





Two foxl2 paralogs, foxl2 (also named foxl2a) and foxl3 (also named foxl2b), were considered as fish-specific duplicates. Both belong to the Forkhead box family of transcription factors, which play important roles in regulating reproduction involved in sexual differentiation and regulation of primordial germ cell and gonadal somatic cell development. We isolated the complete foxl3 cDNA from the rice field eel (Monopterus albus), which undergoes a natural female-to-male sex change via an intersex stage during its life cycle. The deduced amino acid sequence of M albus Foxl3 exhibited high identity with that of the European sea bass (Dicentrarchus labrax, 82.9%). The foxl3 expression levels in gonads were increased during the natural sex change process in M albus. The relative expression level in the testis was greater than 40-fold greater than in the ovary (P < .05). A dual-luciferase assay confirmed that the miR-9, but not the miR-430 family, bound to the foxl3 3′ untranslated region of M albus. Foxl3 was primarily expressed in granulosa cells and previtellogenic follicles in the ovary and in spermatogonia and Sertoli cells in the testis. In conclusion, Foxl3 and miR-9 may be involved in physiological processes that promote oocyte degeneration in the ovotestis and stimulating spermatogenesis in spermatogonia in M albus.
Forkhead box (FOX) proteins are transcription factors that are essential for the regulation of genes involved in cell growth, proliferation, differentiation, and longevity (1, 2). The defining feature of FOX proteins is the Forkhead box, a sequence of 80–100 amino acids forming a motif that binds to DNA (3). Human FOX gene family consists of at least 43 members, which belonged to 18 FOX subfamilies A-R (1, 2, 4).
FOX protein L subfamilies consist of Forkhead box L1 (Foxl1) and Forkhead box L2 (Foxl2). Foxl1 is involved in proteoglycan biosynthesis through the regulation of the Wnt/β-catenin pathway, providing a novel link in mesenchymal/epithelial cross talk in the gut (5). Foxl2 has been identified as an ovary-specific upstream regulator of the cytochrome P450 aromatase promoter responsible for activating cyp19a expression, together with additional partners such as nr5a1 (sf1) and cAMP (6). Cyp19a is the terminal enzyme in the steroidogenic pathway and is responsible for converting androgens to estrogens (7). FOXL2 negatively regulates the steroid synthase transcriptional activator CYP17 by interaction with SF1 protein in granulosa cells (8). Furthermore, the typical follicular structure of the ovary resembled that of seminiferous tubules in the testis in conditional Foxl2-knockout mature female mice (9), suggesting that testicular development is actively repressed throughout the female life cycle (10). These results identified Foxl2 as a critical factor for sex differentiation in mammals and other vertebrates and for preventing transdifferentiation of adult ovaries into testes.
Baron et al (11) found that a divergent gene foxl3 (also named foxl2b) sequence, whose Forkhead domain belonged to the class L2 and was shown to be a paralog of foxl2 (also named foxl2a). Two foxl2 paralogs, which were considered as fish-specific duplicates (12), have been described in various teleost species (13) including rainbow trout (Oncorhynchus mykiss) (11), European sea bass (Dicentrarchus labrax) (12), and Japanese medaka (Oryzias latipes) (14). Genome searches have indicated that other, as-yet-unreported, teleost species may carry a foxl3 gene, suggesting that foxl3 has been lost in eutherians (13). Sex-related differential expression of foxl2 and foxl3 has also been found in European sea bass (12). Foxl2 plays a pivotal role in the development and maintenance of ovarian function in teleosts such as the Nile tilapia (Oreochromis niloticus) (15), Japanese medaka (16), and the rice field eel (Monopterus albus) (17). However, studies of Foxl3 are currently lacking, and more information is required to improve our understanding of the role of the foxl3 gene in vertebrates, particularly in relation to sexual differentiation (13).
MicroRNAs (miRNAs) are endogenous small RNAs that can regulate target mRNAs by binding to sequences in the 3′ untranslated region (UTR) (18). miRNAs have been shown to be involved in a variety of functions, including developmental transitions, neuronal patterning, apoptosis, adipogenesis, metabolism, and hematopoiesis, in different organisms (19). The expression of miRNAs and their biogenetic pathways are associated with sexual differentiation and regulation of primordial germ cell and gonadal somatic cell development (20, 21). The identification of specific miRNAs and their target genes has highlighted their importance in the development and function of testes and ovaries (21). We previously suggested that foxl2 may be a potential target of the miR-430 family in the rice field eel (22). Moreover, Foxl2 was expressed abundantly in granulosa cells, and that its elevated expression level was observed in the ovary of rice field eels (17). A study of Foxl3 function reported that spermatogenesis is suppressed in Foxl3-knockout female Japanese medaka (14), suggesting a possible role in testis physiology. However, studies on Foxl3 function in teleosts are limited, and its posttranscriptional regulatory networks remain unknown until now.
The rice field eel is a teleost from the family Synbranchidae, order Synbranchiformes. The individuals are always females when younger than 2 years. After spawning, the females will undergo a natural female-to-male sex change via an intersex period of about 2 years during their life cycle (23, 24). In this study, we investigated the involvement of foxl3 in testis differentiation and development in rice field eels by isolating the full-length foxl3 cDNA and analyzing its tissue-specific expression pattern, the cellular localization of Foxl3 protein at different sexual stages. In addition, we also predicted the potential miRNAs regulating foxl3 in rice field eels.
Materials and Methods
Fish and gonad histology
Approximately 40 rice field eels were purchased from Baishazhou Agriculture and Sideline Products Market. The standard length and weight of each individual, the proportion of the three categories (female, intersex, and male) are shown in Supplemental Table 1 and Supplemental Figure 1. The rice field eels were raised temporarily in a large water tank (4 × 2 × 1.5 m) at 24°C ± 0.5°C with a 14-hour light, 10-hour dark photoperiod for 1 week. The experimental procedures were approved by the Institutional Animal Care and Use Committee of Huazhong Agricultural University (Wuhan, China).
Gonads were collected from each rice field eel. One part was immediately frozen in liquid nitrogen and stored at −80°C before RNA extraction, and the other part was stored in Bouin’s fixative for histological analysis to determine the sexual stage, as indicated previously (24).
RNA extraction and reverse transcription
Total RNA was extracted using a total RNA extraction kit (Omega Bio-Tek) according to the manufacturer’s instructions. Genomic DNA contamination was removed using RNase-free deoxyribonuclease I (Thermo Fisher Scientific). The quality of the extracted RNA was examined using a NanoDrop 2000c UV-Vis spectrophotometer (Thermo Fisher Scientific) by calculating the ratio of absorbance at 260 and 280 nm. The integrity of the RNA was visualized by electrophoresis on a 1% agarose gel.
Total RNA (1.5 μg) was reverse transcribed using HiScript Q reverse transcription SuperMix for quantitative real-time RT-PCR (qPCR) (+gDNA wiper) according to the manufacturer’s instructions (Vazyme Biotech). These cDNAs were used for gene cloning and the semi- or quantitative real-time RT-PCR.
Cloning of full-length foxl3 cDNA
Partial foxl3 cDNA fragments were amplified using specific primers designed based on conserved regions of homologous sequences between the rice field eel and European sea bass (accession number JQ772483) using Primer3Plus. All primer sequences used in this study are listed in Supplemental Table 2.
A PCR was carried out with 2 μL cDNA temple in a reaction volume of 50 μL containing 1 μL 10 μM each primer, 25 μL master mix (Vazyme Biotech), and 21 μL double-distilled H2O (ddH2O). The PCR products had electrophoresis performed on a 1% agarose gel and were purified using an agarose gel purification kit (Omega Bio-Tek). The purified product was then ligated with the pMD18-T vector (Takara), and the ligation mixtures were used to transform competent Escherichia coli DH5α. Selection for transformed colonies was achieved using LB ampicillin (100 mg/mL)/X-Gal/IPTG plates. White colonies were purified and sequenced by Tianyi Huiyuan Bioscience and Technology Company.
5′-rapid amplification of cDNA ends (RACE) and 3′-RACE PCRs were performed using a SMARTer RACE cDNA amplification kit (CLONTECH) following the manufacturer’s instructions. Gene-specific primers and nested primers for 5′-RACE and 3′-RACE fragments were designed from partial foxl3 cDNA fragments. Touchdown and nested PCRs were used to amplify the target fragments, and the PCR products were sequenced as described above. The full-length cDNAs were assembled by alignment using the SeqMan program of Lasergene software package (DNASTAR).
Sequencing and phylogenetic analysis
The Foxl3 amino acid sequences were deduced using SeqEdit, Lasergene software and aligned with sequences for other species using the Clustal W method by MegAlign, Lasergene software. The protein sequence identities (percentages) with other species were calculated. A phylogenetic tree was constructed using the maximum likelihood method based on the JTT matrix-based model in MEGA version 6.0 (25), using 1000 bootstrap replicates.
Tissue distribution of foxl3 expression by semiquantitative RT-PCR
Total RNA was extracted from blood, heart, spleen, kidney, brain, muscle, liver, ovary, ovotestis, and testis from the rice field eels, and cDNAs were synthesized as described above.
PCR amplification was performed with gene-specific primers designed based on the cloned nucleotide sequence. Semiquantitative RT-PCRs were performed in 20 μL with 10 μL AceQ qPCR SYBR Green master mix (Vazyme Biotech), 1.5 μL 10-fold diluted cDNA, 0.5 μL each primer (10 μM), and 7.5 μL ddH2O. Semiquantitative RT-PCRs were carried out in a MasterCycler (Eppendorf) to analyze the tissue distribution of foxl3 mRNA.
Amplification conditions were as follows: 7 minutes at 95°C, followed by 30 cycles of 10 seconds at 95°C, 30 seconds at 60°C, and 30 seconds at 72°C, and a final extension cycle of 5 minutes at 72°C. rpl17 (accession number KC011267) as an internal control (26) was amplified with 25 PCR cycles to calibrate the expression levels of foxl3. All PCR products were subjected to electrophoresis in 1.5% (wt/vol) agarose gels and the bands were visualized by staining with GelRed (Vazyme Biotech).
foxl3 expression in gonads by quantitative real-time RT-PCR
qPCRs were performed to determine the foxl3 mRNA expression levels in ovary, ovotestis, and testis. Reactions were carried out in 20 μL in triplicate with 10 μL AceQ qPCR SYBR Green master mix, 1.5 μL 10-fold diluted cDNA, 0.5 μL each primer (10 μM), and 7.5 μL ddH2O using a Rotor-Gene 2000 (QIAGEN).
Amplification conditions were as follows: 7 minutes at 95°C, followed by 40 cycles of 10 seconds at 95°C, 30 seconds at 60°C, and 30 seconds at 72°C. Reactions for the reference gene rpl17 (accession number KC011267) were included on each plate (26). Melting-curve analysis was performed for each primer pair to confirm that the specific products were amplified. Cycle threshold (Ct) values used in the calculations were the means obtained from each cDNA in triplicate, and relative foxl3 expression levels were calculated using the comparative threshold 2-ΔΔCt method (27).
Transcriptome source and differential expression analysis
The transcriptome data of rice field eels (accession number SRR1993742, SRR1993743, and SRR1993744 for the ovary, ovotestis, and testis, respectively; SRR1993745, SRR1993746, and SRR1993747 for the brain of female, intersex, and male, respectively) was used in this study from Sequence Read Archive (National Center for Biotechnology Information (NCBI; Bethesda, MD).
Raw data were first processed to generate clean reads before assembly. In this step, reads with adapters, reads containing greater than 10% ploy-N, and low-quality reads were discarded. All the downstream analyses were based on the clean reads. The high-quality reads were assembled into unigenes using Trinity with min_kmer_cov as 2 and other parameters as default settings (28). Unigenes were first aligned by Blastx (E-value < 10−5) to protein databases of Nr, Nt, Swiss-Prot, Pfam, Kyoto Encyclopedia of Genes and Genomes, and Clusters of Orthologous Groups, retrieving proteins with the highest sequence similarity with the given unigenes along with their protein functional annotations. The best aligning results were defined according to the feedback E-value, and the first hits were used for further analysis.
Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted by the edgeR program package through one scaling normalized factor (29). Different sequencing samples data were normalized in RPKM/FPKM (reads or paired end fragments per kilobase of exon model per million mapped reads).
Foxl3 polyclonal antibody and immunohistochemistry
A rabbit anti-Foxl3 polyclonal antibody was developed against the 80-mer peptide located at the N terminus of the rice field eel Foxl3 protein, using the Custom Antibody Development Service of ABclonal Biotech Co, Ltd. The antibody was purified by affinity chromatography. The cellular location of the Foxl3 protein in the rice field eel gonads was then determined by immunostaining using the anti-Foxl3 polyclonal antibody.
Sections were prepared as described above. After deparaffinization and dehydration, the sections were washed with citric acid buffer (0.1 M citric acid and sodium citrate [pH 6.0]), incubated in 3% (vol/vol) H2O2 and 10% (vol/vol) normal goat serum to block nonspecific binding, and then incubated overnight at 4°C with the anti-Foxl3 polyclonal antibody as primary antibody (diluted 1:800). After incubation with the horseradish peroxidase-labeled secondary antibody (antimouse/rabbit, Maixin KIT-9901), the sections were exposed to 3,3′-diaminobenzidine and stained with hematoxylin to visualize the nuclei in the gonadal tissues and then observed under a fluorescence microscope (Eclipse H600L; Nikon). Negative control sections were treated in the same way but using Tris-buffered saline instead of the primary antibody.
microRNomics source and differential expression analysis
The microRNomics data of rice field eels (accession numbers SRR3630968, SRR3598542, and SRR3630970 for the female, intersex, and male, respectively) that we submitted to NCBI Sequence Read Archive were used in this study. The differential expression of miRNA during developmental stages in the rice field eel was analyzed according to our previous study (22).
Briefly, the low-quality reads, adaptor sequences, and the sequences that were greater than 30 nt and less than 18 nt were discarded before analysis. All of the remaining high-quality sequences were analyzed by basic local alignment search tool (BLAST) against the Rfam database and the GenBank noncoding RNA database to discard rRNA, tRNA, small cytoplasmic RNA, small nuclear RNA, and small nucleolar RNA. After normalization, the level of the expression of the conserved miRNAs among the three sexual stages of the rice field eel was performed using DEGseq (30). Different sequencing samples data were normalized in TPM (transcripts per million).
miRNA-mRNA target prediction and plasmid construction
All mature miRNA sequences were obtained by sequencing small RNAs from rice field eels. The foxl3 3′UTR sequence was used for miRNA-mRNA target prediction using PITA (31), RNA22v2 (32), RNAhybrid (33), and miRanda3.3 softwares (34).
The foxl2 or foxl3 3′UTR fragment of the rice field eel was amplified by PCR using primers, and the foxl2 or foxl3 3′UTR was ligated with pMD18-T as a plasmid template, respectively. pSiCHECK-2 vector (Promega) was digested by XhoI (Thermo Fisher Scientific), and the PC-amplified linear fragments were recombined with the linear pSiCHECK-2 vector by the ClonExpress II kit (Vazyme Biotech) to get Psi-foxl3-wild-type (Psi-foxl3-WT) or Psi-foxl2-wild-type (Psi-foxl2-WT) plasmid, respectively. Briefly, the recombination reaction was performed in 10 μL with 40 ng purified PCR amplification, 50 ng linear pSiCHECK-2 vector, 2 μL 5× CE II buffer and 1 μL Exnase II. The mixture was incubated at 37°C for 30 minutes followed by 5 minutes on ice, and then transformed into E coli DH5α competent cells for recombination.
The mutant-type (Mt) plasmids were constructed from the foxl3 or foxl2 3′UTR wild-type (WT) plasmids by generating mismatches within the complementary sequence of the miRNA seed region (Figure 1). All mutation plasmids were amplified from the foxl3 or foxl2 pMD18-T plasmid by a PCR-based protocol, according to the Fast Mutagenesis kit (Vazyme Biotech). The mutant PCR fragments were recombined with pSiCHECK-2 vector to get the Psi-foxl3-miR-430a mutant-type (Psi-foxl3-Mta), Psi-foxl2-miR-430b mutant-type (Psi-foxl2-Mtb), or Psi-foxl3-miR-9 mutant-type (Psi-foxl3-Mt9) plasmids by the ClonExpress II kit (Vazyme Biotech), respectively.
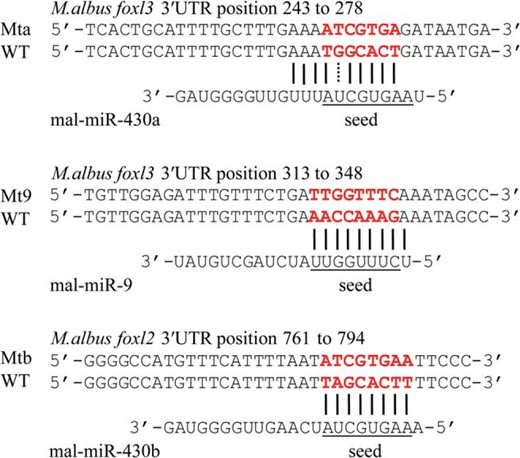
Prediction of miR-9 and miR-430a with potential target gene foxl3 of M albus. Underlining indicates seed region of miR-9, miR-430a, or miR-430b of M albus. Red font indicates the complementary sequence of the seed region between WT and Mt sequence. Prediction of miR-430b with potential target gene foxl2 was cited from our previous study (22).
All constructs were sequenced to verify the sequences.
Dual-luciferase assay
miRNA mimics and miRNA-Lacz (miRNA mimic negative control) were synthesized by Shanghai GenePharma Co, Ltd. miRNA-Lacz suitable for dual-luciferase assay were chosen by predicting the binding sites between miRNA-Lacz and Renilla or firefly luciferase gene in pSiCHECK-2 vector and between miRNA-Lacz and each plasmid using the software mentioned above to ensure there were no binding sites between them. The sequences are shown in Supplemental Table 2.
Human embryonic kidney (HEK)-293T cells were cultured in DMEM (HyClone) with 10% fetal bovine serum under a humid environment with 5% CO2 at 37°C overnight. Cells were plated in a 48-well plate at a density of 2 × 105 for 4–6 hours before transfection. The transfection complex comprised 500 ng reporter constructs, 1.25 μL miRNA mimic or miRNA-Lacz (as a negative control) (20 μM), 1.5 μL Lipofectamine 2000 (Invitrogen), and 50 μL OPTI-MEM (Life Technologies) in each 1.5-mL tube. After standing at room temperature for 5 minutes, 10 μL of the transfection complex was mixed with HEK293T cells in the 48-well plate and incubated with 5% CO2 at 37°C. Each transfection reaction was repeated in triplicate.
Luciferase assays were performed 24 hours after transfection using a dual-luciferase reporter assay kit (Promega). Renilla luciferase activity was normalized by firefly luciferase activity after measurement using a fluorescence detector (Lumat LB 9507; Berthold Technologies).
Statistical analysis
All data were presented as mean ± SD, and the significance of the differences compared with controls were analyzed using SPSS software (version 21.0 for Mac; SPSS Inc). Significant differences were examined by a one-way ANOVA followed by a Tukey’s honestly significant difference test. Differences were considered statistically significant at P < .05.
Results
Gonad histology
Gonad histology (Figure 2) revealed that gonads of female-stage eels contained oocytes at all known developmental stages (ie, from primary growth stage oocytes to mature eggs). Some atretic or degraded oocytes were detected in intersex gonads, together with signs of testicular tissue differentiation, whereas spermatocytes, spermatogonia, and even some mature sperm were present in the late intersex stage. In male-stage eels, the ovarian tissue had been almost completely absorbed and changed into testicular tissue. The testes produced mature sperm with normal male function.
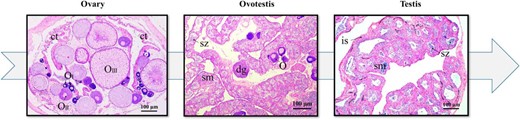
Typical gonad characteristics at different stages of M albus gonadal development. Gonad histology revealed M albus sex change from female (oocytes at all known developmental stages) to male (mature sperm in the gonad) via intersex by hematoxylin and eosin staining of the ovary, ovotestis, and testis. Scale (in each graph), 100 μm. ct, connective tissue; dg, degenerating oocyte; is, interstitial tissue; O, oocytes; OI, oocytes at stage I; OII, oocyte at stage II; OIII, oocyte at stage III; sm, seminiferous; sz, spermatozoa.
Rice field eel foxl3 cDNA sequence and phylogeny
The nucleotide and deduced amino acid sequences of the isolated foxl3 cDNA are shown in Supplemental Figure 2. The cDNA was 1656 bp long and contained a 795-bp open reading frame encoding a protein of 264 amino acids, with a 259-bp 5′UTR and 602-bp 3′UTR, containing a typical polyadenylation signal (AATAAA). The nucleotide and amino acid sequences showed 84% and 81% identity with the European sea bass (accession numbers JQ772483 and AFV13295) by BLASTN and BLASTP (NCBI web servers), respectively.
The predicted amino acid sequence of Foxl3 was aligned with those of Foxl2 and Foxl3 predicted proteins from other species (Figure 3). Multiple alignments showed a highly conserved domain in Foxl3 and Foxl2 corresponding to the characteristic transcription factor Forkhead domain. The Forkhead domain of Foxl2 was conserved, but that in Foxl3 was more divergent. The most notable feature of the rest of the amino acid sequences was the deletion of several fragments in Foxl3 compared with Foxl2 (Figure 3).

Amino acid sequence comparison of Foxl2 and Foxl3 among M albus, D labrax, D rerio, and O latipes. The solid line indicates the Forkhead domain; the black box indicates the specific amino acid residues of Foxl2. NCBI GeneBank accession numbers are as follows: Foxl3 family, D labrax, AFV13295; D rerio, AAI62838; O latipes, XP 004070713; and Foxl2 family, M albus, AGM34484; D labrax, AGS36082; D rerio, NP 001038717; O latipes, NP 001098358
The N-terminal region of Foxl3 was more highly conserved than the C-terminal region, especially in the Forkhead domain (Supplemental Figure 3). Rice field eels Foxl3 exhibited high amino acid sequence identity with other teleosts, including rainbow trout (O mykiss, 64.2%), Japanese medaka (O latipes, 68.8%), zebrafish (Danio rerio, 53.0%), and European sea bass (D labrax, 82.9%).
We constructed a phylogenetic tree for 11 species based on the Foxl1, Foxl2, and Foxl3 amino acid sequence (Figure 4). Foxl1, Foxl2, and Foxl3 were segregated into three different branches. The respective bootstrap values were 100, 99, and 99. Rice field eels Foxl3 clustered with European sea bass and Japanese medaka (bootstrap 98). These results suggest that two foxl2 paralogs, foxl2 and foxl3, are present in the rice field eel genome.
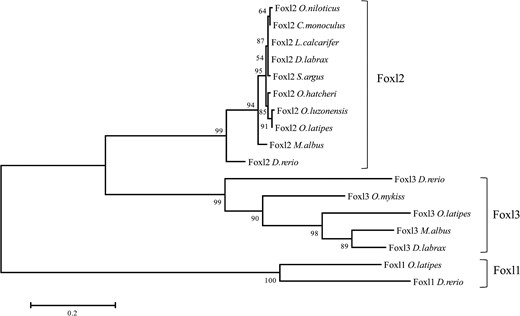
Phylogenetic tree based on proteins of the forkhead family transcription factors from selected teleosts using maximum likelihood method. NCBI GeneBank accession numbers include the following: Foxl2 family, O niloticus, NP 001266707; Cichla monoculus, AFU74229; Lates calcarifer, AKI32579; D labrax, AGS36082; Scatophagus argus, AHZ08826; Odontesthes hatcheri, ACL80211; Oryzias luzonensis, BAH05020; O latipes, NP 001098358; M albus, AGM34484; D rerio, NP 001038717; Foxl3 family, D rerio, AAI62838; O mykiss, NP 001117956; O latipes, XP 004070713; D labrax, AFV13295; and Foxl1 family, O latipes, NP 001116391; D rerio, NP 957278. Note: the numbers at each branch indicate the bootstrap values obtained with 1000 replicates.
Tissue distribution and gonad expression of foxl3 in the rice field eel
foxl3 mRNA was expressed in various tissues, as demonstrated by semiquantitative RT-PCR. Expression levels in the testis were higher than in the ovary, but there was no obvious difference between the testis and ovotestis (Figure 5A). Elevated levels of foxl3 mRNA expression were also observed in the spleen, kidney and brain, whereas lower levels were present in the blood and heart, and foxl3 was absent in muscle and liver tissues.
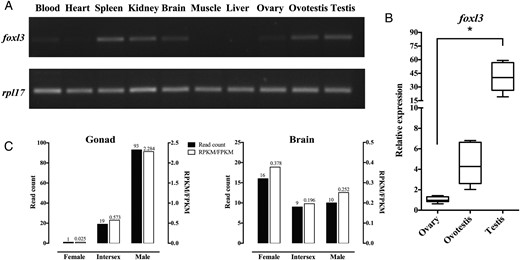
Differential expression of foxl3 mRNA in different tissues of M albus. A, Expression of foxl3 and rpl17 mRNA in different tissues of M albus by RT-qPCR. B, Relative expression of foxl3 mRNA during the different stages of gonadal development in M albus. Ct values were normalized using rpl17 as the endogenous control. Data were represented mean ± SD (n = 6). *, P < .05 compared with expression levels in the ovary. C, Reads count and RPKM/FPKM value of gonad and brain from M albus transcriptome. Note: RPKM/FPKM indicated reads or paired-end fragments per kilobase of exon model per million mapped reads.
We analyzed foxl3 expression levels during the three stages (ie, ovary, ovotestis, and testis) of the rice field eel gonad by qPCR (Figure 5B). Expression levels were increased during the natural sex change process, with higher expression levels (>40-fold) in the testis compared with the ovary (P < .05).
We extracted differential expression data from the rice field eel transcriptome (Figure 5C). The foxl3 normalized data (RPKM/FPKM value) were 0.025, 0.573, and 2.284 (1, 19, and 93 read count) in the ovary, ovotestis, and testis, respectively. The trend in foxl3 expression during sex change based on transcriptome analysis was consistent with the results from other experimental platforms (semiquantitative RT-PCR and qPCR). Taken together, these data indicated that foxl3 expression levels demonstrated significant sexual dimorphism in the rice field eel gonad.
Location of Foxl3 protein in the gonad
We determined the cellular localization of Foxl3 protein by immunohistochemical analysis of different-stage gonad tissue sections from rice field eels. In the ovary, Foxl3 was localized primarily in the granulosa cells of the follicular layers and previtellogenic follicle but not in mature oocytes (Figure 6, A and B). The protein was abundant in the ovotestis, especially in degenerating oocytes and seminiferous tubules (Figure 6, D and E). In the testis, Foxl3-positive signals were detected in the seminiferous lobules containing cysts with germ cells, especially in spermatogonia and Sertoli cells but not in spermatocytes, spermatozoa, and interstitial tissue (Figure 6G, H). No positive signals were observed in the negative controls (Figure 6, C, F, and I).
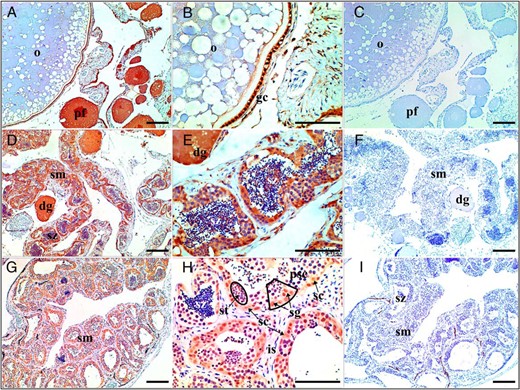
Immunohistochemical localization of Foxl3 at different stages of M albus gonadal development. Location of Foxl3 protein in the ovary (A and B), ovotestis (D and E), and testis (G and H) are shown. Negative control with no added anti-Foxl3 antibodies (C, F, and I) are also shown. Scale in each graph, 100 μm. Black box shows the process of spermatogenesis from spermatogonia, spermatocyte first order, and spermatocyte second order to spermatid. dg, degenerating oocyte; gc, granulosa cell; is, interstitial tissue; o, oocyte; pf, previtellogenic follicle; psc, spermatocytes; sg, spermatogonia; sc, Sertoli cells; sm, seminiferous; st, spermatid; sz, spermatozoa.
miRNA-mRNA target prediction
The potential miRNAs regulating foxl3 in rice field eels were predicted by PITA, RNAhybrid, RNA22v2, and miRanda3.3 softwares. The predicted binding sites of miRNAs on the foxl3 3′UTR are shown in Figure 7A and Supplemental Table 3. All miRNAs were distributed in two main parts (127–357 bp and 478–600 bp) in the foxl3 3′UTR. mal-miR-212 and mal-miR-153b were predicted to target foxl3 by all four softwares used in this study, whereas mal-miR-21, mal-miR-9, and seven other miRNAs were recognized as miRNAs potentially binding the foxl3 3′UTR by three softwares.
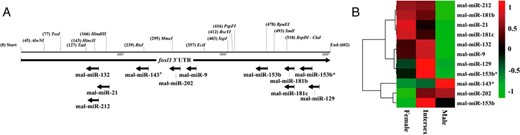
The predicted binding sites and differential expression of potential miRNAs on the foxl3 3′UTR of M albus. A, The horizontal lines indicate a 602-bp foxl3 3′UTR sequence of M albus, with the restriction enzyme sites. Each arrow indicates that the binding sites of potential miRNAs is predicted by at least three softwares. B, Hierarchical cluster analysis of predicted miRNA expression. Expression levels of each miRNA were normalized in transcripts per million. The differential expression of miR-21 and miR-9 was cited from our previous study (22).
We also extracted differential expression data for these miRNAs from the rice field eel microRNomics. Among the 11 miRNAs, mal-miR-212, mal-miR-181b, mal-miR-21, mal-miR-181c, mal-miR-132, mal-miR-9, mal-miR-129, and mal-miR-153b* shared similar expression patterns, with normalized levels being higher in females and intersexes compared with males. In contrast, normalized values for mal-miR-143*, mal-miR-202, and mal-miR-153b were lower in females compared with intersexes and males (Figure 7B).
Dual-luciferase activity analysis
Luciferase activity was significantly decreased as a result of mal-miR-9 binding to foxl3 3′UTR wild-type plasmid, compared with the mutant and control group (P < .05). However, luciferase activity was unaffected by cotransfection of HEK293T cells with mal-miR-430a and foxl3 3′UTR wild-type plasmid (Figure 8A). Binding of mal-miR-430b to the foxl2 3′UTR wild-type plasmid resulted in an obvious decrease in luciferase activity (Figure 8B) (P < .05). None of the mutant plasmids and negative control had any effect on luciferase activity.
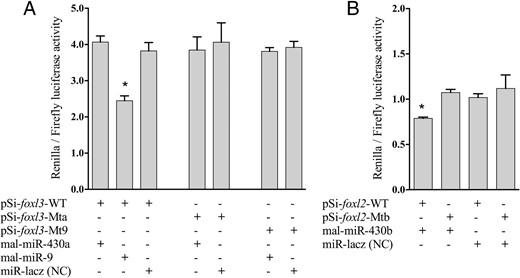
Dual-luciferase assays for validating the interaction of miR-9 with potential target foxl3 in M albus. Psi-foxl3-WT, pSiCHECK-2 foxl3 WT plasmid; Psi-foxl2-WT, pSiCHECK-2 foxl2 WT plasmid; Psi-foxl3-Mta, pSiCHECK-2 foxl3 miR-430a Mt-type plasmid; Psi-foxl2-Mtb, pSiCHECK-2 foxl2 miR-430b Mt-type plasmid; Psi-foxl3-Mt9, pSiCHECK-2 foxl3 miR-9 Mt-type plasmid; miR-Lacz (NC), the miRNA mimic negative control. A, Dual-luciferase assay for validating the interaction between the miR-9 and foxl3 3′UTR. B, Dual-luciferase assay for validating the interaction between the miR-430b and foxl2 3′UTR. Luciferase activity of Renilla was normalized by firefly luciferase activity. Data were presented the mean ± SD (n = 3). *, P < .05 compared with mutant and negative control (NC) groups.
Discussion
foxl2 and foxl3 were identified by BLAST analysis of the rice field eel transcriptome, in agreement with evidence for the duplication event in fish (35). However, Foxl3 was subsequently lost in eutherians as a result of evolution (13), leading to the presence of a single gene (foxl2) in land vertebrates, instead of the duplicated forms of foxl2 in teleosts. During the evolution of ray-finned (actinopterygian) fish, a whole-genome duplication event occurred: the fish-specific genome duplication, probably before the origin of teleosts (36, 37), resulting in an extra set of paralogs for all fish genes. These extra paralogs could serve as a backup for the indispensable ones, a situation known as redundancy (38). Based on the above information, there is an alternative explanation that the common ancestor of land vertebrates has never had a Foxl3 because it has appeared in the teleosts as a result of the fish-specific genome duplication. Additionally, there are different function for extra gene copies with individual regulatory elements existing (38). Therefore, it has a lot of fun among the way of gene regulation and variant gene expression of fish compared with that of mammals.
In this study, we confirmed the existence of foxl3, a foxl2 paralog, in rice field eels. foxl2 and foxl3 were shown to be expressed specifically in the ovary of rainbow trout but displayed different temporal expression patterns (11). Furthermore, gonadal expression patterns of foxl2 and foxl3 showed significant sexual dimorphism in the European sea bass (12). The expression pattern of foxl3 during the natural sex change process in rice field eels was opposite to that of foxl2, according to our previous study (22). Foxl2 plays a pivotal role in the development and maintenance of ovarian function in the rice field eel (17), whereas foxl3 may be involved in testis physiology (12). However, a study of Foxl3 in XX and XY Japanese medaka gonads suggested that Foxl3 was dispensable for testis development but suppressed spermatogenesis in females, given that ovaries produced functional sperm in XX Foxl3-knockout gonads (14), similar to the situation for FOXL2 in mammalian ovaries. FOXL2 is critical for preventing transdifferentiation of adult ovaries to testes by actively repressing the Sertoli cell-promoting gene Sox9 (9). Lack of FOXL2 function may also be the underlying cause of female-to-male sex reversal in goats (39). The above-mentioned data indicate that Foxl2 and Foxl3 are likely to have complementary roles during gonadal differentiation (40).
foxl3 was expressed during oocyte entry into meiosis in rainbow trout, and foxl2 and foxl3 expressions were regulated by androgens or estrogen, indicating a direct link between estrogens and foxl2 expression in fish (11, 41). When entering adulthood, decreased estradiol levels suppressed foxl2 and cyp19a1 expression via a short positive feedback loop (42). FOXL3 maintained female reproductive potential by keeping primordial follicles in oocytes to preserve the resting-follicle pool (39). According to our previous study of Foxl2 (17), Foxl3 and Foxl2 in the ovary were primarily expressed in granulosa cells and previtellogenic follicles in rice field eels. Foxl2 and Foxl3 have been suggested to have similar functions in the ovary, including granulosa cell differentiation and oocyte maintenance, whereas Foxl3 also contributes to oocyte degeneration in the ovotestis. However, there is a difference between the cellular localizations of Foxl3 and Foxl2 in the testis. Spermatogonia originate from primordial germ cells (PGCs), which migrate to the genital lamella in which they differentiate into spermatogonia (43). Sertoli cells express androgen receptors, which increase the T concentration in the seminiferous tubules to stimulate spermatogenesis (44, 45). Unlike mammals and other fish, the male germ cells in rice field eels may originate from PGCs in the peripheral layer of the ovary (46). The role of Foxl3 in the rice field eels might thus be to maintain PGCs in the peripheral layer and support their migration to the genital lamella in the ovotestis and their differentiation into spermatogonia. However, Foxl3 is dispensable for the process, whereby spermatocytes develop into spermatozoa via spermatids in rice field eels.
Eight miRNAs were identified as potentially binding foxl3, thus opposing foxl3 expression during gonadal development in rice field eels. The complex relationship between miRNAs and mRNAs means that a single miRNA may target several hundred genes, and a single gene may also be targeted by multiple miRNAs (21). It is therefore possible that all the identified miRNAs target foxl3 in the rice field eel.
Notably, miR-21 and miR-9 were among the top 10 highly expressed miRNAs during the different gonadal development stages of rice field eels in our previous study (22). miR-21 was also the principal read count in both the ovary and testis in Tibetan pigs (47). The expression of miR-9 was more abundant in ovaries than that in testes of Nile tilapia (48). These predominant miRNAs may be responsible for regulating the stages of sexual development in the rice field eel. The Let-7 family represents a highly expressed miRNA family with biological functions including regulation of cell proliferation and differentiation (49). Myc proto-oncogene protein (MYC) is a target of Let-7 (50) and miR-21 (51), and MYC protein plays an important role in regulating cell growth (up-regulating rRNA and proteins), apoptosis (down-regulating BCL-2), and differentiation (52, 53). Estrogen receptor-α also binds regulatory regions upstream of miR-21 (54), which might be involved in the negative regulatory loop controlling estradiol-induced levels of MYC proteins (55). In this study, we predicted that miR-21 may be a highly expressed miRNA involved in the regulation of foxl3 in the rice field eel. Previous evidence indicated a physiological association between seasonal anovulation in mares and increased follicular miRNA levels, with miR-21, miR-132, and miR-212 regulating ovarian follicle development in vitro (56). In this study, these miRNAs bound to the foxl3 3′UTR, suggesting that foxl3 expression was regulated by sex hormone-regulated miRNAs.
miR-9 is highly expressed in the brain in vertebrates in which it is involved in regulating the differentiation of neuronal cells (57). RE1-silencing transcription factor (REST) represses gene expression related to the nervous system in nonneuronal cells (58, 59), and evidence suggests that REST and CoREST are target genes of miR-9 (58). foxl3 expression was also detected in the brain in the current study, suggesting that foxl3 and miR-9 may be involved in the development of the brain and nervous system in rice field eels. miR-181a and miR-9 suppressed SIRT1 protein expression during embryonic stem cell differentiation in mice (60). miR-9 and miR-11 have also been shown to be expressed during each developmental period in Caenorhabditis elegans, but their levels are reduced in the adult, which may reflect a maternal contribution from germ cells or sex-specific expression (61). A recent study showed that miR-9 inhibits male steroid production through the down-regulation of Hsd11b activity in the Nile tilapia females (48), suggesting miR-9 may be involved in the steroid hormone biosynthesis pathway and regulatory network in sex differentiation of rice field eels.
miR-430 has been shown to facilitate deadenylation and clearance of maternal mRNAs during early embryogenesis (62). The transcription factor Foxl2 has been characterized as an ovary-specific upstream regulator of the cyp19a1a promoter (6). miR-430b targets foxl2 3′UTR, indicating that miR-430b might thus repress the ovary-specific transcription of foxl2 in the testis of rice field eels. However, miR-430a does not target foxl3 3′UTR, and miR-430b and miR-430c have no binding site with foxl3 3′UTR, suggesting that lower foxl3 expression in the ovary is not regulated by the miR-430 family. The transcription factor foxl3 might thus act as a testis related regulator.
In conclusion, we demonstrated miR-9 binding to the foxl3 3′UTR in rice field eels using a dual-luciferase assay. miR-9 expression levels were relatively high in females and intersexes but decreased when the gonads developed into testes during the sex change process. This expression trend was opposite to that for foxl3. These results suggested that foxl3 was posttranscriptionally regulated by miR-9, not the miR-430 family, in the rice field eel. Foxl3 and miR-9 may be involved in physiological processes in ovary granulosa cells, promoting oocyte degeneration in the ovotestis and stimulating spermatogenesis in spermatogonia. Further studies are needed to clarify the mechanisms responsible for Foxl3 regulation, particularly in relation to the natural sex change process in the rice field eel.
Acknowledgments
We thank Jianguo Su’s group for their assistance in fulfilling the present study. We also thank Gailing Yuan for providing the plasmid vector in the present study. We also thank Wei Chi for compiling perl scripts and extracting the data from the transcriptome.
This work was supported by the Twelfth 5-Year National Key Science and Technology Research Program of China (Project 2012BAD25B06); the Special Fund for Agro-Scientific Research in the Public Interest (Project 201003076); and the Fundamental Research Funds for the Central Universities (Project 2662015PY119).
Disclosure Summary: The authors have nothing to disclose.
Appendix
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| Foxl3 | MDTEAKSPADQGVRLLDISSNSPPPEPQGAEETGQLEKPPYSYVALIAMAIKDSQHKRQTLSGIYDYIISKFPYYEKNKK | Foxl3 | ABclonal Biotech Co, Ltd. | Polyclonal antibody | 1:800 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| Foxl3 | MDTEAKSPADQGVRLLDISSNSPPPEPQGAEETGQLEKPPYSYVALIAMAIKDSQHKRQTLSGIYDYIISKFPYYEKNKK | Foxl3 | ABclonal Biotech Co, Ltd. | Polyclonal antibody | 1:800 |
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| Foxl3 | MDTEAKSPADQGVRLLDISSNSPPPEPQGAEETGQLEKPPYSYVALIAMAIKDSQHKRQTLSGIYDYIISKFPYYEKNKK | Foxl3 | ABclonal Biotech Co, Ltd. | Polyclonal antibody | 1:800 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| Foxl3 | MDTEAKSPADQGVRLLDISSNSPPPEPQGAEETGQLEKPPYSYVALIAMAIKDSQHKRQTLSGIYDYIISKFPYYEKNKK | Foxl3 | ABclonal Biotech Co, Ltd. | Polyclonal antibody | 1:800 |
Abbreviations
- Ct
cycle threshold
- ddH2O
double-distilled H2O
- FOX
Forkhead box
- Foxl1
Forkhead box L1
- Foxl2
Forkhead box L2
- HEK
human embryonic kidney
- miRNA
microRNA
- Mt
mutant type
- MYC
Myc proto-oncogene protein
- NCBI
National Center for Biotechnology Information
- PGC
primordial germ cell
- qPCR
quantitative real-time RT-PCR
- RACE
rapid amplification of cDNA ends
- REST
RE1-silencing transcription factor
- UTR
untranslated region
- WT
wild type.
References
Author notes
Author contributions include the following: Y.G. conceived this study, designed and performed the experiments, analyzed the data, and prepared the manuscript; D.J. and Q.H. collected the material for the study and analyzed the data; and D.L. supervised and conceived this study, analyzed the data, and edited the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}