





Homoeothermic species engage thermogenic components of cellular metabolism to both maintain and raise core body temperature (Tc) when exposed to cold, feeding, infection, and certain classes of drugs (sympathomimetics). Body heat production stems from two main thermogenic mechanisms: 1) obligatory thermogenesis, or the heat produced as a collective byproduct of the inefficiency of all cellular metabolic reactions, and 2) facultative thermogenesis (also referred to as adaptive or nonshivering thermogenesis [NST]), the additional heat generation required along with obligatory thermogenesis to rapidly increase body temperature. Thyroid hormone (TH) is the well-established principal endocrine regulator of both obligatory and facultative thermogenesis. Here we aim to provide a brief overview of the mechanisms by which TH regulates facultative thermogenesis and to highlight recent studies describing a role for TH in the regulation of thermogenic mitochondrial uncoupling proteins. For a broader overview of the role of the endocrine control of thermoregulation, we would refer readers to the excellent review by Silva (1).
Facultative thermogenesis is controlled by both a temporary shivering (muscle contraction) component and by more sustained nonshivering mechanisms. NST is regulated primarily by both the circulating levels of TH and the acute activity of the sympathetic nervous system (SNS) and mainly occurs in brown adipose tissue (BAT) and skeletal muscle (2). The primary role of TH in NST regulation is a permissive one; it chronically sets the capacity of thermogenic and metabolic responses to SNS stimulation through its transcriptional regulation of a vast repertoire of metabolic genes (3–5). In contrast, the SNS mainly controls the acute activity of thermogenic signaling pathways culminating in the activation of heat-generating proteins downstream of β-adrenergic receptors (6, 7). Owing to the fundamental importance of TH in human thermoregulation, hyper- and hypothyroid patients are heat and cold intolerant, respectively (8). Similarly, hypothyroid animal models demonstrate that the TH is absolutely required for a normal thermogenic response to both cold and sympathomimetic stimulation (9).
TH is synthesized in the thyroid gland and controlled by thyroid peroxidase activity that regulates the iodination, coupling, and ultimately proteolysis of tyrosine residues on thyroglobulin to release the THs, T4 and T3, into the bloodstream (10). The lesser active T4 is released from the thyroid gland at higher concentrations than the more active T3 and is locally converted to T3 in target tissues by the actions of tissue-specific deiodinases (11–13). Two genes, THRA and THRB, are responsible for the expression of distinct thyroid hormone receptors (TRs), each of which are alternatively spliced to produce multiple isoforms, TRα1, TRα2, TRβ1, and TRβ2, respectively (14–16). With the exception of TRα2, which does not bind T3 and functions to repress T3 actions, TR isoforms mediate distinct functions (both stimulatory and repressive) in response to and in the absence of T3 (2). Integral to their functions as transcriptional regulators, TRs bind other nuclear hormone receptors, coactivators, and corepressors, the details of which have been reviewed elsewhere (2). In addition to its transcriptional regulation, recent work has also revealed that TH may regulate cell signaling pathways nongenomically (17, 18). However, it is not yet established whether and how TH may influence body temperature apart from its role as a ligand for thyroid hormone receptor-dependent gene transactivation.
Uncoupling protein (UCP) mediators of thermogenesis
Mitochondrial uncoupling proteins are highly conserved, nuclear-encoded, six-transmembrane-spanning members of the mitochondrial solute carrier family localized to the inner mitochondrial membrane (19). The prototypical uncoupling protein, UCP1 (previously referred to as thermogenin), was first characterized more than 3 decades ago as the first protein established to mediate cold-, SNS-, and TH-stimulated BAT thermogenesis (6, 20, 21). UCP1 functions ostensibly as a mitochondrial inner membrane proton carrier that, when activated, dissipates the electrochemical proton gradient, decreasing the proton motive force used to synthesize ATP. Proton leak regulated by UCP1 thereby uncouples the proton gradient from ATP synthesis, with the free energy released as heat (22, 23). UCP1 is activated by free fatty acids released from SNS-dependent lipolysis in both white adipose tissue (WAT) and BAT (24–27). In turn, UCP1 activation increases fatty acid oxidation to compensate for respiratory inefficiency and decreased mitochondrial membrane potential (28). UCP1 has classically been thought to be the primary, if not only, physiologically relevant thermogenic protein in mammals. However, its expression is largely, if not exclusively, confined to BAT and beige adipocytes (discussed later) (29), and because of its relatively small size in most adult large mammals, the significance of BAT as a general mediator of NST in humans is debated.
In 1997, 2 decades after UCP1 was initially characterized, the UCP1 homologs UCP2 and UCP3 were identified with relatively broader expression relative to UCP1 (30–32). UCP2 appears to have wide expression in the brain and periphery, including in dividing cells, and its levels are induced by TH in heart and skeletal muscle (33–35). In contrast, UCP3 expression is more restricted to skeletal muscle, heart, and BAT and as we have recently observed in murine epidermis (30, 36, 37). Whereas UCP1 has been established as a physiological mediator of cold thermogenesis, the specific thermogenic functions of UCP2 and UCP3 are currently debated (38). Genetic overexpression and knockout studies in both cells and tissues have demonstrated that these novel UCPs are, like UCP1, also associated with markers of proton leak, including decreased mitochondrial membrane potential and mitigation of reactive oxidant generation, and both homologs are activated by free fatty acids (28, 36, 39, 40). Observations that UCP3 protein is particularly enriched in skeletal muscle led to the early assumption that it functions in a similar manner to BAT UCP1 to control NST, and this was supported by reports of strong induction of UCP3 by TH in skeletal muscle (5, 30, 33, 41). However, unlike UCP1 knockout mice (42), cold-exposed UCP3-null mice (under normal dietary conditions) were able to maintain their body temperatures similar to wild-type mice (43). This observation guided the vast majority of researchers in the field to conclude that UCP3 is not a significant thermoregulatory mediator in mice. Whereas this finding confirmed the necessity and sufficiency of UCP1 in cold-induced NST, it did not rule out the possibility that UCP3 mediates NST in response to other stimuli and the regulation by TH. Consistent with this hypothesis, UCP3 knockout animals administered T3 produce less heat than wild-type animals (44).
The role of UCP2 as an effector of thermogenesis and TH actions has not been established. Although there are reports of TH induction of UCP2 mRNA in skeletal muscle, whether UCP2 protein is present in muscle or BAT mitochondria or corresponds to changes in mRNA levels, is unclear. Interestingly, very recent work suggests that UCP2 may function in the export of C4 metabolites from mitochondria and may actually thereby decrease mitochondrial oxidative functions, suggesting that UCP2 actions could actually oppose thermogenesis (45). More work is needed to understand the significance of TH regulation of UCP2 and its pertinence to NST. Two other brain-expressed homologues have been identified (UCP4 and the brain mitochondrial carrier protein 1 [BMCP1/UCP5]), both of which have more distant homology (<40% amino acid) with UCP1–3 (46–48). These isoforms have not been well studied with respect to TH regulation but are thought to act as regulators of mitochondrial biogenesis, calcium flux, and neurotransmission (49). It is entirely conceivable that UCP4 and UCP5, like UCP2, could act as central regulators of body temperature circuitry and in at least some neuronal cell types be regulated by TH.
TH and the regulation of UCP1-dependent thermogenesis in BAT
Regulation of BAT thermogenesis by T3 is initiated by the actions of type II deiodinases (D2) expressed locally that convert T4 to T3 and, importantly, that are inhibited by high T4 levels (12, 50, 51). The expression of D2 in BAT is induced by norepinephrine in response to cold exposure and is mediated by the activation of β1/β2/β3 adrenergic receptors, leading to the elevation of cAMP and protein kinase A activity (9, 11, 13). Consistent with the requirement for T3 in cold-induced BAT thermogenesis, mice lacking D2 exhibit impaired thermogenesis and hypothermia in response to cold exposure despite markers of chronic SNS stimulation (52). Similarly, the treatment of rodents with norepinephrine (NE), the main acute mediator of BAT thermogenesis, failed to produce an increase in BAT heat generation in hypothyroid animals, but NST could be rescued by subchronic T3 or T4 replacement (8, 53). Importantly, the adrenergic receptors as a class are induced by TH, supporting the notion that TH and adrenergic signaling synergize to control the maximal output of heat during BAT-regulated NST (54–56). In support of this, β-less mice lacking all β-adrenergic receptors (β1/β2/β3 [TKO]) have reduced UCP1 expression and D2 activity in BAT as well as lowered Tc and reduced cold tolerance that can be rescued by acute T3 administration in a manner partially dependent on skeletal muscle UCP3 (44, 57). Notably, however, high levels of T4 may suppress BAT function while stimulating thermogenic activity in skeletal muscle in both rodents and humans (58).
The TR receptors have divergent roles in the regulation of BAT NST. Whereas TRα appears to be most important for the regulation of adrenergic sensitivity, TRβ is critical for the induction of UCP1 gene expression (5, 59). TRα−/− mice (lacking both TRα1 and TRα2) exhibit significantly decreased basal body temperatures that are further decreased by acute cold exposure (60, 61). In contrast, TRβ−/− (lacking both TRβ1 and TRβ2) have normal body temperatures and cold responses (60, 62). Both UCP1 and the master mitochondrial biogenesis regulatory transcription factor, peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) mRNA levels are induced in BAT upon adrenergic stimulation, and PGC1α has been demonstrated to bind and activate TRβ/retinoic acid receptor heterodimers (63). Interestingly, compared with the wild type, TRα1−/− mice show no difference in mRNA levels of UCP1 or PGC1α in BAT at 22°C or after acute cold exposure, suggesting that TRα1 is dominant for NST activation but is not required for induction of UCP1 or PGC1α under these conditions (64). In contrast, the TRβ receptor drives the up-regulation of PGC1α and other mitochondrial metabolic machinery components in BAT in response to T3 production (65).
Recent studies have also demonstrated the presence of UCP1-containing cells in sc WAT in mice and humans stimulated by cold, β-AR agonists, or peroxisome proliferator-activated receptor-γ activators (66–69). These brown-in-white (Brite) or beige adipocytes are present in low numbers compared with white adipocytes and, when fully stimulated, express levels of UCP1 and display uncoupled respiration similar to brown adipocytes (69). Although most evidence suggests that SNS stimulation is the primary effector of beige adipocyte activation, recent studies suggests that T3 treatment can also induce UCP1 expression and increase oxygen consumption in human multipotent adipocytes (70). The T3-mediated regulation of metabolism in WAT has been well studied in mouse and human models and have demonstrated the requirement of T3 for lipogenesis and triglyceride storage and ultimately adipocyte differentiation (71). Additionally, T3 is required for the SNS-mediated elevation of cAMP levels, protein kinase A activation, and lipase activity in WAT, leading to free fatty acid release in response to cold exposure or other thermogenic stimuli (56). These studies indicate the necessity for T3 in the metabolic regulation of white adipocytes to maximize the β-AR stimulation and suggest that beige precursors in WAT depots may also respond to T3 as part of the browning process.
TH in skeletal muscle NST
Unlike rodents and hibernators in which BAT has a significant metabolic function across development, many large mammals, including humans, retain only a small amount, if any, BAT into adulthood. Indeed, despite recent observations suggesting that BAT may play a greater role in human thermoregulation than previously thought, its small size relative to total body weight in most adults has challenged the BAT-centric view of human NST (24). The role of TH in BAT function may also differ between humans and rodents. In rats, there is an absolute dependence and linear relationship between TH levels and sympathomimetic thermogenesis (72). However, a recent positron emission tomography-computed tomography study using 18F-fluorodeoxyglucose as a BAT activity tracer in normal and hyperthyroid human patients failed to detect any amounts of BAT activity in the hyperthyroid state, but instead skeletal muscle glucose uptake was significantly augmented in these patients, suggesting a role for skeletal muscle as a significant site of TH-regulated thermogenesis in humans (36, 73, 74). A similar study showed that humans administered the thermogenic sympathomimetic drug ephedrine failed to show an activation of BAT (75).
The contribution of skeletal muscle to NST is most apparent in mice lacking all β-adrenergic receptors (TKO), which have decreased Tc and cold intolerance that can be rescued upon acute T3 stimulation (44, 57). These mice lack BAT activity but have increased UCP3 expression in skeletal muscle (SKM) upon T3 administration, suggesting that TH actions in SKM may be sufficient to rescue cold tolerance in the absence of SNS stimulation and BAT thermogenesis (44, 57). Similar to the regulation of BAT UCP1, TH is essential for a maximal thermogenic response in skeletal muscle (36, 76). Indeed, euthyroid skeletal muscle generates more heat than hypothyroid muscle (77). This effect may result in part from the TH-dependent regulation of free fatty acid (FFA) levels in SKM, leading to the activation of uncoupling proteins. Indeed, SKM mitochondria from hyperthyroid rats display increased proton leak that is abolished when FFAs are removed (78). Additionally, because of its size in humans, skeletal muscle also contributes significantly to NST responses through its tonic control of body temperature via obligatory thermogenesis. A current model for TH-mediated SKM heat production is one in which TH regulates the expression of thermogenic machinery, and SNS stimulation activates the machinery via increased D2 activity and FFA release from WAT, which activates UCP3 and stimulates non-UCP-mediated thermogenesis (discussed below) (5, 36, 40, 52, 79). The role of SNS stimulation in the promotion of NST through UCP3 activity in SKM is supported by phenotypes in UCP3−/− mice. The finding that sympathomimetic drugs (3,4-methylenedioxy-methamphetamine, methamphetamine) fail to elicit a thermogenic response in UCP3−/− mice demonstrates that there are conditions in which UCP1 expression cannot compensate for lack of UCP3. Sympathomimetic agents also fail to elicit thermogenic responses in hypothyroid mice, suggesting that in skeletal muscle, as in BAT, SNS stimulation is insufficient to compensate for a lack of normal TH levels and that TH is necessary for a full thermogenic response in SKM as it is in BAT (72). Finally, unpublished data from our laboratory indicates that SKM-specific overexpression of UCP3 is sufficient to rescue a thermogenic response to methamphetamine normally lost in UCP3−/− mice.
UCP-independent mechanisms of TH mediated thermogenesis
As noted above, the only established physiological NST mechanism involves the activation of brown adipocyte UCP1. However, in addition to evidence that skeletal muscle UCP3 may also participate in NST, UCP-independent NST mechanisms can also play a role in TH-regulated thermoregulation (1, 80). For example, the glycerol 3-phosphate shuttle, dihydroxyacetone phosphate is reduced to glycerol 3-phosphate (G3P) in the cytosol, and G3P is then reoxidized to dihydroxyacetone phosphate by mitochondrial glycerol 3-phosphate dehydrogenase (mGPD), a flavin adenine dinucleotide-linked enzyme that donates electrons directly to complex III of the respiratory chain (81, 82). Because the G3P shuttle's electrons bypass complex I for ATP synthesis, the process is inherently inefficient, and therefore thermogenic (1). The G3P shuttle is a predominant metabolic pathway in skeletal muscle and BAT and is stimulated by TH (1). Knockout of the mitochondrial GPD (mGPD−/−) in mice leads to decreased energy turnover (intake/oxygen consumption) associated with increases in serum T3 and T4 levels as well as chronic SNS stimulation of BAT (83, 84). These data suggest that these animals have a reduced energy turnover due to cold stress from reduction of non-UCP-mediated heat generating mechanisms in thermogenic tissues. Furthermore, these animals display a TH-dependent, increased expression of UCP3 in both BAT and skeletal muscle, indicating a UCP3-dependent compensation mechanism in these tissues during both obligatory and facultative thermogenesis (84). Another example of a UCP-independent NST pathway has been demonstrated recently linking TRα1 with control of vascular function as a mechanism to regulate heat dissipation and conservation. Mice with a heterozygous mutation in TRα1 (R384C) exhibit decreased Tc at night despite increased heat production from BAT. In this model, decreased Tc results from increased heat dissipation from the tail in response to a failure of arterial constriction in the tail after SNS stimulation (85). These data indicate a clear role for TH and TRs in thermoregulation outside BAT and SKM and independent of UCP actions.
Concluding remarks and future directions
An overview of the various mouse models and their phenotypes relevant to TH and UCP actions are summarized in Table 1. In response to cold exposure, NE released from the hypothalamus binds β3-adrenergic receptors in BAT, WAT, and skeletal muscle, increasing FFA release, the known substrates/second messengers for thermogenesis along with an induction/activation of UCP1/3 and other potential thermogenic genes. Simultaneously, SNS stimulation activates D2 deiodinases in BAT and skeletal muscle that increase T3 levels, leading to the transactivation via TRs of thermogenic genes that ultimately govern the thermogenic capacities of BAT and skeletal muscle (Figure 1). The activities of both UCP1 and UCP3 are stimulated by FFAs, and increased TH signaling in white adipose tissue is required for lipolysis and the liberation of FFAs and may be associated with beige adipocyte activation. Moreover, skeletal muscle UCP3 is induced by conditions of increased fatty acid oxidation demand, including exercise, fasting, increased TH signaling, and obesity. In addition, UCP3 expression is increased in BAT and skeletal muscle, in a TH-dependent manner, upon the reduction of additional thermogenic pathways. More work in genetically modified mice harboring tissue-selective deletions in TH receptors and UCPs subjected to thermoregulatory and metabolic challenges will be necessary to more clearly dissect the roles of UCPs as determinants of TH functions.
Mouse Models of TH and UCP Thermogenesis
| Genetic Mouse Models | Obligatory Thermogenic Phenotype | Facultative Thermogenic Phenotype | References |
|---|---|---|---|
| UCP1−/− | Normal baseline temperature | Hypothermia in response to cold | Enerback et al (1997) (42) |
| Gong et al (2000) (36) | |||
| Golozoubova et al (2006) | |||
| UCP3−/− | Normal baseline temperature | Normal thermogenesis in response to cold, hypothermia in response to sympathomimetics | Mills et al (2003) (72) |
| Vidal-Puig et al (2000) (43) | |||
| Flandin et al (2009) (44) | |||
| TRα1−/− | Reduced baseline temperature | Normal thermogenesis in response to cold | Wikström et al (1998) (61) |
| Golozoubova et al (2003) | |||
| TRa 0/0 | Reduced baseline temperature | Hypothermia in response to cold | Marrif et al (2005) (64) |
| Pelletier et al (2008) (86) | |||
| TRβ−/− | Normal baseline temperatures | Normal response to thermogenesis | Johnsson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| TRα1/TRβ−/− | 0.4ºC decrease in core body temperature | Hypothermia in response to cold | Johansson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| D2−/− (D2KO) | Normal baseline temperature | Cold exposure leads to increased SNS stimulation of BAT but mice are hypothermic | Schneider et al (2001) (87) |
| Impaired embryonic BAT differentiation results in defective BAT function in adult mice | de Jesus et al (2001) (50) | ||
| Christoffolete et al (2004) (52) | |||
| Hall (2010) (88) | |||
| Castillo et al (2011) (89) | |||
| β1/β2/β3−/− (TKO) | 1.2ºC decrease in core body temperature | Hypothermia in response to cold, rescued by acute T3 administration | Jimenez et al (2002) (57) |
| Flandin et al (2009) (44) |
| Genetic Mouse Models | Obligatory Thermogenic Phenotype | Facultative Thermogenic Phenotype | References |
|---|---|---|---|
| UCP1−/− | Normal baseline temperature | Hypothermia in response to cold | Enerback et al (1997) (42) |
| Gong et al (2000) (36) | |||
| Golozoubova et al (2006) | |||
| UCP3−/− | Normal baseline temperature | Normal thermogenesis in response to cold, hypothermia in response to sympathomimetics | Mills et al (2003) (72) |
| Vidal-Puig et al (2000) (43) | |||
| Flandin et al (2009) (44) | |||
| TRα1−/− | Reduced baseline temperature | Normal thermogenesis in response to cold | Wikström et al (1998) (61) |
| Golozoubova et al (2003) | |||
| TRa 0/0 | Reduced baseline temperature | Hypothermia in response to cold | Marrif et al (2005) (64) |
| Pelletier et al (2008) (86) | |||
| TRβ−/− | Normal baseline temperatures | Normal response to thermogenesis | Johnsson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| TRα1/TRβ−/− | 0.4ºC decrease in core body temperature | Hypothermia in response to cold | Johansson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| D2−/− (D2KO) | Normal baseline temperature | Cold exposure leads to increased SNS stimulation of BAT but mice are hypothermic | Schneider et al (2001) (87) |
| Impaired embryonic BAT differentiation results in defective BAT function in adult mice | de Jesus et al (2001) (50) | ||
| Christoffolete et al (2004) (52) | |||
| Hall (2010) (88) | |||
| Castillo et al (2011) (89) | |||
| β1/β2/β3−/− (TKO) | 1.2ºC decrease in core body temperature | Hypothermia in response to cold, rescued by acute T3 administration | Jimenez et al (2002) (57) |
| Flandin et al (2009) (44) |
Abbreviation: D2KO, mice lacking D2.
Mouse Models of TH and UCP Thermogenesis
| Genetic Mouse Models | Obligatory Thermogenic Phenotype | Facultative Thermogenic Phenotype | References |
|---|---|---|---|
| UCP1−/− | Normal baseline temperature | Hypothermia in response to cold | Enerback et al (1997) (42) |
| Gong et al (2000) (36) | |||
| Golozoubova et al (2006) | |||
| UCP3−/− | Normal baseline temperature | Normal thermogenesis in response to cold, hypothermia in response to sympathomimetics | Mills et al (2003) (72) |
| Vidal-Puig et al (2000) (43) | |||
| Flandin et al (2009) (44) | |||
| TRα1−/− | Reduced baseline temperature | Normal thermogenesis in response to cold | Wikström et al (1998) (61) |
| Golozoubova et al (2003) | |||
| TRa 0/0 | Reduced baseline temperature | Hypothermia in response to cold | Marrif et al (2005) (64) |
| Pelletier et al (2008) (86) | |||
| TRβ−/− | Normal baseline temperatures | Normal response to thermogenesis | Johnsson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| TRα1/TRβ−/− | 0.4ºC decrease in core body temperature | Hypothermia in response to cold | Johansson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| D2−/− (D2KO) | Normal baseline temperature | Cold exposure leads to increased SNS stimulation of BAT but mice are hypothermic | Schneider et al (2001) (87) |
| Impaired embryonic BAT differentiation results in defective BAT function in adult mice | de Jesus et al (2001) (50) | ||
| Christoffolete et al (2004) (52) | |||
| Hall (2010) (88) | |||
| Castillo et al (2011) (89) | |||
| β1/β2/β3−/− (TKO) | 1.2ºC decrease in core body temperature | Hypothermia in response to cold, rescued by acute T3 administration | Jimenez et al (2002) (57) |
| Flandin et al (2009) (44) |
| Genetic Mouse Models | Obligatory Thermogenic Phenotype | Facultative Thermogenic Phenotype | References |
|---|---|---|---|
| UCP1−/− | Normal baseline temperature | Hypothermia in response to cold | Enerback et al (1997) (42) |
| Gong et al (2000) (36) | |||
| Golozoubova et al (2006) | |||
| UCP3−/− | Normal baseline temperature | Normal thermogenesis in response to cold, hypothermia in response to sympathomimetics | Mills et al (2003) (72) |
| Vidal-Puig et al (2000) (43) | |||
| Flandin et al (2009) (44) | |||
| TRα1−/− | Reduced baseline temperature | Normal thermogenesis in response to cold | Wikström et al (1998) (61) |
| Golozoubova et al (2003) | |||
| TRa 0/0 | Reduced baseline temperature | Hypothermia in response to cold | Marrif et al (2005) (64) |
| Pelletier et al (2008) (86) | |||
| TRβ−/− | Normal baseline temperatures | Normal response to thermogenesis | Johnsson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| TRα1/TRβ−/− | 0.4ºC decrease in core body temperature | Hypothermia in response to cold | Johansson et al (1999) (62) |
| Golozoubova et al (2003) | |||
| D2−/− (D2KO) | Normal baseline temperature | Cold exposure leads to increased SNS stimulation of BAT but mice are hypothermic | Schneider et al (2001) (87) |
| Impaired embryonic BAT differentiation results in defective BAT function in adult mice | de Jesus et al (2001) (50) | ||
| Christoffolete et al (2004) (52) | |||
| Hall (2010) (88) | |||
| Castillo et al (2011) (89) | |||
| β1/β2/β3−/− (TKO) | 1.2ºC decrease in core body temperature | Hypothermia in response to cold, rescued by acute T3 administration | Jimenez et al (2002) (57) |
| Flandin et al (2009) (44) |
Abbreviation: D2KO, mice lacking D2.
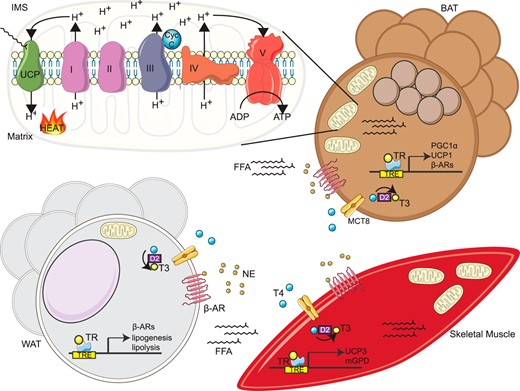
Tissue specific mechanisms of TH-mediated thermogenesis. In response to cold exposure, NE released from SNS nerve terminals binds β3-adrenergic receptors (β-AR) in BAT and WAT, increasing local and systemic FFA release along with an induction/activation of UCP1/3 and other potential thermogenic genes in BAT and skeletal muscle, respectively. Simultaneously, SNS stimulation activates D2 deiodinases in BAT and skeletal muscle that increase T3 levels, leading to the transactivation via TRs of thermogenic genes that ultimately govern the thermogenic capacities of BAT and skeletal muscle including PGC1α and mGPD. In BAT, UCP1 is activated by FFA release to transport protons from the mitochondrial intermembrane space (IMS) to the matrix, dissipating the proton gradient to produce heat. Similarly, we propose that skeletal muscle NST is activated in part by the uptake of FFA released from SNS-stimulated WAT lipolysis and UCP3 activation. MCT8, monocarboxylate transporter 8.
Acknowledgments
This work was funded in part by the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- BAT
brown adipose tissue
- D2
type II deiodinase
- FFA
free fatty acid
- G3P
glyceraldehyde 3-phosphate
- mGPD
mitochondrial glycerol 3-phosphate dehydrogenase
- NE
norepinephrine
- NST
nonshivering thermogenesis
- PGC1α
peroxisome proliferator-activated receptor-γ coactivator 1-α
- SKM
skeletal muscle
- SNS
sympathetic nervous system
- Tc
core body temperature
- TH
thyroid hormone
- TKO
mice lacking all β-adrenergic receptors
- TR
thyroid hormone receptor
- UCP
uncoupling protein
- WAT
white adipose tissue.

{kind=link}