





Gastrin, acting via the cholecystokinin-2 receptor (CCK2R), activates its own promoter in a positive-feed-forward loop that may result in hypergastrinemia. Activity of the gastrin promoter is also stimulated by exogenous Zn2+ ions. Here, the role of intracellular zinc and calcium signaling in the gastrin positive-feed-forward loop was investigated. Gastrin promoter activity was measured in the human gastric carcinoma cell line AGS-CCK2R and in Jurkat cells transfected with various gastrin promoter-luciferase constructs after treatment with gastrin in the presence and absence of zinc- and calcium-chelating agents. The free intracellular zinc ion concentrations were measured in the same cells with the fluorescent indicator FluoZin-3. Cell proliferation and migration/invasion were measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide cell proliferation assay and in Boyden chamber assays, respectively. The zinc chelator N,N,N,N-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN) abolished gastrin-stimulated gastrin promoter activity, and the inhibition was completely reversed by exogenous Zn2+ ions. In contrast, the calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) potentiated gastrin-stimulated gastrin promoter activity. Treatment with gastrin increased the intracellular concentration of free Zn2+ ions, and the increase was blocked by TPEN, but not by BAPTA-AM. TPEN also inhibited the stimulation of cell proliferation and migration/invasion by gastrin, but BAPTA-AM had no effect. These results, which are the first report of the existence of Zn2+ signaling downstream of CCK2R activation, suggest that zinc chelation therapies may be effective in counteracting gastrin-dependent tumor growth.
The hormone gastrin is secreted by G cells in the gastric antrum and regulates gastric acid secretion in the stomach (1). Several different forms of gastrin peptides are generated from the prohormone progastrin of 80 amino acids, which is processed to nonamidated glycine-extended gastrins such as glycine-extended gastrin17 (termed Ggly), and amidation of glycine-extended gastrins gives rise to amidated gastrin17 (termed gastrin or Gamide). The acid-stimulatory and mitogenic effects of amidated gastrin are mediated via the cholecystokinin-2 receptor (CCK2R), which is a member of the G protein-coupled 7 transmembrane domain receptor (GPCR) family (2). In contrast, several candidates have been identified for the receptor(s) for nonamidated forms of gastrin (3–5).
Up-regulation of the expression of gastrin has been previously demonstrated in many cancers, particularly gastrointestinal (GI) cancers, and its role in tumor progression and metastasis is now well documented (2, 6). A key remaining question is the mechanism by which the expression of gastrins is up-regulated in tumors. Recently, our group demonstrated for the first time that hypoxia (1% O2) up-regulated gastrin gene expression in the human gastric adenocarcinoma cell line AGS and the human colorectal cancer cell line LoVo (7, 8). However, the magnitude of up-regulation induced by the commonly used hypoxia mimetic cobalt chloride (CoCl2) was much greater than that induced by true hypoxia (7). Comparison of the effect of other metal ions led to the unanticipated but remarkable discovery that exogenous Zn2+ ions also induced gastrin gene expression in carcinoma cell lines of various origin including stomach (AGS), colon (DLD1, HCT116, HT29, and SW480), and prostate (LNCaP), by as much as 90-fold in the case of HCT116 cells (9). Deletional mutation of the gastrin promoter identified an 11-bp zinc-response element, which contained a putative E-box motif necessary for Zn2+-dependent gastrin induction (9). More recently, using Transcription activator-like effector nucleases, a luciferase reporter gene was inserted downstream of the endogenous human gastrin promoter, and the construct was transfected into SW480 colon cancer cells. Using these luciferase reporter Gastluc SW480 colon cancer cells, we have shown that both Zn2+ and Co2+ ions activate the endogenous gastrin gene promoter in vitro and in vivo and that activation is dependent on the mitogen-activated protein kinases (MAPK), but not the Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), pathway (10).
Previously, we demonstrated that amidated gastrin, acting via the CCK2R, activates its own promoter and that the increase in mRNA transcription leads to an increase in gastrin peptide expression (11). This autoregulation was termed a gastrin positive-feed-forward loop, and a similar phenomenon has since been demonstrated in the regulation of glucagon (12) and hepcidin (13). The gastrin positive-feed-forward loop may be important as it could accelerate the development of GI cancer and explain in part the previously reported positive relationship between circulating gastrin concentrations and cancer incidence (14).
Gastrin-dependent calcium signaling has been described in numerous cell types endogenously expressing the CCK2R and in stably transfected cell lines (reviewed in Ref. 15). For example, gastrin induces AKT phosphorylation and inhibits apoptosis of AR4–2J pancreatic adenocarcinoma cells downstream of CCK2R activation via calcium-dependent pathways (16). In contrast, gastrin inhibited DNA synthesis and increased apoptosis of Mz-ChA-1, HuH-28, and TFK-1 cells, but both effects were inhibited by the calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (17). Thus, the significance of calcium signaling in response to CCK2R activation remains poorly understood. Moreover Ca2+-sensitive probes such as calcium green 1, fura 2, and fluo 3 interact with Zn2+, even in the presence of high concentrations of Ca2+ (18). Using the T-lymphocyte cell line Jurkat, it has been shown that the calcium signal measured using fluo 4 and fura 2 probes was completely inhibited by the Zn2+ chelator N,N,N,N-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN) (19). A critical implication of these results is that data obtained using Ca2+ probes may need to be reexamined to determine whether results previously attributed to Ca2+ could, in part, be due to Zn2+ (20).
In this study, the gastrin-response element within the gastrin promoter sequence has been characterized using various gastrin promoter-luciferase constructs after treatment with gastrin in the human gastric carcinoma cell line AGS-CCK2R. Furthermore, the potential roles of Zn2+ and Ca2+ ions in the gastrin positive-feed-forward loop and in signaling downstream from the CCK2 receptor were investigated by determining the gastrin promoter activity in AGS-CCK2R cells and in Jurkat cells transfected with a gastrin promoter-luciferase construct, after treatment with gastrin in the presence and absence of Zn2+- and Ca2+-chelating agents. The free intracellular zinc ion concentrations were measured in the same cells with the fluorescent indicator FluoZin-3. To determine whether zinc ions were involved in the biological response to gastrin stimulation, proliferation and migration/invasion were measured in the presence and absence of chelating agents.
Materials and Methods
Cell culture
In our laboratory, wild-type AGS cells do not express the CCK2R as determined by radioligand-binding assays. Also, these cells do not respond to gastrin in assays of proliferation or migration. Generation of AGS-CCK2R has been described previously (21). Human gastric carcinoma (AGS-CCK2R), colonic carcinoma (Colo320-CCK2R), and T-lymphocyte cells (Jurkat), were cultured in RPMI 1640 medium. The medium was supplemented with 7.5% fetal bovine serum, 100-U/mL penicillin, 100-pg/mL streptomycin, and 10mM HEPES (Invitrogen). All cells were maintained at 37°C in a humidified incubator with 95% air and 5% CO2. Untransfected wild-type cells were obtained from ATCC, and the cell passage numbers were kept less than 25.
RNA preparation and quantitative PCR
Cells were seeded at a density of 2.5 × 105 per well in growth media 1 day before the treatment. Treatments were carried out in serum-free medium for the indicated times. After treatment, total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized from isolated RNA with the SuperscriptII First Strand Synthesis system (Invitrogen) and was then used for real-time PCR amplification using an ABI PRISM 7700 Sequence Detector (Applied Biosystems) and TaqMan chemistry. The following primers were used: hGastrin forward, 5′-CCGCAGTGCTGAGGATGAG-3′; hGastrin reverse, 5′-GGAGGTGGCTAGGCTCTGAA-3′; and hGastrin MGB probe, 5′-CTAACAATCCTAGAACCAAG-3′. Gene expression was normalized to 18S rRNA as follows. Firstly, the Δ cycle threshold (CT) (CTgastrin − CT18S) values for untreated control and various treated samples were determined. Using this ΔCT values, the ΔΔCT (ΔCTtreated − ΔCTuntreated control) were calculated. The fold increase in gastrin mRNA expression was then calculated using the formulae 2−ΔΔCT.
Plasmid constructs
The gastrin-promoter Luciferase vector (1,300pGASLuc), which contains 1300 bp of the upstream promoter region and the first exon of the human gastrin gene, was a kind gift from Professor J. Merchant (University of Michigan, Ann Arbor, MI). Constructs with further deletions and mutations of the gastrin promoter in the pGL4.10 luciferase vector have been described previously (11).
Gastrin promoter assays
Cells were transfected using a Neon cell transfection system (Invitrogen) according to the manufacturer’s instructions. Briefly 2.0 × 106 cells were transfected with 3.5 μg of gastrin promoter-luciferase construct and 0.7 μg of control pGL4.74[hRLuc/TK] vector expressing hRLuc luciferase protein. Cells were pretreated for 1 hour with either TPEN or BAPTA-AM before the addition of gastrin. Cells were incubated 24 hours after transfection with gastrin for 16 hours, and the promoter activity was determined using a luciferase assay kit (Promega). Briefly, cells were lysed with 60–70 μL/well Reporter Lysis buffer and Firefly luciferase activity was measured with a FLUOstar Optima plate reader (BMG Labtech). Relative luciferase activity was normalized to the total protein content of each well as determined with a Bradford protein assay kit (Bio-Rad).
Intracellular free Zn2+ measurements
Cells (2.5 × 103 cells in 100 μL/well) were seeded onto black 96-well plates and incubated in 5% CO2 for 24 hours at 37°C. FluoZin-3 (AM, cell permeant, F-24195; Life Technologies) was diluted in Hanks’ balanced salt solution to a final concentration of 5μM (50 μL/well) and added to the 96-well plates, which were covered with foil to protect them from light. Cells were incubated for 30 minutes before the dye was removed and replaced by Hanks’ balanced salt solution for 15 minutes. Gastrin at a final concentration of 1μM was directly added to the cells. The resulting fluorescence was recorded on a MicroLumi XS luminometer (Harta Instruments). Minimum fluorescence (Fmin) and maximum fluorescence (Fmax) were determined by incubating with zinc chelator TPEN (10μM) or ZnCl2 (500μM), respectively. The dissociation constant (Kd) of FluoZin-3 for Zn2+ ions is 15nM according to the manufacturer’s instruction manual. The concentration of free Zn2+ ions (nM) was calculated as:
Western blot analysis
Cells were washed once with ice-cold PBS and lysed with 0.1- to 0.2-mL preboiled sodium dodecyl sulfate lysis buffer. Proteins were separated by SDS-PAGE and transferred onto Hybond-C Extra nitrocellulose membrane (GE Healthcare). Stripping the first antibody off the Western blot analysis and subsequently reprobing with a second antibody against the same protein leads to artifacts and false negatives in our chemiluminescent detection method, which in turn makes quantitation difficult and unreliable; a fact highlighted by various companies selling antibodies. To avoid these well-recognized pitfalls of stripping and reprobing the same membrane, 2 gels were run simultaneously using equal amounts of the same cell extracts. After transfer, 1 membrane was probed with antibody against total protein (eg, AKT), and the second membrane with antibody against the corresponding phosphoprotein (eg, phospho-AKT). Further, as AKT and ERK1/2 are well separated from one another during gel electrophoresis, each membrane was cut in 2. Half of 1 membrane was then used for detection of AKT, and the corresponding other half for detection of ERK1/2. Bands were visualized in a LAS 3000 Image Reader (Fujifilm), with an ECL Advance Western Blotting Detection kit (GE Healthcare). Band densities were analyzed with MultiGauge software (Fujifilm), and the ratio of phosphoprotein over total protein was calculated. Thus, the blots shown for total ERK and total AKT in figure 5 below are actually from the same membrane and therefore show the same curvature, whereas the blots shown for phospho-ERK1/2 and phospho-AKT in figure below are from a different membrane and therefore show a different curvature.
Measurement of cell proliferation
Cell proliferation was measured using a colorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) as previously described (22). In brief, 1.2 × 105 cells/well were plated into a 12-well plate in culture medium containing 7.5% fetal bovine serum. The next day, the medium was replaced with serum-free medium containing various treatments and cells were incubated 36 hours with gastrin. At the end of incubation, 10 μL of MTT solution (5 mg/mL in PBS) were added to each well and incubated at 37°C for 2 hours. The medium was then removed carefully without disturbing the insoluble formazan crystals. 200 μL of 0.04M HCl in isopropanol was added to each well to solubilize the crystals, and the absorbance was read at 570 nm using a spectrophotometer. Readings from wells that received control medium and no MTT treatment were used as blanks. Cells were pretreated for 1 hour with either TPEN or BAPTA-AM before the addition of gastrin.
Migration/invasion assay
Migration/invasion was measured using a modified Boyden chamber assay. Cells at a density of 5 × 104 cells in 250-μL aliquots of serum-free culture medium were seeded into upper chambers containing polyethylene terephthalate filter membranes (8-μm pore size; Becton Dickinson) coated with fibronectin. The upper chambers were inserted into tissue culture wells, and 750-μL aliquots of serum-free culture medium containing gastrin with and without TPEN or BAPTA-AM were added to the lower chambers. After incubation overnight at 37°C, nonmigratory cells on the upper surface of the membranes were removed with a cotton swab, and cells that had migrated through the membrane pores and invaded the underside of the membranes were fixed with 90% methanol, stained with hematoxylin and eosin, and counted under a microscope.
Statistical analysis
Data are presented as mean ± SEM. Statistical significance for single comparisons of normally distributed data was determined by Student’s t test or, for data that were not normally distributed, by Mann-Whitney rank sum test. Multiple comparisons for normally distributed data were performed by one-way ANOVA followed by the Bonferroni correction. For multiple comparisons of data that was not normally distributed a nonparametric Kruskal-Wallis ANOVA test was performed. All statistics were analyzed with the program SigmaStat (Jandel Scientific).
Results
Characterization of the gastrin-response element within the gastrin promoter
Sequential deletion of the human gastrin promoter previously showed that responsiveness to stimulation with exogenous gastrin was dependent on elements within the first 365 bp of the promoter (11). To identify the gastrin-response element more precisely, the responsiveness to exogenous gastrin of a panel of gastrin promoter mutants, previously generated to investigate responsiveness to exogenous Zn2+ ions, was analyzed (9). Treatment with 50nM gastrin of AGS-CCK2R cells transfected with the plasmid 365pGASLuc significantly stimulated the gastrin promoter activity by 5.7 ± 0.8-fold (Figure 1A), and deletion of promoter sequences comprising blocks 1 to 4 had no effect on the stimulation of gastrin promoter activity by 50nM gastrin. In contrast, deletion of the promoter sequence contained in region 5 (−163 and −109 bp) resulted in a significant reduction in gastrin promoter activity.
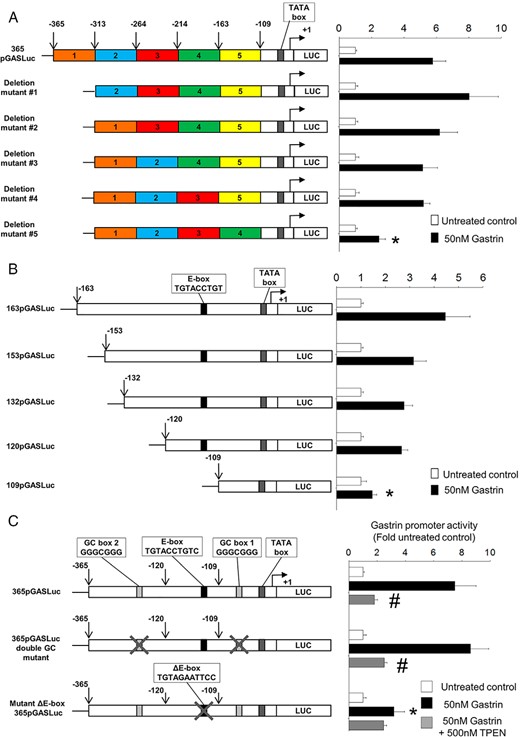
Mutation of the zinc-response element in the proximal gastrin promoter or zinc chelation completely abolishes gastrin-induced gastrin promoter activity. A, Gastrin promoter activity was measured in AGS-CCK2R cells transfected with the gastrin promoter luciferase construct 365pGASLuc or with one of the 5 mutants with block deletions of approximately 50 bp generated from the 365pGASLuc construct. Cells were treated with 50nM gastrin for 16 hours. The firefly luciferase activity of each construct was expressed as a ratio of the corresponding value for untreated cells. Values are expressed as the mean ± SEM of at least 3 separate experiments. *, P < .05 vs 365pGASLuc. B, Gastrin promoter activity was measured in AGS-CCK2R cells transfected with 163pGASLuc, 153pGASLuc, 132pGASLuc, 120pGASLuc, or 109pGASLuc gastrin promoter luciferase constructs. *, P < .05 vs 163pGASLuc. C, Gastrin promoter activities were determined in the presence or absence of 500nM TPEN in AGS-CCK2R cells transfected with either the wild-type 365pGASLuc construct, or the ΔE-box365pGASLuc mutant, or the 365pGASLuc double GC mutant, in which both the GC box sequences (GGGCGGG) were mutated. *, P < .05 vs 365pGASLuc; #, P < .05 vs gastrin-treated wild-type 365pGASLuc.
Because the gastrin-response element lay within the sequence between −163 and −109 bp, gastrin promoter activity was measured using a panel of shorter gastrin promoter luciferase constructs to narrow down the region further (Figure 1B). The significant reduction of gastrin promoter activity from 2.7 ± 0.2-fold in AGS-CCK2R cells transfected with the 120pGASLuc construct to 1.4 ± 0.2-fold in AGS-CCK2R cells transfected with the 109pGASLuc construct indicated that the gastrin-response element lay within the 11 nucleotides between −120 and −109 bp in the gastrin promoter (Figure 1B). Our previous analysis revealed the presence of a putative E-box consensus sequence (CANNTG), which regulates gastrin expression in response to exogenous Zn2+ ions, within the 11 nucleotides between −120 and −109 bp (9). To investigate whether the putative E-box sequence also mediates the effect of gastrin, AGS-CCK2R cells were transfected with the plasmids 365pGASLuc or ΔE-box 365pGASLuc, and gastrin promoter activity was measured by luciferase assay. Mutation of the E-box sequence significantly reduced the stimulation of gastrin promoter activity by 50nM gastrin (Figure 1C). In contrast, mutation of the 2 GC boxes in the 365pGASLuc plasmid had no effect on gastrin responsiveness.
The observation that the same E-box mediated induction of gastrin promoter activity by either exogenous Zn2+ ions or gastrin suggested that Zn2+ ions might be involved in the gastrin positive-feed-forward loop downstream of CCK2R activation. To test this hypothesis, gastrin promoter activity of AGS-CCK2R cells transfected with the 365pGASLuc plasmid and stimulated by 50nM gastrin was measured with and without the zinc chelator TPEN. Treatment of AGS-CCK2R cells transfected with the 365pGASLuc plasmid with 50nM gastrin significantly stimulated the gastrin promoter activity by 7.5 ± 1.5-fold. However, in the presence of 500nM TPEN, the gastrin promoter activity was significantly reduced to only 1.8 ± 0.2-fold (Figure 1C). A similar inhibition of gastrin-stimulated gastrin promoter activity in the presence of 500nM TPEN was also observed with the 365pGASLuc double GC mutant construct, but in contrast, TPEN did not significantly reduce gastrin promoter activity in AGS-CCK2R cells transfected with the ΔE-box 365pGASLuc plasmid.
Inhibition of gastrin expression in the presence of TPEN is reversed by Zn2+ ions
Although TPEN has a high affinity for Zn2+ ions, it has also been shown to chelate ferric ions. To test whether the inhibition of the gastrin feed-forward loop by TPEN was specifically mediated by zinc chelation, the ability of exogenous Zn2+, Fe3+ or Ca2+ ions to reverse the inhibition of gastrin promoter activity by TPEN was investigated using AGS-CCK2R cells transfected with the 365pGASLuc construct. As shown in Figure 2A, 50nM gastrin stimulated the gastrin promoter activity by 10.1 ± 1.1-fold, and the stimulation was reduced to 2.0 ± 0.2-fold in the presence of 500nM TPEN. Because the inhibition of gastrin stimulation by 500nM TPEN was almost completely reversed in the presence of 5μM exogenous Zn2+ ions to 9.1 ± 1.4-fold, the inhibition of gastrin promoter activity in the presence of TPEN was likely due to zinc chelation. These results were replicated in Jurkat cells which endogenously express CCK2R (Figure 2B).
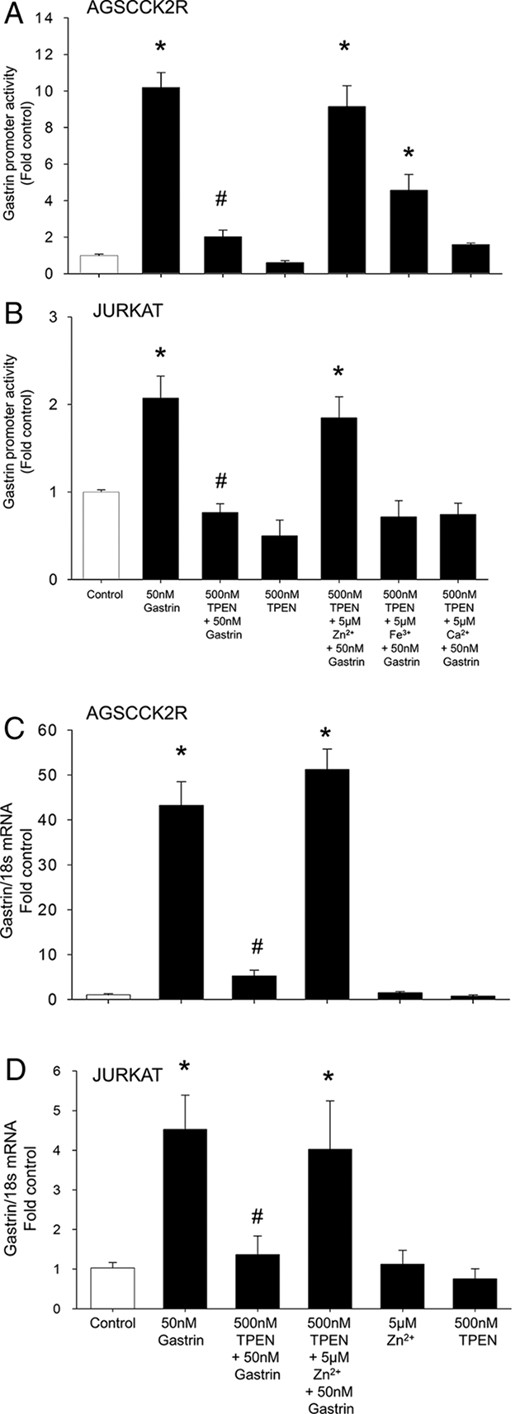
TPEN inhibition of gastrin promoter activity and gastrin mRNA expression is reversed in the presence of exogenous Zn2+ ions. Gastrin promoter activity was measured in AGS-CCK2R cells (A) or Jurkat cells (B) transfected with 365pGASLuc in the presence of 500nM TPEN in combination with either Zn2+, Fe3+ (ferric ammonium citrate), or Ca2+ ions. Values are expressed as the mean ± SEM of at least 3 separate experiments. *, P < .05 vs 365pGASLuc; #, P < .05 vs gastrin-treated 365pGASLuc. Gastrin mRNA expression was measured by real-time PCR in AGS-CCK2R cells (C) or Jurkat cells (D) transfected with 365pGASLuc in the presence of 500nM TPEN in combination with either Zn2+, Fe3+, or Ca2+ ions. Gastrin mRNA was expressed relative to the amount of 18S rRNA. Values (mean ± SEM of at least 3 separate treatments) were expressed as the fold increase relative to the corresponding untreated cells. *, P < .05 vs untreated control; #, P < .05 vs 50nM gastrin-treated cells.
The inhibition of gastrin promoter activity by TPEN in AGS-CCK2R cells transfected with the 365pGASLuc construct was partially reversed to 4.5 ± 0.9-fold in the presence of 5μM exogenous Fe3+ ions but unaffected by Ca2+ ions (Figure 2A). Previously, we have shown that treatment of AGS cells with exogenous Fe3+ ions does not stimulate gastrin expression (9), and therefore, the partial reversal of gastrin promoter activity in the presence of Fe3+ ions may be due to intracellular binding of Fe3+ ions to TPEN, which may in turn prevent TPEN from chelating intracellular Zn2+ ions. In Jurkat cells, neither ferric nor Ca2+ ions were able to reverse the inhibition of gastrin promoter activity by TPEN (Figure 2B).
To determine whether inhibition of gastrin promoter activity by TPEN leads to a change in gastrin gene transcription, expression of gastrin mRNA in AGS-CCK2R and Jurkat cells was measured after treatment with gastrin in the presence or absence of TPEN. In both AGS-CCK2R (Figure 2C) and Jurkat cells (Figure 2D), gastrin gene transcription was activated by 50nM gastrin, by 43.2 ± 10- and 4.5 ± 0.9-fold, respectively. The increase in gastrin gene transcription was significantly inhibited in the presence of 500nM TPEN, and the inhibition was reversed in the presence of exogenous Zn2+ ions (Figure 2, C and D).
The calcium chelator BAPTA-AM does not inhibit gastrin expression in AGS CCK2R cells
TPEN has a high affinity for Zn2+ ions but can also bind Ca2+ ions, although with very low affinity (23). Because activation of the CCK2R stimulates calcium signaling (15), the possible involvement of Ca2+ ions in the gastrin positive-feed-forward loop was investigated by measurement, in the presence or absence of the cell permeable calcium chelator BAPTA-AM, of the gastrin-stimulated gastrin promoter activity using AGS-CCK2R cells transfected with the 365pGASLuc construct. As shown in Figure 3, in contrast to the 4.4 ± 1.4-fold increase in gastrin promoter activity in cells treated with 50nM gastrin only, in the presence of 500nM or 1μM BAPTA-AM, gastrin stimulated the promoter activity by 7.4 ± 1.4- or 17.1 ± 3.1-fold, respectively. In conclusion BAPTA-AM did not inhibit the stimulation of gastrin promoter activity by 50nM gastrin. On the contrary, there was a dose-dependent increase in gastrin promoter activity in the presence of BAPTA-AM.
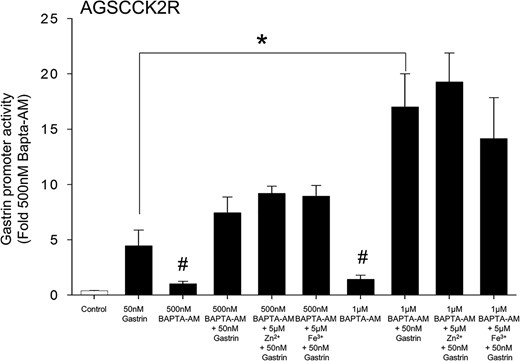
Augmentation of gastrin-induced gastrin promoter activity in the presence of the intracellular calcium chelator BAPTA-AM. Gastrin promoter activity was measured in the presence of BAPTA-AM at the indicated concentration in AGS-CCK2R cells transfected with the 365pGASLuc plasmid. Values (mean ± SEM of at least 3 separate treatments) were expressed as the fold increase relative to cells treated with 500nM BAPTA-AM. #, P < .05 vs control.
Increase in intracellular free Zn2+ ions after CCK2R activation in AGS-CCK2R cells
To investigate whether activation of the CCK2R by gastrin increases the level of intracellular free Zn2+ ions, the change in fluorescence of the zinc-specific dye FluoZin-3, which change is directly proportional to the increase in intracellular free Zn2+ ions, was measured over time after the treatment of AGS-CCK2R cells or Jurkat cells with gastrin. As seen in Figure 4A, gastrin increased the fluorescence of FluoZin-3 in a dose-dependent manner in both the CCK2R-transfected AGS-CCK2R cells and in Jurkat cells, which express the CCK2R endogenously. Treatment of AGS-CCK2R cells with a maximal dose of 1μM gastrin resulted in the concentration of intracellular free Zn2+ ions increasing by 2.5 ± 0.2-fold after 30 minutes (Figure 4B) and 5.1 ± 0.9-fold after 2 hours (Figure 4C). Importantly, treatment of the cells with 500nM TPEN blocked the increase in intracellular free Zn2+ ions, but 500nM BAPTA-AM had no effect. Furthermore, the inhibition in the rise of free Zn2+ ions after treatment with 500nM TPEN was reversed in the presence of 5μM ZnCl2. The concentration of intracellular free Zn2+ ions in cells treated with 500nM TPEN only remained unchanged at 0.95 ± 0.1-fold, compared with 1.0 ± 0.15-fold in control untreated cells. The basal concentration of Zn2+ ions in untreated AGS-CCK2R was 0.6 ± 0.12nM. Although treatment of AGS-CCK2R cells with 5μM ZnCl2 caused a similar increase of 2.1 ± 0.2-fold in intracellular free Zn2+ ions as treatment with 1μM gastrin (2.5 ± 0.2-fold) (Figure 4B), it failed to increase the expression of gastrin mRNA (Figure 2C). Using FluoZin-3 in combination with the zinc chelator TPEN, an increase in intracellular free Zn2+ ions downstream of the CCK2 receptor was also observed in the human colorectal carcinoma cell line Colo320-CCK2R (Figure 4D).
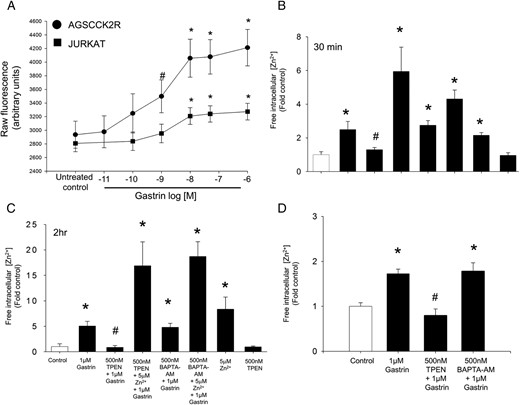
CCK2R activation by gastrin increases intracellular free Zn2+. A, The first panel shows the raw fluorescence increase in AGSCCK2R or Jurkat cells preloaded with FluoZin-3 after the treatment with increasing doses of gastrin for 45 minutes. The raw fluorescence readings for Fmin (10μM TPEN) and Fmax (500μM ZnCl2) were 2062 ± 142 and 22 586 ± 114, respectively, for AGS-CCK2R cells. Similarly for Jurkat cells, the raw fluorescence reading for Fmin and Fmax were 486 ± 60 and 5887 ± 149, respectively. *, P < .05 vs untreated control by one way ANOVA; #, P < .05 vs control by t test. The concentration of intracellular free Zn2+ ions was determined using the FluoZin-3 fluorescent probe in gastric AGS-CCK2R cells treated with 1μM gastrin for either 30 minutes (B) or 2 hours (C), or in colorectal Colo-CCK2R cells treated with 1μM gastrin for 30 minutes (D). Fluorescence was measured, and Fmin and Fmax were determined by addition of 10μM TPEN and 500μM ZnCl2, respectively. The free Zn2+ concentration (nM) was calculated as described in Materials and Methods, and values (mean ± SEM of at least 3 separate treatments) were expressed as the fold increase relative to untreated cells.
Zinc ions downstream of CCK2R activation do not induce ERK1/2 phosphorylation
Although binding of gastrin to the CCK2R activates both ERK1/2 and AKT signaling pathways in AGS-CCK2R cells, we have previously shown that only phosphorylation of ERK1/2 is important for the gastrin positive-feed-forward loop (11). On the other hand, Zn2+ ions have been shown to activate phosphorylation of both ERK1/2 and AKT (9), possibly via inhibition of protein tyrosine phosphatases (PTPs) (24, 25). To investigate the possibility that activation of ERK1/2 downstream of CCK2R activation is dependent on the mobilization of Zn2+ ions and that inhibition of the gastrin positive-feed-forward loop by TPEN is due to inhibition of phosphorylation of ERK1/2, the phosphorylation of ERK1/2 was measured after stimulation by gastrin in the presence or absence of TPEN or BAPTA-AM (Figure 5). Treatment of AGS-CCK2R cells with 50nM gastrin for 2 hours stimulated phosphorylation of ERK1/2 by 11.8 ± 1.2-fold (Figure 5A). In the presence of 500nM TPEN, gastrin-stimulated phosphorylation of ERK1/2 was significantly lower at 7.3 ± 1.5-fold, but unexpectedly, in the presence of 5μM BAPTA-AM, gastrin-stimulated phosphorylation of ERK1/2 was significantly higher at 24 ± 7-fold, a value approximately twice the stimulation observed with gastrin alone. There was no change in the magnitude of phosphorylation of AKT stimulated by gastrin in the presence of either TPEN or BAPTA-AM. Even though treatment of AGS-CCK2R cells with 5μM ZnCl2 resulted in a similar increase in intracellular free Zn2+ ions to the increase induced by 50nM gastrin (Figure 4A), the failure to stimulate gastrin expression (Figure 2C) was consistent with the observation that 5μM ZnCl2 did not stimulate phosphorylation of ERK1/2.
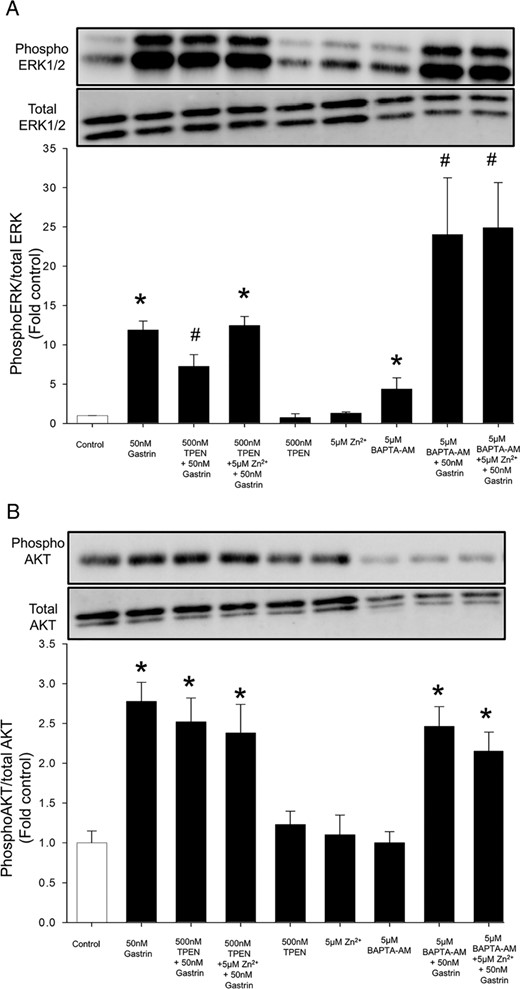
Zinc chelation does not inhibit gastrin stimulation of the MAPK or PI3K pathway. Phosphorylation of ERK1/2 (A), a mediator of the MAPK pathway, and of AKT (B), a mediator of the PI3K pathway, was measured by Western blot analysis as described in Materials and Methods after treatment of AGS-CCK2R cells with 50nM gastrin for 2 hours either in the presence of the zinc chelator TPEN or the calcium chelator BAPTA-AM. Band densities were determined and are presented as the ratio of densities of phosphorylated to total protein. Values are expressed as the mean ± SEM of at least 3 separate experiments. *, P < .05 vs untreated control cells; #, P < .05 vs 50nM gastrin-treated cells.
CCK2R-dependent release of paracrine growth factors is inhibited by zinc chelation
To determine whether the increase in intracellular free Zn2+ ions stimulated by activation of CCK2R by gastrin results in a biological response, the effect of gastrin on proliferation of AGS-CCK2R cells was examined. As shown in Figure 6A, treatment of AGS-CCK2R cells with gastrin at concentrations greater than 10nM for 36 hours resulted in a small but significant inhibition of proliferation. The effect of gastrin on proliferation of AGS-CCK2R cells has been controversial, with one study reporting stimulation (26), and another inhibition (27). In our hands, the effects of gastrin with or without TPEN were not of sufficient magnitude to say with certainty whether or not they were dependent on Zn2+ ions.
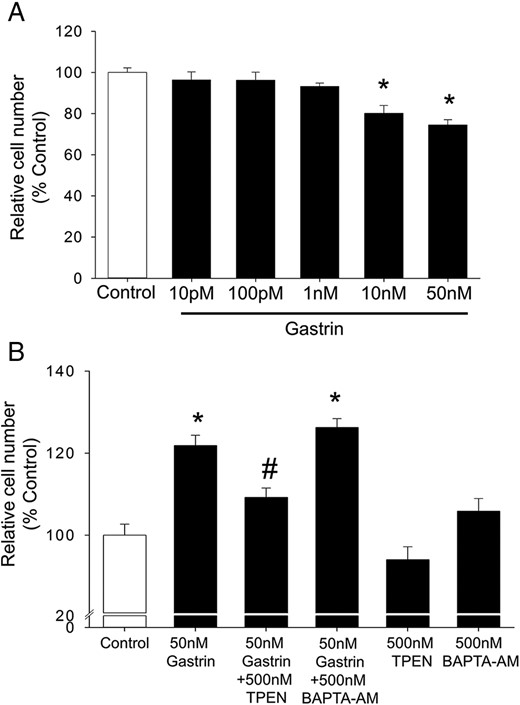
Gastrin directly inhibits cell proliferation but stimulates paracrine growth effects. A, Proliferation of AGS-CCK2R cells was assessed by MTT assay after exposure to the indicated concentrations of gastrin for 36 hours in a serum-free medium. Values are expressed as a percentage of untreated cells and are mean ± SEM of at least 3 separate experiments. *, P < .05 vs control. B, Proliferation of quiescent wild-type AGS cells was assessed after exposure to the conditioned media collected after the treatment of AGS-CCK2R cells with gastrin in the presence of absence of TPEN or BAPTA-AM. Values are expressed as a percentage of cells treated with control conditioned medium and are the mean ± SE of 3 separate treatments. *, P < .05 vs control; #, P < .05 vs 50nM gastrin-treated cells.
Previously, gastrin, acting via the CCK2R, has been shown to stimulate the production of multiple paracrine mediators of proliferation (27). To determine whether the paracrine proliferative responses attributed to gastrin-dependent release of paracrine growth factors were dependent on Zn2+ ions, AGS-CCK2R cells were treated with 50nM gastrin in the presence or absence of either TPEN or BAPTA-AM for 24 hours, and conditioned medium was collected. Wild-type AGS cells were then treated with the conditioned medium for 36 hours, and cell proliferation was measured by MTT assay. As shown in Figure 6B, conditioned medium acquired after treatment of AGS-CCK2R cells with 50nM gastrin significantly increased proliferation of wild-type AGS cells. In contrast, the observation that conditioned medium acquired in the presence of 50nM gastrin and 500nM TPEN increased proliferation of wild-type AGS cells to a significantly lesser extent suggested that gastrin-induced secretion of paracrine mediators of proliferation was dependent on CCK2R-stimulated signaling by Zn2+ ions. On the other hand, as shown in Figure 6B, conditioned medium acquired after treatment of AGS-CCK2R cells with 50nM gastrin in the presence of 500nM BAPTA-AM simulated paracrine proliferation as effectively as conditioned medium acquired in the presence of 50nM gastrin only.
Zinc chelation suppresses gastrin-induced cell migration/invasion
Although gastrin has previously been shown to stimulate migration and invasion of AGS-CCK2R cells, the role of zinc signaling in this process is unknown. To investigate whether gastrin-stimulated migration/invasion was inhibited by zinc chelation, migration/invasion of either AGS-CCK2R or Jurkat cells was assayed in a Boyden chamber in the presence and absence of the zinc chelator TPEN. Treatment of AGS-CCK2R cells with 50nM gastrin resulted in morphological changes and a significant increase in migration/invasion to 155 ± 10% compared with untreated AGS-CCK2R cells (Figure 7, A and B). The fact that stimulation of migration/invasion in AGS-CCK2R cells by gastrin was reduced to 115 ± 13% in the presence of 500nM TPEN suggested that intracellular Zn2+ ions play an important role downstream of the CCK2R (Figure 7B). Furthermore, the inhibition by 500nM TPEN of gastrin-stimulated migration/invasion in AGS-CCK2R (Figure 7B) and Jurkat cells (Figure 7C) was reversed in each case by exogenous Zn2+ ions. In contrast, the fact that 500nM BAPTA-AM had no effect on the stimulation of migration/invasion in AGS-CCK2R cells by gastrin (Figure 7B) suggested that intracellular Ca2+ ions are not essential for gastrin-simulated migration.
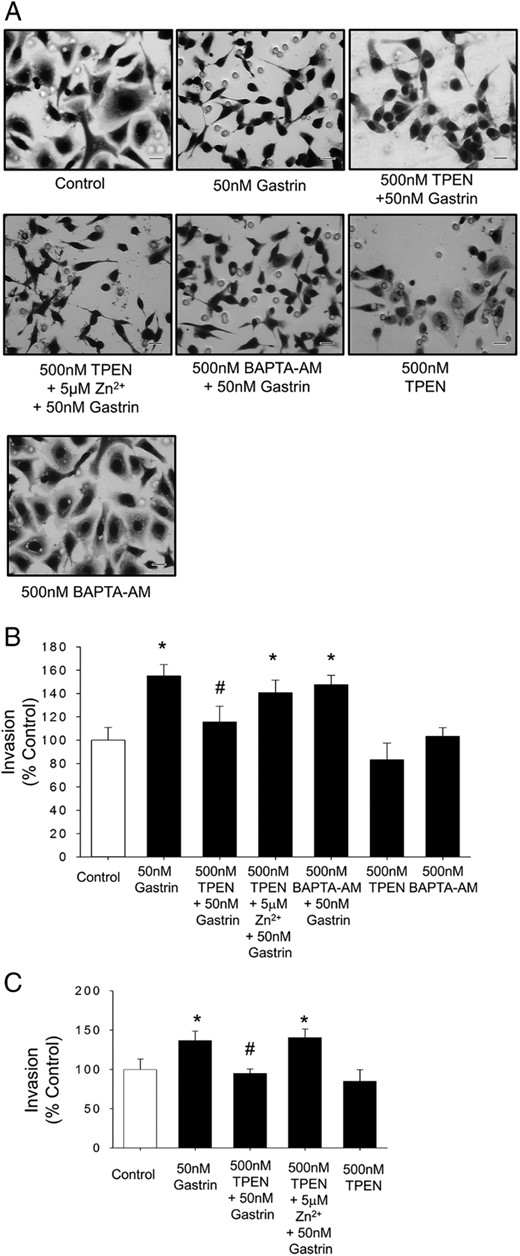
Zinc chelation inhibits gastrin-stimulated migration. A, Morphological changes in AGS-CCK2R cells after the treatment with 10nM gastrin in a Boyden chamber transwell assay in the presence or absence of TPEN or BAPTA-AM. Scale bars, 20 μm. B and C, Migration/invasion of AGS-CCK2R cells (B) or Jurkat cells (C) after treatment with 50nM gastrin in the presence or absence of TPEN was assessed by transwell assay. Values are the mean ± SEM of at least 3 separate experiments and are expressed as a percentage of untreated control cells. *, P < .05 vs control; #, P < .05 vs 50nM gastrin-treated cells.
Discussion
We have previously demonstrated that gastrin acting via the CCK2R increases its own expression via a positive-feed-forward loop (11). In a more recent publication, we reported that an 11-bp sequence containing an E-box motif located between −120 and −109 bp in the gastrin proximal promoter acted as the Zn2+-response element, which regulates gastrin expression in response to exogenous Zn2+ ions (9). In this study, the same 11-bp sequence containing the putative E-box was shown to be responsible for the activation of the gastrin promoter downstream of CCK2R activation by gastrin. The fact that either treatment with exogenous Zn2+ ions or activation of the CCK2R stimulated gastrin promoter activity via the same 11-bp sequence led to the hypothesis that occupation of the CCK2 receptor by gastrin activated downstream intracellular zinc signaling. Here, we also report that the cell-permeable Zn2+ chelator TPEN inhibited the induction of gastrin promoter activity by gastrin in AGS-CCK2R cells and in Jurkat T-lymphocyte cells (which express endogenous CCK2R). The observation that TPEN inhibition was reversed by addition of Zn2+ ions confirmed that intracellular Zn2+ ions are required for activation of the gastrin positive-feed-forward loop.
Zn2+ ions have recently been implicated in information transfer within and between cells (28), and extracellular stimuli have been postulated to increase the intracellular [Zn2+] (the so-called “Zn wave”) (29, 30), which then acts as an intracellular signaling molecule. In this study, using the Zn2+-specific fluorescent probe FluoZin-3 in combination with the Zn2+ chelator TPEN an increase in intracellular free Zn2+ downstream of the CCK2R activation was demonstrated in both gastric AGS and colonic Colo320 cells. Although G protein-coupled receptors (eg, 5HT1A and 5HT2A serotonin receptors) (31) and cytokine receptors (32) have previously been shown to mobilize Zn2+ ions in response to agonist stimulation, the current study is the first to demonstrate that activation of CCK2R leads to an increase in intracellular free Zn2+ ions.
Activation of intracellular calcium signaling has been described in numerous cell types naturally expressing endogenous CCK2R as well as in stably transfected cell lines (15). However, the mechanisms regulating calcium signals in response to CCK2R activation remain poorly understood (15). Previously, calcium chelation has been shown to inhibit gastrin-induced cancer cell proliferation (33–35). In the current study, the observation that gastrin promoter activity was blocked in the presence of the Zn2+ chelator TPEN (Figure 3) while increased in the presence of the calcium chelator BAPTA-AM in a dose-dependent manner suggested a previously unrecognized role for Zn2+ ions in response to CCK2R activation. A possible mechanism is that depletion of intracellular Ca2+ ions facilitates the up-regulation of gastrin expression downstream of CCK2R activation. BAPTA-AM has previously been shown to stimulate expression of the plasminogen activator inhibitor I gene (36) and carbonic anhydrase 9 (37) via hypoxia-inducible factor-1 (HIF1). However, a role for HIF1 in the induction of gastrin expression by BAPTA-AM can be ruled out because, although cobalt and hypoxia induce expression of both HIFs and gastrin, gastrin expression is increased via a HIF-independent pathway (7).
Zinc ions have previously been shown to modulate signal transduction pathways downstream of activation of various receptors (38, 39). For example, intracellular Zn2+ ions inhibit PTP (24, 25), which in turn results in augmented phosphorylation of ERK1/2 (40). Previously, we had shown that treatment of AGS cells with extracellular Zn2+ ions induced phosphorylation of ERK1/2 in a time-dependent manner (9), and that activation of phosphorylation of ERK1/2 is essential for stimulation of gastrin expression by exogenous Zn2+ ions (9). Therefore the possibility that the increase in phosphorylation of ERK1/2 downstream of CCK2R activation by gastrin might be instigated by Zn2+-dependent inhibition of PTPs was investigated. However, contrary to the above hypothesis, the observation that gastrin was able to stimulate phosphorylation of ERK1/2 in the presence of the zinc chelator TPEN suggested that the increase in phosphorylation of ERK1/2 was not regulated by intracellular free Zn2+ ions in the case of CCK2R. However, a small but significant decrease in the phosphorylated form of ERK1/2 in the presence of TPEN plus gastrin compared with gastrin alone suggest that free intracellular Zn2+ ions may decrease the rate of dephosphorylation of ERK/1/2 via inhibition of PTPs and hence maintain ERK1/2 in a phosphorylated form.
Interestingly, the depletion of intracellular calcium by BAPTA-AM not only increased basal ERK1/2 phosphorylation but also doubled the magnitude of the stimulation of phosphorylation of ERK1/2 by gastrin. In contrast, BAPTA-AM did not affect either basal or gastrin-stimulated AKT phosphorylation. These observations are in agreement with our previous report that, although gastrin activated both ERK1/2 and AKT phosphorylation, only inhibitors of the ERK pathway were able to block the gastrin positive-feed-forward loop, and our conclusion that ERK signaling is indeed the main pathway responsible for the increased gastrin expression (11). Moreover the increased gastrin promoter activity in BAPTA-AM-treated cells may be due to the increased phosphorylation of ERK1/2 in the presence of BAPTA-AM in AGS cells. Our observation that BAPTA-AM increases phosphorylation of ERK1/2 is not an isolated one as various studies have shown that BAPTA-AM can increase basal phosphorylation of ERK1/2 (41–43).
Zinc is an essential heavy metal and zinc deficiency is associated with a range of pathological conditions, including retarded growth, and delayed wound healing and tissue repair (28, 44). Zn2+ ions are critical for cell proliferation, cell cycle regulation, differentiation and apoptosis (45). To determine whether the increase in free intracellular Zn2+ ions downstream of CCK2R activation by gastrin had biological significance, 2 measures of bioactivity were assessed: proliferation and migration/invasion. The direct effect of gastrin on proliferation of AGS-CCK2R cells has been controversial, with one study reporting stimulation (26), and another inhibition (27). Our results are consistent with the inhibition reported by Varro et al (27). In addition to the direct inhibition of proliferation of AGS-CCK2R cells by gastrin, Varro et al have previously shown that gastrin stimulates proliferation of cocultured AGS cells that do not express the CCK2R by a paracrine pathway involving the EGF receptor (27). We have now shown that stimulation of the release of paracrine factors by gastrin is dependent on Zn2+ ions using the zinc chelator TPEN (Figure 6B). In contrast to proliferation, gastrin has been consistently shown to stimulate migration of AGS-CCK2R cells (46, 47). The observation that TPEN significantly inhibited gastrin-stimulated migration in both AGS-CCK2R and Jurkat cells, and that the inhibition was reversed in the presence exogenous Zn2+ ions, suggested that the gastrin-stimulated increase in free intracellular Zn2+ ions is an essential event in CCK2R-mediated migration.
Various studies have shown the existence of calcium signaling downstream of the CCK2R (15). BAPTA-AM is commonly used as a cell-permeable Ca2+ chelator to chelate cytosolic calcium and to elucidate the downstream effect of Ca2+ signaling. The fact that BAPTA-AM at micromolar concentrations inhibited gastrin-stimulated proliferation in CHO-CCK2R cells (35), and also gastrin-stimulated histamine release in isolated rat stomach (48), was taken as evidence for a direct involvement of intracellular Ca2+ ions in CCK2R signaling. Interestingly, BAPTA has been shown to bind Zn2+ with high affinity, and the Kd for Zn2+/BAPTA binding is 7.9nM compared with a Kd of 110nM for Ca2+/BAPTA binding (49). This observation suggests that the biological effects of BAPTA-AM, previously attributed to chelation of intracellular Ca2+ ions, could, in part, be due to chelation of Zn2+ ions. The data presented here that BAPTA-AM at nanomolar concentrations does not inhibit gastrin-simulated migration or paracrine proliferation but that, in contrast, the zinc chelator TPEN at the same concentration significantly inhibited both processes, suggest an important role of zinc signaling downstream of the CCK2R. Calcium has previously been shown to be necessary but not sufficient for FcϵRI-induced zinc signaling (50), and the increase in intracellular Zn2+ ions downstream of CCK2R activation may still possibly be calcium dependent. However, investigation of the precise details of the involvement of Ca2+ ions is beyond the scope of the current study.
In a recently published study, we demonstrated that Zn2+ ions induce gastrin transcription via an 11-bp DNA sequence, which contains an E-box motif (9). The current study demonstrates that the positive feed-forward loop, which induces gastrin expression downstream of its receptor, CCK2R (11), is mediated by the same 11-bp DNA sequence containing the E-box motif. Furthermore, our data indicate that stimulation of the CCK2R by gastrin results in increased cell migration and paracrine proliferation, which is dependent on an increase in intracellular free Zn2+ ions. As shown in Figure 8, a novel connection is therefore proposed between gastrin, zinc signaling, and the development of GI cancers with special emphasis on the involvement of E-box-binding transcription factors. An increase in intracellular free Zn2+ ions (the Zn wave), in response to either CCK2R activation by gastrin or uptake of exogenous extracellular Zn2+ ions via zinc transporters, may modulate the downstream activation of the E-box-binding transcription factor, which controls the expression of gastrin mRNA in GI cancer cells. The release of more gastrin into the surrounding medium would ultimately result in an autocrine increase in proliferation and migration, and would stimulate the development of GI cancers. However, the involvement of Ca2+ ions downstream of CCK2R remains unclear and further research is warranted (49).
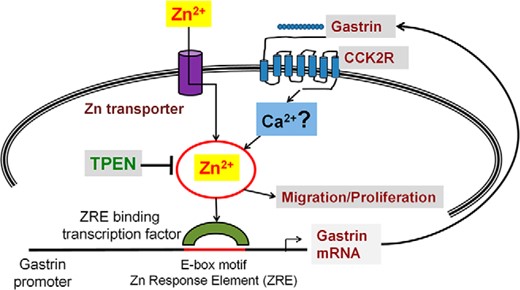
Gastrin, the CCK2R and zinc signaling. An increase in intracellular free Zn2+ ions (the Zn wave), in response to either CCK2R activation by gastrin, or uptake of exogenous extracellular Zn2+ ions via Zn2+ transporters, modulates the downstream activation of the E-box-binding transcription factor, which controls the expression of gastrin mRNA in GI cancer cells. The release of more gastrin into the surrounding medium ultimately results in an autocrine increase in proliferation and migration and stimulates the development of GI cancers. The involvement of Ca2+ ions downstream of the CCK2R remains unclear and further research is warranted.
Recognition of the biological importance of Zn2+ ions is continually expanding (28, 51), and this article demonstrates for the first time that activation of the CCK2 receptor by gastrin stimulates an increase in free intracellular Zn2+ ions (the so-called Zn wave). There is continuing interest in the role of gastrin and the related peptides progastrin and gastrin-gly as growth factors in GI cancer. Numerous studies have demonstrated an association between Helicobacter pylori infection and hypergastrinemia and gastric cancer. However, the clinical use of the CCK2R antagonist gastrazole has not resulted in an increase in survival in cancer patients (52). Targeting zinc signaling directly might be a superior approach, which will inhibit not only the gastrin positive-feed-forward loop that leads to hypergastrinemia but also gastrin-stimulated migration and paracrine growth effects. Further, because activation of zinc signaling is now increasingly being recognized as a feature common to various receptors and growth factors, therapeutics directed against Zn2+ ions might be effective not only for gastrin-dependent cancers of the GI tract but also for tumors of diverse origin that utilize growth factors other than gastrin.
Appendix
See Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Phospho-p44/42 | Phospho-p44/42 MAPK | Cell Signaling, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody 9101 | Rabbit | 1:3000 | |
| Total p44/42 | Total p44/42 MAPK | Cell Signaling, p44/42 MAPK (Erk1/2) antibody 9102 | Rabbit | 1:3000 | |
| Phospho-AKT | Phospho-AKT | Cell Signaling, phospho-Akt (Ser473) (D9E) XP rabbit mAb 4060 | Rabbit | 1:3000 | |
| Total AKT | Total AKT | Cell Signaling, Akt (pan) (11E7) rabbit mAb 4685 | Rabbit | 1:3000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Phospho-p44/42 | Phospho-p44/42 MAPK | Cell Signaling, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody 9101 | Rabbit | 1:3000 | |
| Total p44/42 | Total p44/42 MAPK | Cell Signaling, p44/42 MAPK (Erk1/2) antibody 9102 | Rabbit | 1:3000 | |
| Phospho-AKT | Phospho-AKT | Cell Signaling, phospho-Akt (Ser473) (D9E) XP rabbit mAb 4060 | Rabbit | 1:3000 | |
| Total AKT | Total AKT | Cell Signaling, Akt (pan) (11E7) rabbit mAb 4685 | Rabbit | 1:3000 |
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Phospho-p44/42 | Phospho-p44/42 MAPK | Cell Signaling, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody 9101 | Rabbit | 1:3000 | |
| Total p44/42 | Total p44/42 MAPK | Cell Signaling, p44/42 MAPK (Erk1/2) antibody 9102 | Rabbit | 1:3000 | |
| Phospho-AKT | Phospho-AKT | Cell Signaling, phospho-Akt (Ser473) (D9E) XP rabbit mAb 4060 | Rabbit | 1:3000 | |
| Total AKT | Total AKT | Cell Signaling, Akt (pan) (11E7) rabbit mAb 4685 | Rabbit | 1:3000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Phospho-p44/42 | Phospho-p44/42 MAPK | Cell Signaling, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody 9101 | Rabbit | 1:3000 | |
| Total p44/42 | Total p44/42 MAPK | Cell Signaling, p44/42 MAPK (Erk1/2) antibody 9102 | Rabbit | 1:3000 | |
| Phospho-AKT | Phospho-AKT | Cell Signaling, phospho-Akt (Ser473) (D9E) XP rabbit mAb 4060 | Rabbit | 1:3000 | |
| Total AKT | Total AKT | Cell Signaling, Akt (pan) (11E7) rabbit mAb 4685 | Rabbit | 1:3000 |
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- BAPTA-AM
1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- CCK2R
cholecystokinin-2 receptor
- CT
cycle threshold
- Fmax
maximum fluorescence
- Fmin
minimum fluorescence
- GI
gastrointestinal
- HIF1
hypoxia-inducible factor-1
- Kd
dissociation constant
- MAPK
mitogen-activated protein kinases
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- TPEN
N,N,N,N-tetrakis-(2-pyridylmethyl)-ethylenediamine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}