium
- SMO
smoothened.
References
Author notes
H.Z. and C.Z. contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}