





The hippocampus, a brain region critical for learning, memory and emotional processing, maintains its capacity to undergo structural plasticity throughout life. Hippocampal structural plasticity can be modulated by a number of intrinsic and extrinsic factors. This study investigated the effects of adiponectin, an adipocyte-derived hormone, on dendritic growth, arborization, and spinogenesis in mature granule neurons of the hippocampal dentate gyrus generated during embryonic (early-born) or early postnatal (late-born) stages. We found that adiponectin deficiency reduced dendritic length, branching and spine density of granule neurons. The reduction was more evident in early-born granule neurons than in late-born granule neurons. Intracerebroventricular infusion of adiponectin for 1 week increased of dendritic spines and arbor complexity in late-born granule neurons. Moreover, adiponectin deficiency decreased the production of adult-born new granule neurons through suppressing neural progenitor cell proliferation and differentiation, whereas intracerebroventricular adiponectin infusion increased the proliferation of neural progenitor cells in adult dentate gyrus. These results suggest that adiponectin plays an important role in dendritic spine remodeling and neurogenesis in the dentate gyrus.
The hippocampus maintains the ability to undergo structural reorganization and continuously generate new neurons during adulthood. Remodeling of dendritic spines and synapses of existing neurons and adult neurogenesis in the hippocampus are critical for memory and emotional processing (1–3). Aberrant dendritic spine remodeling and disrupted adult neurogenesis are thought to contribute to the pathophysiology of depression and cognitive deficits (4–6).
Dendrites elongate and branch, and spines along dendrites constantly change in number and shape in response to a number of extracellular stimuli. Dendritic spines are the major postsynaptic sites of excitatory synapses and highly plastic in the hippocampus (7). Remodeling of dendritic spines and synapses is a fundamental process for reshaping neural circuits. Changes in dendritic spine density and morphology have been associated with hippocampal-dependent learning and memories (8–10). The acquisition of new memories increases dendritic spine density in the hippocampus in a variety of learning tasks (8, 9, 11–14). Specifically, spine density in the dentate gyrus of the hippocampus has been shown to increase after passive avoidance training (15) and spatial learning (9). Long-term potentiation, a presumed cellular substrate for learning and memory, induces the formation of new dendritic spines in the dentate gyrus (16). Chronic stress causes retraction of dendritic trees and loss of dendritic spines in hippocampal neurons, accompanied by cognitive deficits and depression-related behaviors (17–21). Similar dendritic spine changes were also seen after administration of the stress hormone corticosterone (22–24). On the other hand, antidepressant treatment can promote dendritic growth and spine formation (25–28) and reverse stress-induced changes in dendritic spine loss (29–31).
The dentate gyrus is one of the two brain regions where neurogenesis occurs throughout adulthood (32, 33). Adult neurogenesis gives rise to new granule neurons that integrate into existing dentate gyrus circuits to influence hippocampal-dependent functions (34–36). This is a multistep process that can be regulated by numerous factors associated with mood and cognitive states (37–39). Hippocampal-dependent learning increases the number of adult-born neurons (40, 41), whereas exposure to different stressors and stress hormones reduces hippocampal neurogenesis (42–45). Antidepressants share the common feature of increasing hippocampal neurogenesis and block stress-induced suppression of neurogenesis (36, 46–48). Studies have shown that ablation of hippocampal neurogenesis impairs contextual fear conditioning, spatial and object recognition memory, and pattern separation (49–51). Moreover, mice deficient in hippocampal neurogenesis show abnormal responses to stress and impaired negative feedback control of the hypothalamo-pituitary-adrenal (HPA) axis (52–54), supporting a role for adult neurogenesis in stress-related disorders.
Adiponectin is secreted by adipocytes and circulates in the blood as trimeric, hexameric, and high-molecular-weight multimeric forms (55). The trimeric and hexameric forms are detectable in the cerebrospinal fluid (CSF) of humans and rodents (56–59). Intravenous injection of adiponectin increases adiponectin level in CSF of wild-type (WT) animals and adiponectin-deficient mice (57, 59), indicating that adiponectin enters the brain from circulation (60). We have shown that adiponectin haploinsufficiency increases susceptibility to stress-induced depressive-like behaviors, and central infusion of adiponectin produces antidepressant-like effects (61). The role of adiponectin in regulating depression-related behavior has been confirmed by two recent studies (62, 63). Moreover, we have recently found that adiponectin in the dentate gyrus regulates contextual extinction learning (64). Whether these behavioral effects of adiponectin are associated with dendritic spine remodeling and adult neurogenesis are unknown. Anatomical data revealed that adiponectin receptor (AdipoR)1 and AdipoR2 are highly expressed in the dentate gyrus (61, 65), suggesting that adiponectin directly acts on dentate gyrus neurons and influences their functions. In this study, we investigated the effects of adiponectin deficiency and central infusion of exogenous adiponectin on two forms of structural plasticity in the dentate gyrus, ie, morphological modifications of dendritic spines of existing neurons and generation of new neurons.
Materials and Methods
Animals
Adult WT male C57BL/6J mice were purchased from The Jackson Laboratory. Heterozygous adiponectin gene knockout mice on a C57BL/6J genetic background originally obtained from Dr Philipp Scherer's laboratory (University of Texas Southwestern Medical Center, Dallas, TX) (66) were intercrossed to generate adiponectin-deficient (Adipo−/−) mice, adiponectin-haploinsufficient (Adipo+/−) mice, and WT littermates. Mice were housed in groups of 5 under a 12-hour light, 12-hour dark cycle (lights on at 9 am) with ad libitum access to food and water. All experiments were conducted on adult male mice at the age of 9–12 weeks. All procedures were carried out in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committees of University of Texas Health Science Center at San Antonio.
Chemicals
Recombinant mouse adiponectin (R&D Systems) was dissolved in artificial CSF (137mM NaCl, 2.7mM KCl, 0.5mM MgCl2 · 6H2O, 0.9mM CaCl2 · 2H2O, 1.5mM KH2PO4, and 8.1mM Na2HPO4) at a concentration of 0.5 μg/μL. Bromodeoxyuridine (BrdU) (Sigma-Aldrich) was dissolved in sterile saline at a concentration of 5 mg/mL before use and administrated ip at a dose of 50 mg/kg.
Intracerebroventricular (ICV) cannulation and infusion
Stereotaxic surgery was performed as previously described (61). A 23-gauge guide cannula was implanted into the lateral ventricle (0.2 mm posterior, 1.1 mm lateral, and 2.7 mm ventral to the bregma). After surgery, mice were individually housed, handled daily, and allowed to recover for 7 days. ICV infusion was performed on conscious, unrestrained, and freely moving mice in their home cage in order to avoid the peripheral effects. On the experimental day, a 33-G stainless-steel injector connected to a 5-μL syringe was inserted into the guide cannula and extended 1 mm beyond the tip. Adiponectin (0.5 μg/μL) or artificial CSF was infused in a volume of 2 μL over 2 minutes. The injector tip was held in place for additional 5 minutes after the end of infusion to avoid backflow.
Nissl staining
Brain sections (20 μm) were mounted onto glass slides coated with 0.5% gelatin and stained with 0.1% cresyl violet solution (0.1-g cresyl violet acetate dissolved in 100-mL distilled H2O with 0.25-mL glacial acetic acid) for 10 minutes. Sections were then rinsed with distilled H2O, dehydrated in a series of ascending concentrations of ethanol, and cleared in xylene before coverslipping in Permount.
Golgi staining and 3-dimensional reconstruction of Golgi-impregnated dentate granule neurons
Golgi-Cox staining randomly labels a small proportion of neurons so that the detailed morphology of neurons can be analyzed. This method has been widely used due to its reliability and sensitivity for demonstrating subtle morphological details of neurons, especially dendritic branches and spines.
Golgi Impregnation
Adipo+/− mice (n = 5), Adipo−/− mice (n = 5) and WT littermate control mice (n = 5) at 9 weeks of age were rapidly decapitated. Golgi staining was performed with a FD Rapid Golgi Stain kit according to the manufacturer's protocol with slight modifications (FD NeuroTechnologies). Briefly, brains were immediately dissected out and immersed in 10-mL impregnation solution, made of equal volume mixture of solution A and solution B. The solution was refreshed after 6 hours of immersion. The brains were then fully impregnated for 10 more days and transferred to solution C for 72 hours. The brains were kept at room temperature in the dark to reduce background staining.
Slice preparation and staining
Golgi-impregnated brains were sectioned into 150-μm-thick coronal sections along the hippocampus axis using a Leica CM1850 cryostat microtome (Leica Microsystems). Sections were immediately mounted onto glass slides coated with 3% gelatin and dried for 48 hours in the dark at room temperature, followed by a staining procedure. Briefly, the sections were rinsed with distilled H2O for 4 minutes and stained with the staining solution, containing 1 part of solution D, 1 part of solution E, and 2 parts of distilled H2O for 10 minutes. After rinsed with distilled H2O for 4 minutes, sections were dehydrated in ascending ethanol series (50%, 75%, 95%, and 100%; 4 min each), followed by being cleared in xylene (3 times, 4 min each time), and covered with Permount.
Dendritic tree morphology analysis
Dentate gyrus granule neurons were reconstructed after Golgi-Cox staining using an Olympus BX51 bright-field microscope equipped with a motorized XYZ stage, video camera and Neurolucida (MicroBrightfield Bioscience) with a ×40 objective. Neurons in the suprapyramidal blade of the dorsal dentate gyrus were selected for reconstruction using the following criteria: 1) they were located in the granule cell layer with intact cone-shaped dendritic tree morphology; 2) the soma and dendritic processes were fully impregnated; and 3) they were relatively isolated and unobscured by other impregnated neurons. Early- and late-born granule neurons were classified according to their soma location and morphology. The soma of early-born neurons is located in the superficial granule cell layer with multiple primary dendrites. The soma of late-born neurons is located in the deeper granule cell layer with one or two primary dendrites. Neurons with the soma located within the inner one-third of the granule cell layer with a single primary dendrite were excluded from the analysis to avoid including adult-born neurons. Six to 10 dentate granule neurons with the soma located within the outer two-thirds of the granule cell layer from each animal were traced and reconstructed. To determine the genotype effects on dendritic tree morphology of dentate granule neurons, 35–44 neurons from 5 Adipo+/− (early-born neurons, n = 19; late-born neurons, n = 18), Adipo−/− (early-born neurons, n = 24; late-born neurons, n = 20), and WT littermate control mice (early-born neurons, n = 16; late-born neurons, n = 19) were analyzed. To test whether exogenous adiponectin regulates dendritic tree morphology, total of 25 neurons (early-born neurons, n = 9; late-born neurons, n = 16) from 4 vehicle-treated animals, and 28 neurons (early-born neurons, n = 10; late-born neurons, n = 18) from 5 adiponectin-treated group were traced and analyzed. The size of soma area, total dendritic length, and branching number were measured from the selected neurons. Dendritic tree complexity was determined using Sholl analysis, by which the number of dendritic branching points intersecting with the concentric circles located 10-μm intervals from the soma was measured. The morphological analyses were performed by an experimenter blinded to the genotype or treatment.
Dendritic spine analysis
The analysis of dendritic spine density was performed by sampling 4 segments (20–40 μm) of distal dendrites (>120 μm from soma) in the outer molecular layer and quantified at ×100 magnification. Total number of 26–29 neurons from 4 to 5 Adipo+/− (early-born neurons, n = 15; late-born neurons, n = 11), Adipo−/− (early-born neurons, n = 14; late-born neurons, n = 14), and WT littermate control mice (early-born neurons, n = 15; late-born neurons, n = 14) were analyzed for each genotype. To determine the effects of adiponectin on dendritic spine formation of dentate granule neurons, spine density were estimated from 28 neurons (early-born neurons, n = 14; late-born neurons, n = 14) in the vehicle-treated group and 32 neurons (early-born neurons, n = 14; late-born neurons, n = 18) in the adiponectin-treated group. Five animals per treatment group were analyzed. Spines were further categorized into 3 subtypes, ie, mature mushroom spines, thin spines, and immature stubby spines (67–69). Mushroom spines were identified as small protrusions with an actin-enriched spherical head connected to a dendrite via a thin neck and the head diameter was at least twice the diameter of the neck; thin spines were identified as short dendritic protrusions with a long, thin neck and without an obvious head and the neck diameter was similar to the spine head diameter in this category of spine subtype; stubby spines were categorized as protrusions that have a very short or nonexistent neck (67–69). All samples were randomly numbered and analyzed by an experimenter blind to the genotype or treatment.
BrdU labeling and immunohistochemistry
BrdU labeling and Ki67 were used to examine proliferation of hippocampal neural progenitor cells. Animals were injected with BrdU (50 mg/kg, ip 3 times in 24-h). Twenty-eight days after BrdU injection, mice were perfused transcardially with cold 0.1M PBS followed by 4% paraformaldehyde in 0.1M PBS (pH 7.4). Brains were removed and postfixed in 4% paraformaldehyde overnight at 4°C, and then transferred to 30% sucrose in PBS. Brains were sectioned into 40-μm coronal sections on a cryostat and stored in cryoprotectant (30% sucrose, 30% ethylene glycol, 1% polyvinyl pyrrolidone, and 0.05M sodium phosphate buffer). Brain sections were pretreated with 10mM Na-citrate (pH 6.0) at 95°C for 10 minutes to recover antigen for Ki67 immunohistochemical staining or 2M HCl at 37°C for 30 minutes to denature DNA for BrdU immunohistochemical staining before the standard immunohistochemical/immunofluorescent procedures were applied. For immunohistochemical staining, brain sections were incubated with 0.3% hydrogen peroxide for 30 minutes, rinsed with PBS and blocked for 1 hour in a blocking buffer (1× PBS, 1% BSA, 3% normal serum, and 0.3% Triton X-100) (70). The sections were then incubated with one of the following primary antibodies overnight at 4°C: 1) rabbit anti-Ki67 (1:1000, Abcam); 2) rat anti-BrdU (1:400; Accurate Chemical); and 3) goat anti-doublecortin (DCX) (1:200; Santa Cruz Biotechnology, Inc). Next day, brain sections were further incubated with a biotinylated secondary antibody (1:400; Vector Laboratories) at room temperature for 1 hour, and subsequently with the ABC reagents (1:100; Vector Laboratories) for 1 hour. Immunoreactivity was visualized using a 3,3′-diaminobenzidine tetrahydrochloride (DAB) kit (Sigma). For triple immunofluorescent staining, brain sections were rinsed with PBS, blocked for 1 hour in the blocking buffer, and coincubated with 3 primary antibodies overnight at 4°C: rat anti-BrdU, mouse anti-neuronal-specific nuclear protein (NeuN), and rabbit anti-glial fibrillary acidic protein (GFAP) (1:1000; Millipore). Next day, the sections were coincubated with the following Alexa Fluor-conjugated secondary antibodies for 4 hours: antirabbit Alexa Fluor 647 goat IgG, antimouse Alexa Fluor 594 goat IgG, and antirat Alexa Fluor 488 goal IgG (1:400; Life Technologies) (Table 1).
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Ki67 | Synthetic peptide conjugated to KLH derived from within residues 1200 − 1300 of Human Ki67 | Anti-Ki67 antibody | Abcam, Ab15580 | Rabbit; polyclonal | 1:1000 |
| Doublecortin (DCX) | epitope mapping at the C-terminus of Doublecortin of human origin | Doublecortin antibody (C-18) | Santa Cruz Biotechnology, Inc, sc-8066 | Goat; polyclonal | 1:200 |
| BrdU | Anti-BrdU antibody [BU1/75 (ICR1)] | Accurate Chemical, YSRTMCA2060GA | Rat; monoclonal | 1:400 | |
| NeuN | Anti-NeuN antibody, clone A60 | Millipore, MAB377 | Mouse; monoclonal | 1:1000 | |
| GFAP | Anti-glial fibrillary acidic protein (GFAP) antibody | Millipore, AB5804 | Rabbit; polyclonal | 1:1000 | |
| Biotinylated antirabbit IgG | Biotinylated goat antirabbit IgG antibody | Vector Laboratories, BA-1000 | Goat | 1:400 | |
| Biotinylated antigoat IgG | Biotinylated horse antigoat IgG antibody | Vector Laboratories, BA-9500 | Horse | 1:400 | |
| Alexa Fluor 488 goat antirat IgG | Goat antirat IgG (H + L) secondary antibody, Alexa Fluor 488 conjugate | Invitrogen, A11006 | Goat | 1:400 | |
| Alexa Fluor 546 goat antimouse IgG | F(ab')2-goat antimouse IgG (H + L) secondary antibody, Alexa Fluor 546 conjugate | Invitrogen, A11018 | Goat | 1:400 | |
| Alexa Fluor 647 goat antirabbit IgG | F(ab')2-goat antirabbit IgG (H + L) secondary antibody, Alexa Fluor 647 conjugate | Invitrogen, A21246 | Goat | 1:400 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Ki67 | Synthetic peptide conjugated to KLH derived from within residues 1200 − 1300 of Human Ki67 | Anti-Ki67 antibody | Abcam, Ab15580 | Rabbit; polyclonal | 1:1000 |
| Doublecortin (DCX) | epitope mapping at the C-terminus of Doublecortin of human origin | Doublecortin antibody (C-18) | Santa Cruz Biotechnology, Inc, sc-8066 | Goat; polyclonal | 1:200 |
| BrdU | Anti-BrdU antibody [BU1/75 (ICR1)] | Accurate Chemical, YSRTMCA2060GA | Rat; monoclonal | 1:400 | |
| NeuN | Anti-NeuN antibody, clone A60 | Millipore, MAB377 | Mouse; monoclonal | 1:1000 | |
| GFAP | Anti-glial fibrillary acidic protein (GFAP) antibody | Millipore, AB5804 | Rabbit; polyclonal | 1:1000 | |
| Biotinylated antirabbit IgG | Biotinylated goat antirabbit IgG antibody | Vector Laboratories, BA-1000 | Goat | 1:400 | |
| Biotinylated antigoat IgG | Biotinylated horse antigoat IgG antibody | Vector Laboratories, BA-9500 | Horse | 1:400 | |
| Alexa Fluor 488 goat antirat IgG | Goat antirat IgG (H + L) secondary antibody, Alexa Fluor 488 conjugate | Invitrogen, A11006 | Goat | 1:400 | |
| Alexa Fluor 546 goat antimouse IgG | F(ab')2-goat antimouse IgG (H + L) secondary antibody, Alexa Fluor 546 conjugate | Invitrogen, A11018 | Goat | 1:400 | |
| Alexa Fluor 647 goat antirabbit IgG | F(ab')2-goat antirabbit IgG (H + L) secondary antibody, Alexa Fluor 647 conjugate | Invitrogen, A21246 | Goat | 1:400 |
Abbreviation: H+L, Heavy and light chain.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Ki67 | Synthetic peptide conjugated to KLH derived from within residues 1200 − 1300 of Human Ki67 | Anti-Ki67 antibody | Abcam, Ab15580 | Rabbit; polyclonal | 1:1000 |
| Doublecortin (DCX) | epitope mapping at the C-terminus of Doublecortin of human origin | Doublecortin antibody (C-18) | Santa Cruz Biotechnology, Inc, sc-8066 | Goat; polyclonal | 1:200 |
| BrdU | Anti-BrdU antibody [BU1/75 (ICR1)] | Accurate Chemical, YSRTMCA2060GA | Rat; monoclonal | 1:400 | |
| NeuN | Anti-NeuN antibody, clone A60 | Millipore, MAB377 | Mouse; monoclonal | 1:1000 | |
| GFAP | Anti-glial fibrillary acidic protein (GFAP) antibody | Millipore, AB5804 | Rabbit; polyclonal | 1:1000 | |
| Biotinylated antirabbit IgG | Biotinylated goat antirabbit IgG antibody | Vector Laboratories, BA-1000 | Goat | 1:400 | |
| Biotinylated antigoat IgG | Biotinylated horse antigoat IgG antibody | Vector Laboratories, BA-9500 | Horse | 1:400 | |
| Alexa Fluor 488 goat antirat IgG | Goat antirat IgG (H + L) secondary antibody, Alexa Fluor 488 conjugate | Invitrogen, A11006 | Goat | 1:400 | |
| Alexa Fluor 546 goat antimouse IgG | F(ab')2-goat antimouse IgG (H + L) secondary antibody, Alexa Fluor 546 conjugate | Invitrogen, A11018 | Goat | 1:400 | |
| Alexa Fluor 647 goat antirabbit IgG | F(ab')2-goat antirabbit IgG (H + L) secondary antibody, Alexa Fluor 647 conjugate | Invitrogen, A21246 | Goat | 1:400 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Ki67 | Synthetic peptide conjugated to KLH derived from within residues 1200 − 1300 of Human Ki67 | Anti-Ki67 antibody | Abcam, Ab15580 | Rabbit; polyclonal | 1:1000 |
| Doublecortin (DCX) | epitope mapping at the C-terminus of Doublecortin of human origin | Doublecortin antibody (C-18) | Santa Cruz Biotechnology, Inc, sc-8066 | Goat; polyclonal | 1:200 |
| BrdU | Anti-BrdU antibody [BU1/75 (ICR1)] | Accurate Chemical, YSRTMCA2060GA | Rat; monoclonal | 1:400 | |
| NeuN | Anti-NeuN antibody, clone A60 | Millipore, MAB377 | Mouse; monoclonal | 1:1000 | |
| GFAP | Anti-glial fibrillary acidic protein (GFAP) antibody | Millipore, AB5804 | Rabbit; polyclonal | 1:1000 | |
| Biotinylated antirabbit IgG | Biotinylated goat antirabbit IgG antibody | Vector Laboratories, BA-1000 | Goat | 1:400 | |
| Biotinylated antigoat IgG | Biotinylated horse antigoat IgG antibody | Vector Laboratories, BA-9500 | Horse | 1:400 | |
| Alexa Fluor 488 goat antirat IgG | Goat antirat IgG (H + L) secondary antibody, Alexa Fluor 488 conjugate | Invitrogen, A11006 | Goat | 1:400 | |
| Alexa Fluor 546 goat antimouse IgG | F(ab')2-goat antimouse IgG (H + L) secondary antibody, Alexa Fluor 546 conjugate | Invitrogen, A11018 | Goat | 1:400 | |
| Alexa Fluor 647 goat antirabbit IgG | F(ab')2-goat antirabbit IgG (H + L) secondary antibody, Alexa Fluor 647 conjugate | Invitrogen, A21246 | Goat | 1:400 |
Abbreviation: H+L, Heavy and light chain.
Cell counting and stereology
Cell number quantification was performed on every sixth section throughout the entire dorsal-ventral axis of hippocampus (from 1.46 mm to 3.40 mm posterior to bregma) using unbiased stereology. Cells from both sides of the hippocampus were counted under ×20 and ×63 oil immersion objectives using a bright-field microscope (Olympus BX51) equipped with a motorized XYZ stage, video camera and Stereoinvestigator (MicroBrightfield Bioscience). Total number of Ki67- and DCX-positive cells in the dentate gyrus were estimated with a slightly modified optical fractionator technique (71). The stereological parameters were predetermined to maintain the low coefficient error [Gundersen (m = 1) CE < 0.05]. Specifically, 5 sections per animal were examined within the contour of the targeted zone in the dentate gyrus using counting frames area of 40 × 40 μm, dissector height (15 μm), and 400 μm matrix spacing (MBF 200 Bioscience) under a ×60 oil objective (numerical aperture = 1.4). A dual vertical guard zone height (2 μm) was set and the thickness of each section was measured with each counting frame (mean = 11.5 ± 0.6). Immunohistochemically stained cells that intersected the exclusion boundaries of the unbiased sampling frame were excluded from counting. Cells that met the counting criteria were counted bilaterally throughout the dentate gyrus. Total number of stained cells was estimated by multiplying the number of counts by the reciprocals of the area section fraction, serial section fraction, and tissue section fraction (72). For Ki67 quantification, only the cells located within the subgranular zone were counted. For DCX quantification, the cells located within the whole dentate gyrus granule cell layer were counted. Because BrdU-positive cells double-labeled for NeuN or GFAP were sparsely distributed, conventional counting was applied. Every sixth hippocampal sections were quantified bilaterally. Colocalization of BrdU and NeuN or GFAP were confirmed with z-stack of the respective cell soma using line-based sequential scan (3-μm interval). The percentage of BrdU-positive cells that were double-labeled for NeuN or GFAP throughout the dentate gyrus was calculated.
Statistical analysis
All results were presented as mean ± SEM. The dendritic morphology, dendritic spine, and neurogenesis data were analyzed using one-way ANOVA followed by Dunnett post hoc tests to determine whether there were significant differences between heterozygous and homozygous adiponectin-deficient mice and WT littermates, and using a 2-tailed t test to compare the means of mice received ICV infusion of adiponectin and vehicle. Sholl analysis and density of spine subtypes were analyzed using two-way repeated measures ANOVA followed by Bonferroni's multiple comparisons tests. Two-way multivariate ANOVA (MANOVA) was used to determine whether there were differences in dendritic tree morphology (soma size, total dendritic length, and branch number) between neuron subtypes (early- vs late-born neurons), followed by Scheffe's post hoc comparisons. P < .05 was considered statistically significant.
Results
Adiponectin deficiency decreases dendritic complexity and spine density of dentate gyrus granule neurons
Adiponectin-deficient mice showed normal growth and body weight as reported previously (66). Gross morphology of the hippocampus was assessed using Nissl-stained brain sections. No overt cytoarchitectural abnormalities were observed in the dentate gyrus of adult Adipo+/− and Adipo−/− mice compared with their WT littermate controls (data not shown).
Granule neurons are the principal cell type in the dentate gyrus, which are generated by a heterogeneous population of precursor cells at different developmental stages (73, 74). The outer two-thirds of the granule cell layer of the dentate gyrus contains granule cells derived from embryonic (early-born) and early-postnatal (late-born) progenitors, and adult-born granule cells are distributed in the inner third of the granule cell layer-subgranular zone (Figure 1A). Golgi staining revealed that the apical dendrites of granule neurons formed a characteristic cone-shaped tree, which extended into the molecular layer (75). Dendritic morphology of granule cells varies depending on the position of their somata in the granule cell layer and along the transverse axis, ie, suprapyramidal vs infrapyramidal blades (76). Golgi-impregnated mature granule neurons located at the outer two-thirds of the granule cell layer in the suprapyramidal blade were reconstructed using Neurolucida to determine the effects of adiponectin deficiency on dendritic spine morphology. There was no significant effect of genotype on soma size of granule neurons (F2,108 = 1.156, P > .1) (Figure 1B). A significant main effect of genotype was revealed for total dendritic length (F2,113 = 14.31, P < .0001) and number of dendritic branches (F2,113 = 15.28, P < .0001). Post hoc analyses indicated that total dendritic length and number of dendritic branches were significantly decreased in both Adipo+/− mice (P < .0001) and Adipo−/− mice (P < .0001) (Figure 1B). Sholl analysis of dendritic intersections showed main effects of genotype (F2,113 = 15.57, P < .0001) and distance from soma (F21,2373 = 434.5, P < .0001) with significant genotype × distance from soma interactions (F42,2373 = 3.551, P < .0001). Further analysis revealed significant reductions in dendritic complexity between 80 and 180 μm from the soma in Adipo+/− and Adipo−/− mice (P < .05 for each 10-μm comparison except from 90–100 μm for Adipo−/− mice) (Figure 1B).
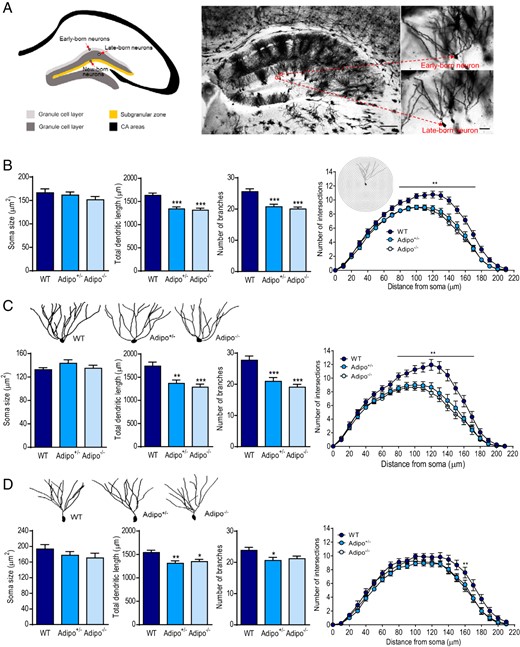
Effects of adiponectin deficiency on dendritic tree morphology of dentate granule neurons. A, left panel, Schematic of adult dentate gyrus illustrating the location of early-, late- and, newborn granule neurons. The early-born neurons are scattered in the superficial granule cell layer (light gray). The late-born cells, shown in dark gray, are located in the deeper granule cell layer. Adult-born granule cells are mainly distributed in the subgranular zone (SGZ) (orange). Right panel, Golgi-impregnated hippocampal neurons from the brain of an adult mouse (scale bar, 125 μm) indicating the location of early- and late-born granule neurons in the dentate gyrus (scale bar, 25 μm). B, The soma size (left), total dendritic length (middle left), number of branches (middle right), and Sholl analysis (right) of dentate granule neurons in Adipo+/−, Adipo−/−, and WT littermate control mice; **, P < .01; ***, P < .001 compared with the WT group. C, upper panel, Representative images of dendritic tracings of early-born dentate granule neurons in mice of different genotypes. Lower panel, Quantification of soma size (left), total dendritic length (middle left), number of branches (middle right), and Sholl analysis (right) of early-born dentate granule neurons; **, P < .01; ***, P < .001 compared with the WT group. D, upper panel, Representative images of dendritic tracings of late-born dentate granule neurons in mice of different genotypes. Lower panel, Quantification of soma size (left), total dendritic length (middle left), number of branches (middle right), and Sholl analysis (right) of late-born dentate granule neurons from mice of the different genotypes; *, P < .05; **, P < .01 compared with the WT group.
Given early- and late-born granule neurons are morphologically and functionally different (73, 77–81), we further grouped the reconstructed granule neurons into 2 subpopulations. Early-born granule neurons showed no significant difference in soma size between 3 genotypes (F2,51 = 1.065, P > .1), but displayed the genotype main effect on both total dendritic length (F2,56 = 9.747, P < .001) and number of dendritic branches (F2,56 = 14.79, P < .0001) (Figure 1C). The total dendritic length was decreased by 21% in Adipo+/− mice (P < .01) and by 26% in Adipo−/− mice (P < .001), and number of dendritic branches was decreased by 24% in Adipo+/− mice (P < .001) and by 31% in Adipo−/− mice (P < .0001). Sholl analysis of dendritic intersections of early-born granule neurons found main effects of both genotype (F2,56 = 9.694, P < .001) and distance from soma (F21,1176 = 229.9, P < .0001) with significant genotype × distance from soma interactions (F42,1176 = 3.856, P < .0001). Dendritic complexity was reduced between 80 and 170 μm from the soma in Adipo+/− and Adipo−/− mice (P < .05 for each 10-μm comparison) (Figure 1C). Similar to early-born granule neurons, late-born granule neurons displayed no genotype effect on soma size (F2,54 = 1.073, P > .1) but significant main effects of genotype on total dendritic length (F2,54 = 5.970, P < .005) and number of dendritic branches (F2,54 = 3.283, P < .05) (Figure 1D). Sholl analysis of late-born granule neurons indicated main effects of both genotype (F2,54 = 7.515, P < .005) and distance from soma (F21,1134 = 254.0, P < .0001) on number of dendritic intersections, but the interaction between genotype and distance from soma was not significant (F42,1134 = 1.010, P > .1). To determine whether early- and late-born granule neurons were differentially affected by adiponectin deficiency, the data were reanalyzed using two-way MANOVA. There was a significant multivariate main effect of genotype (Wilks' λ = 0.699, F6,206 = 6.723, P < .001, partial η2 = 0.164) and neuron subtype (Wilks' λ = 0.743, F3,103 = 11.854, P < .001, partial η2 = 0.257). This indicated that the morphology of dentate granule neurons was affected by both genotype and neuron subtype (early- vs late-born neurons). Then, separate univariate ANOVAs for each variable were performed. There were a marginal genotype × neuron subtype interaction on total dendritic length (F2,105 = 2.770, P = .067) and a significant genotype × neuron subtype interaction on total branch number (F2,105 = 4.584, P < .05), indicating that the effects of adiponectin deficiency on dendritic length and branching of dentate granule neurons depend on neuron subtypes. Adiponectin deficiency caused a more evident reduction of dendritic complexity in early-born granule neurons than late-born granule neurons. These observations suggest that endogenous adiponectin is necessary for maintaining normal dendritic tree morphology of dentate granule neurons and the 2 subtypes of neurons are affected differentially by adiponectin deficiency.
Mature granule neurons receive most of their synaptic inputs on dendritic spines, thus dendritic spines reflecting synaptic connectivity. Two features of spines, density and shape were measured in granule neurons from Adipo+/− mice, Adipo−/− mice, and WT littermate controls. Spine density measurements were taken from distal dendritic branches of granule neurons (>120 μm from soma), where the entorhinal terminals are strictly confined (82, 83). Analysis of total spine densities showed a main genotype effect (F2,80 = 6.163, P < .005) (Figure 2A). Dendritic spines were further morphologically categorized into 3 types: mushroom, thin, and stubby spines according to their head-to-neck ratios (67, 68). There were significant differences among spine subtypes and significant interaction between genotype and spine subtype (genotype: F2,80 = 0.931, P = .0591; spine subtype: F2,160 = 281.7, P < .0001; genotype × spine subtype interaction: F4,160 = 2.447, P < .05) (Figure 2A). Further analysis of total spine densities of early-born granule neurons vs late-born granule neurons showed main genotype effects (F2,41 = 3.398, P < .05 for spines of early-born neurons; F2,36 = 7.655, P < .005 for spines of late-born neurons). After spines were classified into 3 types, early-born granule neurons showed no significant genotype difference in any spine subtype (genotype: F2,41 = 2.257, P > .1; spine subtype: F2,82 = 222.6, P < .001; genotype × spine subtype: F4,82 = 1.113, P > .1) (Figure 2B). In contrast, late-born granule neurons exhibited significant genotype difference and interaction of genotype × spine subtype (genotype: F2,36 = 14.57, P < .001; spine subtype: F2,72 = 325.9, P < .001; genotype × spine subtype: F4,72 = 4.077, P < .005). Post hoc analysis revealed a decrease in mushroom spine density in Adipo+/− mice and a reduction of thin spine density in Adipo−/− mice compared with WT littermates (Figure 2C). These results suggest that endogenous adiponectin exerts differential effects on spine density of dentate granule neurons.
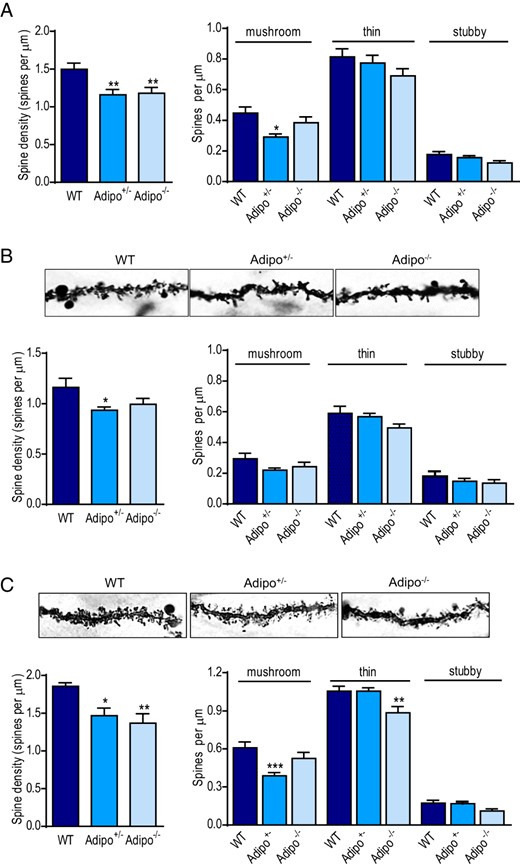
Effects of adiponectin deficiency on total dendritic spine density and spine subtype densities of dentate gyrus granule neurons. A, left panel, Total spine density sampled from the distal dendrites (120 μm from soma) of granule neurons in adult Adipo+/−, Adipo−/−, and WT littermate control mice. Right panel, Quantification of density of each spine type (mushroom, thin, and stubby spines); *, P < .05; **, P < .01 compared with the WT group. B, upper panel, Representative Golgi-stained images of dendritic spines in early-born dentate granule neurons in mice of different genotypes. Lower panel, Quantification of total spine density (left) and densities of each spine type (right) on distal dendrites of early-born dentate granule neurons; *, P < .05 compared with WT group. C, upper panel, Representative Golgi-stained images of dendritic spines on late-born dentate granule neurons in mice of different genotypes. Lower panel, Quantification of total spine density (left) and densities of each spine type (right) on distal dendrites of late-born dentate granule neurons; *, P < .05; **, P < .01; ***, P < .001 compared with the WT group.
ICV infusion of exogenous adiponectin increases dendritic spine densities without affecting dendritic tree structure of dentate granule neurons
To examine whether exogenous adiponectin affects dendritic spine plasticity, full-length adiponectin was administered ICV to avoid confounding results derived from potential peripheral effects. Previous studies have shown that systemic treatment with adiponectin causes metabolic changes mainly through its interaction with 2 receptor subtypes distributed in peripheral tissues (84–89), which may indirectly influence both spinogenesis and neurogenesis in the brain (90, 91). Mice received ICV injection of 1-μg adiponectin once daily for 7 consecutive days (Figure 3A). This dose was chosen based upon the effective doses of centrally administered adiponectin that were reported in previous work (61, 64, 92). Body weight was monitored before, during, and after a 7-day ICV infusion of adiponectin. Repeated measures ANOVA revealed significant main effects of treatment (F1,7 = 24.69, P < .01), time (F7,49 = 2.982, P < .05) and treatment × time interaction (F7,49 = 2.867, P < .05) on body weight. Adiponectin treatment significantly decreased body weight on days 4–7 (Figure 3A). However, accumulative food intake showed no difference during the first 3 days (vehicle, 13.9 ± 2.39 g; adiponectin, 13.0 ± 0.48 g) and 4–7 days (vehicle, 17.4 ± 2.92 g; adiponectin, 16.6 ± 0.64 g) of measurements (treatment, F1,7 = 0.1233, P > .5; treatment × time, F1,7 = 0.0155, P > .5). This is consistent with the previous reports (85, 92). Adiponectin treatment did not alter soma size (t(51) = 1.337, P > .1), total dendritic length (t(51) = 0.3024, P > .1), and number of dendritic branches (t(51) = 1.382, P > .1) of granule neurons compared with vehicle treatment (Figure 3B). Sholl analysis of dendritic intersections indicated a main effect of distance from soma (F21,1092 = 186.2, P < .0001), but not treatment (F1,52 = 0.0009, P > .1) or treatment × distance from soma interaction (F21,1092 = 0.7566, P > .1) (Figure 3B). Further classification of granule neurons into early- and late-born neurons revealed no difference in soma size (early-born neurons: t(17) = 0.4637, P > .1; late-born neurons: t(32) = 1.316, P > .1), total dendritic length (early-born neurons: t(17) = 0.2318, P > .1; late-born neurons: t(32) = 0.6999, P > .1), and number of dendritic branches (early-born neurons: t(17) = 0.7298, P > .1; late-born neurons: t(32) = 1.222, P > .1) between adiponectin-treated and vehicle-treated mice (Figure 3, C and D). Sholl analysis of dendritic intersections revealed no main effect of treatment (early-born neurons: F1,17 = 0.03281, P > .1; late-born neurons: F1,32 = 0.007820, P > .1) but an effect of distance from soma (early-born neurons: F21,357 = 62.33, P < .0001; late-born neurons: F21,672 = 163.9, P < .0001). A significant interaction between adiponectin treatment and distance from soma was observed in late-born granule neurons (F21,672 = 2.339, P < .001) but not in early-born granule neurons (F21,357 = 0.4913, P > .1) (Figure 3, C and D). Two-way MANOVA was performed to determine whether early- and late-born dentate granule neurons were differentially affected by exogenous adiponectin. There was no multivariate main effect of treatment (Wilks' λ = 0.915, F3,47 = 1.258, P > .1, partial η2 = 0.085) or neuron subtype (Wilks' λ = 0.885, F3,47 = 2.033, P > .1, partial η2 = 0.115).
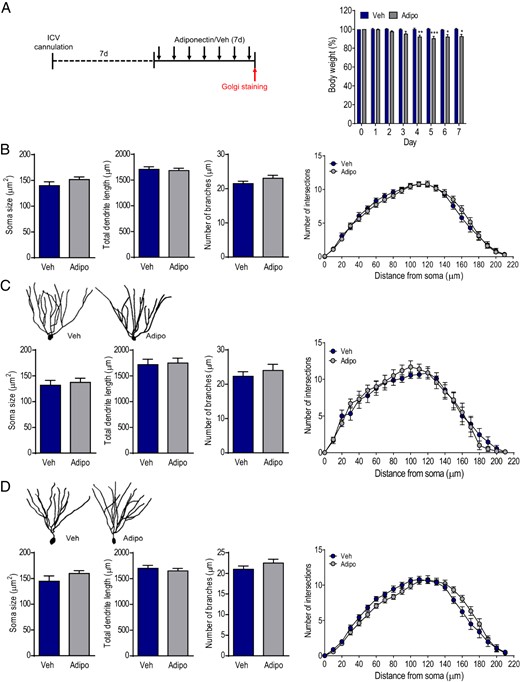
Effects of ICV infusion of adiponectin on dendritic tree morphology of dentate granule neurons in adult WT mice. A, left panel, Experimental timeline. Chronically cannulated mice received intracerebroventricular infusion of mouse recombinant adiponectin daily for 7 consecutive days. Right panel, Changes in body weight over 7 days of treatment; Veh, vehicle; Adipo, adiponectin; *, P < .05; **, P < .01; ***, P < .001 compared with the vehicle-treated group. B, The soma size (left), total dendritic length (middle left), number of branches (middle right), and Sholl analysis (right) of dentate granule neurons from mice treated with adiponectin or vehicle. C, upper panel, Representative images of dendritic tracings of early-born dentate granule neurons in adiponectin- or vehicle-treated mice. Lower panel, Quantification of soma size (left), total dendritic length (middle left), number of branches (middle right), and Sholl analysis (right) of early-born dentate granule neurons in adiponectin- or vehicle-treated mice. D, upper panel, Representative images of dendritic tracings of late-born dentate granule neurons in adiponectin- or vehicle-treated mice. Lower panel, Quantification of soma size (left), total dendritic length (middle left), and number of branches (middle right) of late-born dentate granule neurons showing no difference between 2 treatment groups; Sholl analysis of dendritic intersections (right) indicating a significant interaction between treatment and distance from soma (P < .001).
We next determined spine density in distal dendrites of granule neurons of adiponectin-treated and vehicle-treated mice. The density of total dendritic spines was significantly increased after adiponectin treatment for 7 days (t(58) = 2.368, P < .05) (Figure 4A). Further analysis of spine subtype density by morphology revealed a 29.5% higher density of thin spines in adiponectin-treated mice compared with vehicle-treated mice (treatment: F1,57 = 4.985, P < .05; spine subtype: F2,114 = 241.4, P < .001; genotype × spine subtype: F2,114 = 6.735, P < .005), but no difference in the densities of mushroom and stubby spines (Figure 4A). Dendritic spine density on early- vs late-born granule neurons were quantified. Early-born granule neurons showed no difference in total spine density (t(26) = 0.8195, P > .1). Further analysis of densities of spine subtype revealed no significant changes in any of the spine subtype on early-born granule neurons (treatment: F1,26 = 0.662, P > .1; spine subtype: F2,52 = 97.0, P < .001; treatment × spine subtype: F2,52 = 2.492, P > .05) (Figure 4B). Late-born granule neurons showed a significant increase in total spine density (t(30) = 2.417, P < .05). Further analysis of different spine subtypes showed significant treatment effects and treatment × spine subtype interaction on spine density (treatment: F1,30 = 6.369, P < .05; spine subtype: F2,60 = 141.3, P < .001; treatment × spine subtype: F2,60 = 6.051, P < .005). Post hoc analysis showed a significant increase in thin spine density by adiponectin treatment (P < .001); other spine subtypes remained unaffected (Figure 4C). These data indicate that differential sensitivity of dendritic spines to exogenous adiponectin treatment.
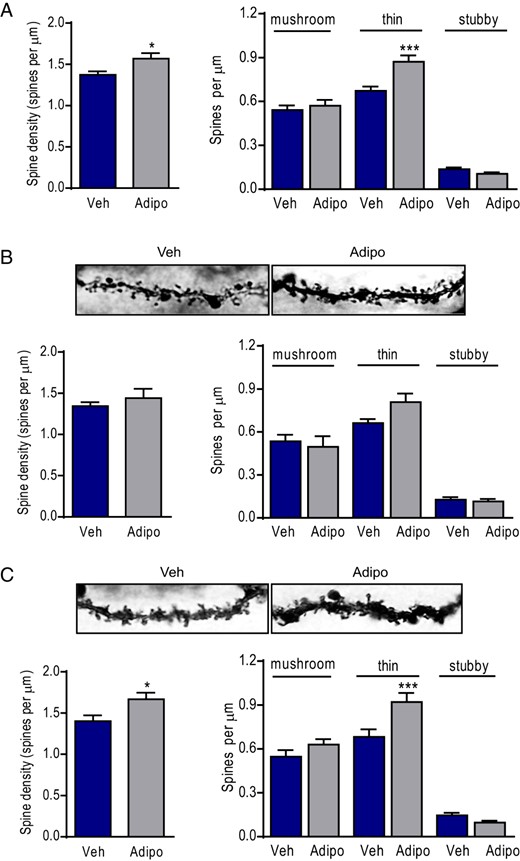
Effects of ICV infusions of adiponectin on total dendritic spine density sampled from the distal dendrites. A, left panel, Total spine density. Right panel, Quantification of density of each spine type (mushroom, thin, and stubby spines) on distal dendrites of dentate granule neuron in adiponectin- or vehicle-treated mice; Veh, vehicle; Adipo, adiponectin; *, P < .05; ***, P < .001 compared with the vehicle-treated group. B, upper panel, Representative Golgi-stained images of dendritic spines on early-born dentate granule neurons from adiponectin- or vehicle-treated mice. Lower panel, Quantification of total spine density (left) and densities of each spine type (right) on distal dendrites of early-born dentate granule neurons in mice infused with adiponectin or vehicle. C, upper panel, Representative Golgi-stained images of dendritic spines on late-born dentate granule neurons from adiponectin- or vehicle-treated mice. Lower panel, Quantification of total spine density (left) and densities of each spine type (right) on distal dendrites of late-born dentate granule neurons in mice infused with adiponectin or vehicle; *, P < .05; ***, P < .001 compared with the vehicle-treated group.
Adiponectin deficiency reduces adult hippocampal neurogenesis
To determine whether endogenous adiponectin is necessary for the production of new-born neurons in the subgranular zone of adult dentate gyrus, the effects of adiponectin deficiency on different stages of neurogenesis were assessed. First, proliferative activity of neural progenitor cells was examined quantitatively by immunohistochemical analysis of Ki67-positive cells in the dentate gyrus from Adipo+/− and Adipo−/− mice in comparison with their WT littermate controls. Ki67 is a proliferation marker that is expressed exclusively in proliferating cells during the G1, S, and G2 phases of the cell cycle (93). The total number of Ki67-positive cells was determined throughout the entire rostral-caudal extent of the dentate gyrus and showed a main effect of genotype (F2,12 = 4.791, P < .05). A significant reduction was observed in the subgranular zone of the dentate gyrus of Adipo−/− mice (P < .05), Adipo+/− mice showed a similar trend approaching but not reaching significance (P = .052) (Figure 5A). This suggests a role of adiponectin in maintaining normal proliferation of neural progenitors in the dentate gyrus.
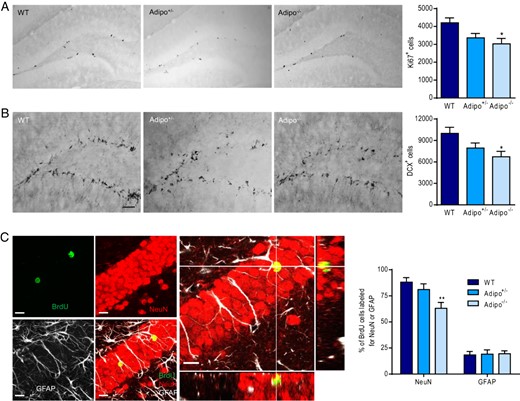
Effects of adiponectin deficiency on adult neurogenesis in the dentate gyrus. A, left panel, Representative images of Ki67-positive cells. Right panel, Quantification of the number of Ki67-positive cells in the dentate gyrus of mice of different genotypes; *, P < .05 compared with the WT group. B, left panel, Representative images of DCX-positive cells. Right panel, Quantification of the number of DCX-positive cells in the dentate gyrus of mice of different genotypes; *, P < .05 compared with the WT group. Scale bar, 50 μm. C, left panel, Representative confocal images of triple labeling showing the colocalization of BrdU with NeuN or GFAP. Right panel, Quantitative analysis of neuronal differentiation by calculating the percentage of BrdU-positive cells double-labeled with NeuN or GFAP; **, P < .01 compared with the WT group. Scale bar, 10 μm.
Newly proliferated cells differentiate into neurons or glia. DCX is one of the earliest neuronal markers to express following cell fate determination, which is found in immature neurons (94). We therefore determined the effect of adiponectin deficiency on neuronal differentiation by using immunohistochemical detection of cells expressing DCX. A significant main effect of genotype was revealed for the total number of DCX-labeled cells in the dentate gyrus (F2,11 = 4.393, P < .05) (Figure 5B), indicating that immature neurons are reduced by adiponectin deficiency. To further determine whether the effects of adiponectin deficiency on neuroblast differentiation are transformed into mature neurons, BrdU was injected into the mice to track down the final fate of proliferated cells in the dentate gyrus. To ensure that the survived new-born cells have characteristic of mature neurons, the percentage of BrdU-labeled cells that colocalized with the mature neuron marker NeuN or the glial marker GFAP were assessed 28 days after BrdU labeling (Figure 5C). There was a main effect of genotype on the percentage of BrdU cells double-labeled with NeuN (F2,12 = 6.240, P < .05) but not the percentage of BrdU cells double-labeled with GFAP (F2,12 = 0.02836, P > .1). Adipo−/− mice displayed a significant reduction in the percentage of BrdU cells double-labeled for NeuN compared with WT littermate controls (P < .01) (Figure 5C). These results suggest that endogenous adiponectin is involved in the determination of neuronal identity of newly proliferated cells in the subgranular zone of the adult dentate gyrus.
ICV infusion of adiponectin promotes adult hippocampal neurogenesis
Our previous studies have shown that adiponectin promotes neurogenesis in vitro (65), however, whether exogenous adiponectin has a similar proneurogenic effect in vivo remains unclear. Mice were given ICV infusion of adiponectin or vehicle daily for 7 consecutive days. In the first 3 days, all mice received ICV injection of BrdU (100 μg/d) to label proliferating cells in the adult dentate gyrus (Figure 6A). Adiponectin treatment didn't significantly affect body weight over 7 days (treatment: F1,8 = 0.0259, P > .5; treatment × time interaction: F7,56 = 1.362, P > .05) (Figure 6A) and accumulating food intake measured day 1–3 (vehicle, 12.6 ± 1.19 g; adiponectin, 10.0 ± 0.86 g) and day 4–7 (vehicle, 16.6 ± 0.79 g; adiponectin, 13.3 ± 1.59) (treatment, F1,8 = 4.865, P > .5; treatment × time, F1,8 = 0.126, P > .5). The total number of Ki67-positive cells in the subgranular zone of the dentate gyrus was increased by 7 days of ICV adiponectin infusion (t(8) = 2.338, P < .05) (Figure 6B). We next examined the effect of adiponectin on differentiation of neural progenitor cells in the dentate gyrus. The percentage of BrdU-positive cells double-labeled for NeuN or GFAP was not significantly altered by adiponectin treatment (NeuN, t(8) = 1.735, P > .1; GFAP, t(8) = 0.576, P > .1) (Figure 6C). However, the total number of BrdU-positive cells that were double-labeled for NeuN was significantly increased in adiponectin-treated mice compared with vehicle-treated mice (t(8) = 2.597, P < .05) (Figure 6C). This was likely due to increased proliferation of neural progenitor cells.
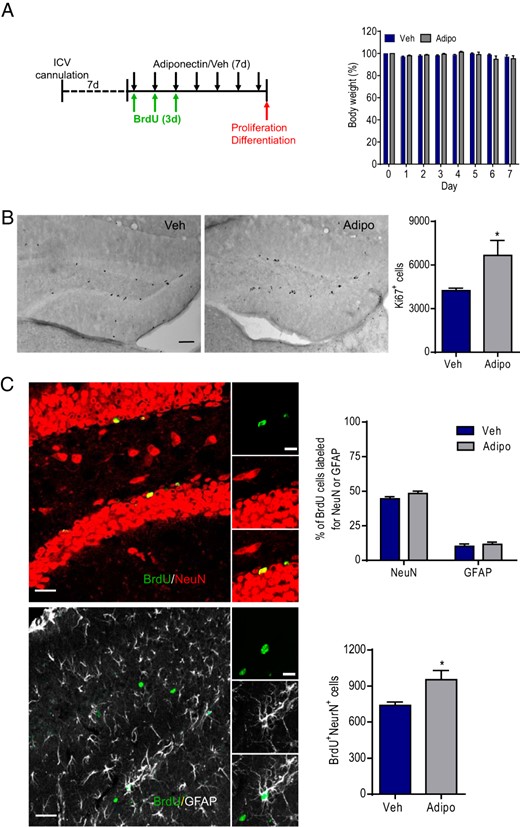
Effects of ICV infusions of adiponectin on adult neurogenesis in the dentate gyrus of WT mice. A, left panel, Experimental timeline. Chronically cannulated mice received ICV infusions of mouse recombinant adiponectin daily for 7 consecutive days. BrdU was infused ICV for the first 3 days. Right panel, Changes in body weight over 7 days of adiponectin treatment. Veh, vehicle; Adipo, adiponectin. B, left panel, Representative images of Ki67-positive cells. Right panel, Quantification of the number of Ki67-positive cells in the dentate gyrus of adiponectin- and vehicle-treated mice; *, P < .05 compared with the vehicle-treated group. Scale bar, 50 μm. C, left panel, Representative confocal images showing the colocalization of BrdU with NeuN or BrdU with GFAP. Right upper panel, Quantitative analysis of neuronal differentiation by calculating the percentage of BrdU-positive cells double-labeled with NeuN or GFAP in mice treated with adiponectin or vehicle. Right lower panel, Quantification of total number of new-born neurons (cells double-labeled for BrdU and NeuN). Scale bar, 10 μm; *, P < .05 compared with the vehicle-treated group.
Discussion
Hippocampal structural plasticity, including dendritic remodeling of existing neurons and neurogenesis, can be modulated by a number of intrinsic and extrinsic factors. In this study, we presented the first evidence that the adipocyte hormone adiponectin regulates dendritic arborization and spinogenesis of existing granule neurons and the production of new granule neurons in the adult dentate gyrus. These studies provide novel insights into how adipose-derived hormones modulate hippocampal neural circuit formation.
Granule neurons in the dentate gyrus receive their major input from the entorhinal cortex and are thought to serve as a gate regulating the excitatory signal flow from the entorhinal cortex into the hippocampus (95, 96). Granule neurons are derived from 3 populations of neural progenitor cells during embryonic and early postnatal development and throughout adulthood. Neurons that are born embryonically settle in the superficial granule cell layer, and cells born postnatally are positioned in the deeper granule cell layer closer to the hilus (97–101). Adult-born new neurons are restricted to the subgranular zone (102, 103). Early- and late-born granule neuron subpopulations in the dentate gyrus exhibit morphological differences. Neurons situated in the superficial granule cell layer possess more primary dendrites and wider dendritic fields relative to neurons located in the deeper granule cell layer (77–79). In spite of substantial differences in morphology and developmental origins of granule neurons in the dentate gyrus, very few studies have given appropriate consideration to position of the cell in the granule cell layer. In the present study, we performed a detailed analysis of dendritic spine morphology of early- and late-born granule neurons to explore the influences of adiponectin on dentate gyrus structural plasticity. We found that dendritic length and number of dendritic branches were decreased in heterozygous (Adipo+/−) and homozygous (Adipo−/−) adiponectin null mutant mice. Adiponectin deficiency also reduced dendritic tree complexity. These morphological changes were more evident in early-born than in late-born granule neurons of Adipo+/− and Adipo−/− mutant mice. By contrast, ICV infusion of exogenous adiponectin for 1 week increased dendritic complexity in late-born granule neurons. These observations suggest that adiponectin promotes dendritic growth and branching with differential sensitivity of early- and late-born granule neurons to the levels of adiponectin. Adiponectin is first detected in serum at embryonic day 16.5 and its concentrations peak at birth and decline postnatally (104). Therefore, adiponectin insufficiency or deficiency in Adipo+/− and Adipo−/− mice would cause a greater reduction in dendritic growth and branching in embryonic-born granule neurons than postnatal-born granule neurons. However, dendrites of late-born granule neurons in WT mice appeared to be more responsive to exogenous adiponectin treatment in adulthood. Whether this is due to relative expression of AdipoRs in early- vs late-born neurons is currently unknown.
Spines on dendrites usually receive excitatory synaptic input, which provides a morphological basis for synaptic plasticity and cellular substrates of neural connectivity (105). During development and in adulthood, changes in number and morphology of dendritic spines accompany the expression of synaptic plasticity (105–107). Dendritic spines can be categorized into mushroom, thin, and stubby types based upon their shape and size (67–69), with the thin type being most common and the mushroom type being the most mature and stable (52, 82). The density of total dendritic spines of dentate granule neurons was decreased in Adipo+/− and Adipo−/− mutant mice. Adipo+/− mice showed a significant decrease in total spine densities in both early- and late-born neurons, whereas only late-born neurons of Adipo−/− mice displayed a significant decrease in the total spine density. When the analysis separated different spine subtypes, no difference was found in early-born neurons. However, late-born neurons exhibited a decreased mushroom spine density in Adipo+/− mice and a decreased thin spin density in Adipo−/− mice. Moreover, the density of total dendritic spines in dentate granule neurons was increased after ICV infusion of adiponectin for 1 week, being attributed mainly to increased spines in late-born neurons. These results indicated that adiponectin was critical for spine formation and maturation, and spines of late-born neurons appear to be more plastic than those of early-born neurons. While adiponectin deficiency does not change body weight, ICV infusion of adiponectin decreases body weight by increasing metabolic rate (92). Because dendritic spine densities in the dentate gyrus have been reported to respond to manipulations of peripheral metabolism (108), it cannot be ruled out that enhanced metabolism by adiponectin may partially contribute to its effects on dendritic spine densities.
Despite extensive studies, it remains unclear about the functional demands on plastic and heterogeneous dendritic spines. It has been suggested that different types of dendritic spines may serve different functions. The responsiveness of thin spines to fast changes in synaptic activity have suggested that they are “learning spines,” whereas the stability and persistence of mushroom spines suggest that they may represent “memory spines” that are responsible for the maintenance of neuronal networks and long-term memory (109, 110). The question then arises as to whether changes in dendritic spines correlate with learning and memory behavioral phenotypes. We have recently shown that contextual extinction learning is impaired in Adipo−/− mice and facilitated by intra-dentate gyrus infusion of adiponectin (64). The effects of adiponectin deficiency and exogenous adiponectin treatment on dendritic spines in dentate granule neurons may contribute to their impact on contextual extinction learning. Moreover, it is known that chronic stress induces dendritic retraction and spine loss, accompanied by depressive-like behaviors (111). We have shown that chronic social defeat stress reduces circulating levels of adiponectin, and adiponectin haploinsuffiency increases susceptibility to stress-induced depression-like behaviors (61). These studies imply a possible link between adiponectin levels, dendritic spines and depression-related behaviors.
It is intriguing to note that mushroom spines in late-born neurons were reduced in Adipo+/− mice, whereas thin spines were decreased in Adipo−/− mice. Apparently, this does not follow a simple gene dosage effect, in that a phenotype of the homozygous mutation would be more severe than that of the heterozygous mutation (112, 113). One explanation could be that reduced levels of adiponectin in Adipo+/− (60% of WT levels) (61) differentially alter the occupancy of AdipoR1 and AdipoR2 receptors. AdipoR2 has high affinity for full-length adiponectin, and AdipoR1 has high affinity for globular adiponectin, a truncated form of this protein (114). Because the vast majority of adiponectin is in the full-length form (55, 115), we speculate that the high-affinity AdipoR2 but not the low-affinity AdipoR1 would remain a significant degree of receptor occupancy at reduced levels of adiponectin in Adipo+/− mice. The specific roles of AdipoR1 and AdipoR2 in regulating dendritic spine remodeling require further investigations in future studies.
Adult-born new granule neurons in the subgranular zone can be integrated into existing neural circuits, representing another form of structural plasticity in the dentate gyrus. Our previous in vitro studies have shown that adiponectin promotes neurogenesis by inducing neural progenitor cell proliferation (65). The present studies examined the in vivo effects of exogenous adiponectin. We found that ICV infusion of adiponectin increased proliferation of neural progenitor cells without affecting neuronal differentiation in the dentate gyrus, which are similar to our in vitro results (65). Adiponectin deficiency reduced number of proliferating cells and immature neurons and decreased neuronal differentiation in the dentate gyrus. These findings suggest that adiponectin is required for both cell proliferation and neuronal differentiation processes. A recent study reported that hippocampal neurogenesis was not altered in Adipo−/− mice (62). The discrepancy could be due to the use of two different lines of Adipo−/− mice (66, 116). The Adipo−/− mice used in this study were deleted for all 3 exons and have been shown to develop insulin resistance when given a high fat diet (66), whereas Adipo−/− mice in Yau et al were only deleted for exon 2 and display no insulin resistance (62, 116). Another possible cause of the discrepancy could result from the different procedures used in these 2 studies. Yau et al labeled proliferating cells during the last 5 days of running and killed the mice after a forced swim test (62). Both running and forced swim have been reported to influence neurogenesis (36, 117). Exposure to these procedures and labeling proliferating cells during these procedures may mask the effects of adiponectin deficiency.
Adult neurogenesis has been implicated in the pathogenesis of depression and therapeutic mechanisms of antidepressants (117). It has been reported that hippocampal neurogenesis is decreased after chronic social defeat (118). Adiponectin levels are reduced by chronic social defeat and correlated with depressive behaviors (61). We speculate that reduced adiponectin levels may mediate at least part of social defeat-suppressed hippocampal neurogenesis and associated behavioral deficits. Moreover, neurogenesis-deficient mice exhibit impaired dexamethasone suppression (54), which is commonly used to test HPA axis feedback in depressed patients (119). Such an impairment of negative feedback to the HPA axis was also observed in adiponectin-haploinsufficient mice (61). However, whether reduced hippocampal neurogenesis contributes to increased susceptibility to stress-induced depressive behavior in adiponectin-deficient mice remains to be determined.
In summary, the dentate gyrus undergo dynamic modifications of dendritic spines of existing and mature granule neurons and continuous generation of new neurons. The adult-born new neurons are incorporated into behaviorally relevant neuronal circuitry throughout life (120–122). The present study demonstrates that adiponectin exerts neurotrophic effects on dendritic spine remodeling and neurogenesis in the dentate gyrus, which represents a new mechanism underlying hippocampal structural plasticity. Given the involvement of adiponectin signaling in stress-related behavioral and neuroendocrine alterations relevant to depression and posttraumatic stress disorder (61, 64), future studies will investigate whether adiponectin treatment can reverse, and adiponectin deficiency can exacerbate, stress-induced structural changes in the dentate gyrus and whether such plastic changes in neuronal circuits underlie behavioral deficits or maladaptations.
Acknowledgments
This work was supported by National Institutes of Health Grants MH076929 and MH100583 (to X.Y.L.).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AdipoR
adiponectin receptor
- BrdU
bromodeoxyuridine
- CSF
cerebrospinal fluid
- DCX
doublecortin
- GFAP
glial fibrillary acidic protein
- HPA
hypothalamo-pituitary-adrenal
- ICV
intracerebroventricular
- MANOVA
multivariate ANOVA
- NeuN
neuronal-specific nuclear protein
- WT
wild type.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}