





Sleep disturbances are associated with type 2 diabetes; therefore, the amelioration of sleep may improve metabolic disorders. To investigate this possibility, we here examined the effects of suvorexant, an antiinsomnia drug targeting the orexin system, on sleep and glucose metabolism in type 2 diabetic mice. Diabetic db/db mice had a longer wakefulness time during the resting period, as compared with nondiabetic db/m+ control mice. The single or 7-day administration of suvorexant at lights-on (ie, the beginning of the resting phase) increased nonrapid eye movement sleep time during the resting phase and, as a consequence, reduced awake time. The daily resting-phase administration of suvorexant for 2–4 weeks improved impaired glucose tolerance in db/db mice without affecting body weight gain, food intake, systemic insulin sensitivity, or serum insulin, and glucagon levels. No changes were detected in the markers of lipid metabolism and inflammation, such as the hepatic triglyceride content and Tnf-α mRNA levels in liver and adipose tissues. The improving effect of suvorexant on glucose tolerance was associated with a reduction in the expression levels of hepatic gluconeogenic factors, including phosphoenolpyruvate carboxykinase and peroxisome proliferator-activated receptor-γ coactivator-1α in the liver in the resting phase. In contrast, the daily awake-phase administration of suvorexant had no beneficial effect on glucose metabolism. These results suggest that the suvorexant-induced increase of sleep time at the resting phase improved hepatic glucose metabolism in db/db mice. Our results provide insight into the development of novel pharmacological interventions for type 2 diabetes that target the orexin-operated sleep/wake regulatory system.
The sleep/wake rhythm in a 24-hour light/dark cycle is one of the most obvious daily rhythms in many species of animals, including humans and rodents (1). Based on the sleep/wake rhythm, energy and nutritional balance markedly change at each time of the day. In order to adapt to these changes, the metabolism of glucose and lipids in peripheral tissues is periodically and proactively modulated by the central circadian system via hormonal and autonomic nerve pathways (2). Therefore, the disruption of biological rhythms due to frequent sleep disturbances (eg, shift work, insomnia, and obstructive sleep apnea) impairs the regulation of metabolism, which, in turn, increases the risk of developing type 2 diabetes (3). A previous study demonstrated that the less of even a single night of sleep resulted in insulin resistance in healthy humans (4). A recent meta-analysis revealed that the effects of sleep disturbances are similar to those of traditional risk factors for diabetes, such as being overweight, a family history, and physical inactivity (5). Moreover, rapid eye movement (REM) sleep and deep non-REM sleep (also referred to as slow-wave sleep) are related to appetite and glucose metabolism, respectively (6). The acute suppression of slow-wave sleep has been shown to reduce insulin sensitivity in healthy humans (7, 8). Thus, the quality and quantity of sleep contribute greatly to the maintenance of normal glucose homeostasis.
Leptin receptor-deficient db/db mice have severe obesity due to hyperphagia and reduced energy expenditure, and display type 2 diabetic characteristics, such as hyperglycemia and insulin resistance (9). They also showed impaired sleep consolidation and an attenuated daily sleep/wake rhythm in association with constant elevations in sympathetic tone (10, 11). Furthermore, patients with type 2 diabetes exhibit disruptions in the sleep architecture (eg, less slow-wave sleep) (12). Moreover, excessively short and long sleep durations have been associated with hyperglycemia and insulin resistance, even in type 2 diabetic patients (13, 14). These findings suggest that abnormal sleep and already developed diabetes exacerbate each other (6), and this vicious cycle may be prevented by the treatment of sleep disturbances.
The daily rhythm of sleep and wakefulness is tightly regulated by multiple neuronal systems in the brain. Orexin, a neuropeptide locally produced in the perifornical and lateral hypothalamic areas, plays a major role in the maintenance of wakefulness (15). The orexin system is activated daily during the awake period and is inactivated during the resting period (16). On the other hand, sleep deprivation causes the overactivation of the orexin system to maintain wakefulness against increased homeostatic sleep pressure (17). The overexpression of orexin has been shown to disturb REM and non-REM sleep (18). Because orexin also promotes food intake and sympathetic-driven hepatic glucose production (19, 20), the excessive and/or untimely activation of the orexin system due to sleep disturbances is considered to impair the regulation of glucose homeostasis (17).
The effects of orexin are mediated by orexin receptor-1 (OX1R) and OX2R. The activation of OX2R is essential to stabilize wakefulness, whereas both OX1R and OX2R contribute to the inhibition of REM sleep (15). Suvorexant is a dual orexin receptor antagonist (DORA) that blocks both OX1R and OX2R and is clinically used in the treatment of insomnia (21). Therefore, we hypothesize that suvorexant may improve glucose regulation in the type 2 diabetic state by restoring the daily sleep/wake rhythm. Chronic daily administration may be required for this purpose, because circadian rhythms in peripheral tissues have been reported to be more slowly reset than that in central circadian system when exposed to a new photoperiod (22).
In order to investigate whether the amelioration of sleep disturbances improves glucose metabolism in the diabetic state, we here employed db/db mice as a model of type 2 diabetes with sleep disturbances and examined the influence of the daily administration of suvorexant on sleep and glucose regulation. This is the first study to attempt to elucidate the therapeutic potential of an antiinsomnia drug targeting the orexin system for type 2 diabetes.
Materials and Methods
Animals
Male C57BLKS/J Iar-+Leprdb/+Leprdb mice (db/db) and C57BLKS/J Iar-m+/+Leprdb mice (db/m+) were purchased from the Institute for Animal Reproduction. Male C57BL/6J mice were purchased from Japan SLC. Mice were maintained under specific pathogen-free conditions at 20°C–26°C, and allowed free access to a standard rodent diet and water under a 12-hour light, 12-hour dark cycle. Zeitgeber time (ZT)0 and ZT12 represent light onset and offset times, respectively. All experimental procedures were approved by the Committee of Animal Experiments at the University of Toyama.
Materials
Suvorexant ([(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone) was synthesized according to our previous studies (23). It was suspended in 0.5% methyl cellulose 400 (Wako Pure Chemicals) just before its oral administration. Orexin A was purchased from Peptide Institute. All other reagents were purchased from Sigma-Aldrich Japan or Wako, unless otherwise indicated.
Experimental design
We examined the effects of suvorexant on the states of sleep and wakefulness (experiment 1) and the states of glucose and energy metabolism (experiment 2) in db/db and db/m+ mice. In addition, treatment dose verification was conducted in C57BL/6J mice (experiment 3).
In experiment 1, suvorexant (30 mg/kg) or vehicle (0.5% methyl cellulose 400) was administered by oral gavage (p.o.) to mice (males, 11 wk old) once daily at ZT0 (ie, the beginning of the resting phase) for 2 weeks. Sleep recordings were performed on days 1 and 7, as described below. On day 14, mice fasted for 6 hours (at ZT0–ZT6) were killed at ZT6 by cervical dislocation, and tissue samples were obtained for biochemical analyses.
Experiment 2 consisted of experiment 2-1 to investigate the chronopharmacological profiles of suvorexant and experiment 2-2 to assess dose dependency and the influence on insulin signaling. In experiment 2-1, mice (males, 11 wk old) received suvorexant (30 mg/kg, p.o.) or vehicle (0.5% methyl cellulose 400, p.o.) once daily at ZT0 or ZT12 for 6 weeks. Glucose and insulin tolerance tests were conducted after 3 and 4 weeks of the treatment, respectively. In figure 5 below, the acute effects of suvorexant on glucose tolerance were examined in a crossover test separated by a 7-day washout period. Daily changes in food intake, locomotor activity, O2 consumption, energy expenditure, and the respiratory quotient (RQ) were analyzed using a metabolism measuring system (MK-5000RQ; Muromachi Kikai) after 4–5 weeks of the drug treatment. Tissue and blood samples were obtained at ZT6 under 6-hour fasting conditions after 6 weeks of the treatment and subjected to biochemical analyses. In experiment 2-2, mice (males, 11 wk old) received suvorexant (10 mg/kg, p.o.) or vehicle once daily at ZT0 for 2 weeks and then received an increased dose of suvorexant (50 mg/kg, p.o.) or vehicle once daily at ZT0 for 3 more weeks. Glucose tolerance tests were conducted after 2 weeks of the treatment with the respective doses of the drug (ie, on d 15 and 29). Insulin signaling in the peripheral tissues was analyzed, as described below, using mice treated with suvorexant (0 or 50 mg/kg) for 3 weeks.
In experiment 3 to evaluate the antagonistic effect of suvorexant against orexin A in vivo, 7-week-old C57BL/6J mice implanted with a guide cannula for intracerebroventricular (icv) injection were prepared, as described previously (24). After a 7-day recovery period, suvorexant (30 mg/kg) or vehicle was administered by p.o. under 2-hour fasting conditions. Fifteen minutes after the administration, 1 μL of orexin A (1 nmol/mouse) or saline was icv injected. Increase in blood glucose levels was analyzed for 2 hours as an indirect index of the central action of orexin A (24).
Sleep recordings
The states of sleep and wakefulness in mice were analyzed by a standard method, as follows. Under 3% isoflurane anesthesia, mice were implanted with electrodes (Pinnacle Technology) for electroencephalogram (EEG) and electromyogram (EMG) recordings. Regarding EEG, epidural electrodes were set by implanting 4 stainless steel screws through holes drilled in the skull (∼1 mm anterior to the bregma or lambda and 1.5 mm left- or right-lateral to the midline) with attaching to the dura mater. Regarding EMG, Teflon-coated stainless steel wires were placed into the neck muscles bilaterally. After 3 days of recovery, mice were moved to a recording cage in a sound-attenuated room. The electrodes were then connected to a data collection system (Pinnacle Technology) through a swivel system that allowed the mice to move freely within the cage and take food and water ad libitum. Mice were treated with suvorexant (30 mg/kg, p.o.) or vehicle, and EEG and EMG signals were recorded for 24 hours on experimental days 1 and 7. The data collected were analyzed using Sleepsign software (Kissei Comtec). The vigilance of every 4-second epoch was automatically classified into 3 stages, ie, wakefulness, REM, and non-REM sleep, according to standard criteria. Each automatically defined stage was reexamined visually, and corrected if necessary. The vigilance states of 4-second epochs were assessed as follows: 1) wakefulness was defined by a high EMG amplitude, low EEG amplitude, and high θ wave activity concomitant with the highest EMG values; 2) REM sleep was defined by a low EMG amplitude, low EEG amplitude, and high θ wave activity; and 3) non-REM sleep was defined by a low EMG amplitude, high EEG amplitude, and high δ wave activity.
Glucose and insulin tolerance tests
In the glucose tolerance test, db/db mice were fasted for 16 hours, and glucose (1.0 g/kg) was then ip injected at ZT6. In the insulin tolerance test, db/db mice were fasted for 2 hours, and insulin (4.0 U/kg, Humulin R; Eli Lilly) was then ip injected at ZT6. Blood samples (<1 μL) were collected from the tail vein, and blood glucose levels were measured using a FreeStyle Lite blood glucose meter (Abbott Japan).
Analysis of serum metabolic parameter levels
Blood samples were obtained from the orbital sinus of mice in experiments 1 and 2-1. In order to prepare serum, blood samples were left to stand overnight at 4°C and then centrifuged (220g, 15 min, 4°C). Serum insulin, glucagon, and corticosterone levels were measured using ELISA kits for insulin (Takara Bio; assay range, 0.156–10 ng/mL; intraassay coefficient of variation [CV], <10%), glucagon (Wako; assay range, 41–10 000 pg/mL; intraassay CV, <5.1%), and corticosterone (Enzo Life Sciences; assay range, 32–20 000 pg/mL), respectively, with a microplate reader (FilterMax F5; Molecular Devices Japan), according to the manufacturer’s instructions. Nonesterified fatty acid levels were analyzed by a colorimetric kit (Wako).
Analysis of hepatic parameter levels
Liver tissue was isolated from mice under 6-hour fasting conditions and stored at − 80°C until used. In order to measure the content of hepatic triglycerides, lipids were extracted according to the standard Bligh and Dyer method, and their levels were colorimetrically analyzed using a triglyceride E-test kit (Wako; assay range, 7.2–1000 mg/dL; CV, <2.0%) with a FilterMax F5 microplate reader (Molecular Devices Japan). The levels of cAMP in the liver were analyzed using a cAMP Parameter Assay kit (R&D Systems; assay range, 3.8–240 nmol/L; intraassay CV, <4.0%), according to the manufacturer’s instructions. The content of hepatic glycogen was analyzed using a glucose assay kit (Sigma-Aldrich; assay range, 0.02–1 mg/mL).
Western blot analysis
The expression or phosphorylation levels of the objective proteins were analyzed by Western blotting, as described previously (25). In the analysis of insulin signaling in experiment 2-2, insulin (4.0 U/kg) or PBS was iv injected into mice at ZT6 under 16-hour fasting conditions (6 h after the last suvorexant administration). Fifteen minutes after the insulin injection, the liver, skeletal muscle, and epididymal adipose tissues were isolated, snap frozen in liquid nitrogen, and stored at −80°C until used. The phosphorylation levels of cAMP-response element-binding protein (CREB) and forkhead box protein O1 (FoxO1) were analyzed in the liver obtained in experiment 2-1. An anti-phospho-Akt (Ser473) antibody, anti-CREB antibody, anti-phospho-CREB (Ser133) antibody, anti-phospho-FoxO1 (Ser256) antibody, and anti-FoxO1 antibody were purchased from Cell Signaling Technology. An anti-Akt1 antibody was from Santa Cruz Biotechnology.
Reverse transcription-PCR
The extraction of RNA, reverse transcription, and quantitative real-time PCR were conducted as described previously (24). Hypothalamus samples were collected in experiment 1, and liver and adipose tissues were obtained in experiment 2-1. Relative mRNA expression levels were determined by calculating the ratio to the 18S rRNA level in each sample. Primer pairs are shown in Supplemental Table 1.
Histochemical analysis
Isolated epididymal white adipose tissues and the liver were fixed with 4% paraformaldehyde and subjected to hematoxylin and eosin staining according to a standard protocol. In the analysis of the size of adipocytes, 4 microscopic images (magnification, ×4) were randomly selected for each mouse, and the areas of adipocytes (>900 cells in total) were measured and averaged using ImageJ software.
Statistical analysis
Data are expressed as means ± SEM. The significance of differences between 2 groups was evaluated by the Student’s t test, and differences between multiple groups were evaluated by a one-way ANOVA with Bonferroni’s test. P < .05 was considered significant.
Results
Effects of suvorexant on the sleep-wake state in db/db mice
In order to elucidate the profile of the sleep architecture in diabetic db/db mice, daily changes in the active wake, non-REM sleep, and REM sleep times were compared with those in nondiabetic db/m+ mice after the treatment with vehicle at ZT0. On day 1, db/m+ mice clearly showed daily changes in all these parameters according to the light/dark cycle (Figure 1, A–C). The extent of changes in the sleep/wake architecture during the light-to-dark transition (from ZT6–ZT12 to ZT12–ZT18) was less in db/db mice than in db/m+ mice (Supplemental Figure 1, A–C), indicating that the daily sleep/wake rhythm was flattened in the diabetic state. Moreover, the total awake time was significantly longer in db/db mice (4210 ± 524 s, n = 6; P < .05) than in db/m+ mice (2436 ± 478 s, n = 5), whereas the total REM sleep time was significantly shorter in db/db mice (105 ± 60 s, n = 6; P < .05) than in db/m+ mice (526 ± 136 s, n = 5) during ZT4–ZT6. Similar abnormalities were observed on day 7. The total awake time during the 12-hour light period was significantly longer (Figure 2, A and B), and the extent of changes in the awake and non-REM sleep time during the light-to-dark transition was slightly less (Supplemental Figure 1, D–F) in db/db mice than in db/m+ mice.
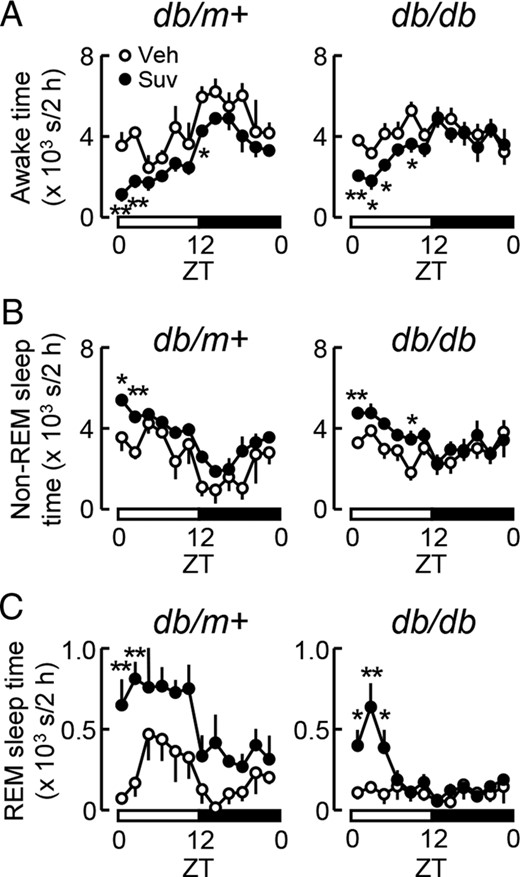
Influence of the single administration of suvorexant on sleep and wakefulness in db/m+ and db/db mice. Suvorexant (closed circles, Suv, 30 mg/kg) or vehicle (open circles, Veh) was administered by p.o. at ZT0 to male db/m+ and db/db mice at 11 weeks old. Mice were then subjected to sleep recordings. A–C, Daily changes in the awake (A), non-REM sleep (B), and REM sleep times (C) in db/db and db/m+ mice treated with suvorexant or vehicle. Values represent the times spent in each sleep/wake state every 2 hours in db/m+ (left) and db/db mice (right). Data were expressed as the mean ± SEM (db/m+, n = 3–4; db/db, n = 6). *, P < .05 and **, P < .01.
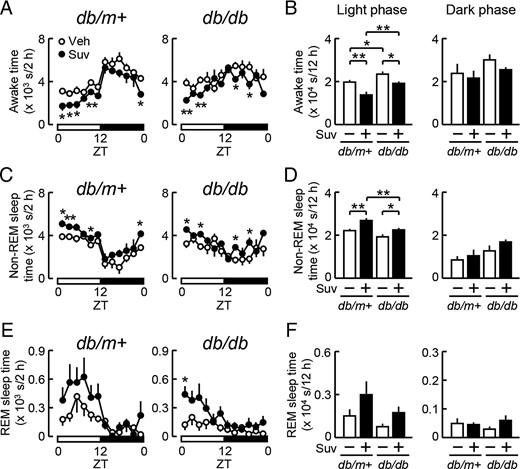
Influence of the daily administration of suvorexant on sleep and wakefulness in db/m+ and db/db mice. Male db/m+ and db/db mice at 11 weeks old were treated with suvorexant (closed symbols, Suv, 30 mg/kg) or vehicle (open symbols, Veh) once daily at ZT0 for 7 days. Mice were then subjected to sleep recordings to analyze awake (A and B), non-REM sleep (C and D), and REM sleep times (E and F). A, C, and E, Daily changes in the awake (A), non-REM sleep (C), and REM sleep times (E) in db/m+ (left) and db/db mice (right). Values represent the times spent in each sleep/wake state every 2 hours. B, D, and F, Total times spent in the awake (B), non-REM sleep (D), and REM sleep states (F) during the 12-hour light (left) and 12-hour dark (right) phase in db/m+ and db/db mice. Data used in A, C, and E were reevaluated in B, D, and F, respectively. Data were expressed as the mean ± SEM (db/m+, n = 3–6; db/db, n = 6). *, P < .05 and **, P < .01.
We then examined the effects of suvorexant on the sleep/wake state. When suvorexant (30 mg/kg) or vehicle was administered at ZT0, acute increases in the non-REM and REM sleep time in association with reductions in the awake time were observed during the light period in both db/m+ and db/db mice on day 1 (Figure 1, A–C). Similarly, after 7 days of the daily administration of suvorexant at ZT0, the non-REM sleep time was increased, and, as a consequence, the awake time was reduced in both mice (Figure 2, A–D). The effects of suvorexant on the total awake and non-REM sleep times reached significant levels exclusively in the 12-hour light phase, although these effects were less potent in db/db mice than in db/m+ mice (Figure 2, B and D). Suvorexant also caused slight increases in REM sleep in the light phase (Figure 2, E and F). These results indicate that the daily administration of suvorexant at ZT0 promoted sleep mainly during the light phase in diabetic and nondiabetic mice.
In order to clarify whether the administration of suvorexant affects the expression of orexin and its receptors, the mRNA levels of preproorexin, Ox1r, and Ox2r were measured in the hypothalamus of db/db mice treated daily with suvorexant (0 or 30 mg/kg, p.o., ZT0) for 14 days. As shown in Supplemental Figure 2, suvorexant had no effect on the levels of these mRNAs at ZT6 under the 6-hour fasting condition.
For validation of the dosage used, we investigated whether suvorexant (30 mg/kg, p.o.) blocks the central effect of orexin A. Orexin A has been shown to acutely increase blood glucose levels by promoting hepatic glucose production via sympathetic nervous system (24). As shown in Supplemental Figure 3, suvorexant inhibited the orexin-A (1 nmol/mouse, icv)-induced glucose elevation without affecting basal glucose levels in normal C57BL/6J mice. Thus, this dose of suvorexant was appropriate to inhibit the central actions of orexin in mice.
Effects of suvorexant on energy balance in db/db mice
The chronic daily administration of suvorexant (30 mg/kg) at ZT0 did not affect body weight gain in db/db mice, at least until 4 weeks (Figure 3A). Daily changes in food intake (Figure 3B) and locomotor activity (Figure 3C) were not affected by this treatment. The levels of O2 consumption (Figure 3D) and energy expenditure (Figure 3E) were significantly reduced in the light phase but not in the dark phase by the suvorexant treatment. These reductions appeared to occur in parallel with the increase in non-REM sleep and the reduction in wakefulness, as shown above (Figure 2). No significant differences were observed in RQ levels between the suvorexant- and vehicle-treated groups (Figure 3F).

Influence of the daily resting-phase administration of suvorexant on the regulation of the energy balance in db/db mice. Suvorexant (closed symbols, Suv, 30 mg/kg) or vehicle (open symbols, Veh) was administered by p.o. at ZT0 for 4–5 weeks to db/db mice (males, 11 wk old). Daily changes in the energy balance were then analyzed using a metabolism measuring system. A, Time course of body weight gain. B, Food intake. Left, Daily changes in the meal size every 2 hours. Right, Total amount during the light and dark phases. C–F, Locomotor activity (C), O2 consumption (D), energy expenditure (E), and RQ (F). Left, Daily changes in respective parameters expressed as average levels during 2-hour measurements. Right, Average levels during the light and dark phases. Data were the mean ± SEM (n = 10–11). *, P < .05 and **, P < .01, determined by the t test. GTT, glucose tolerance test. ITT, insulin tolerance test.
We investigated the impact of the awake-phase administration of suvorexant on the energy balance. Suvorexant (30 mg/kg) was administered daily at the beginning of the dark phase (ZT12) for 4–5 weeks to db/db mice. No changes in body weight gain (Figure 4A), food intake (Figure 4B), locomotor activity (Figure 4C), O2 consumption (Figure 4D), and energy expenditure (Figure 4E) were detected under this condition. However, RQ levels were decreased at ZT0–ZT2, ZT12–ZT14, and ZT16–ZT18 by the suvorexant treatment (Figure 4F). Thus, the influence of suvorexant on the source and utilization of energy in db/db mice appeared to differ between its awake-phase and resting-phase administration.
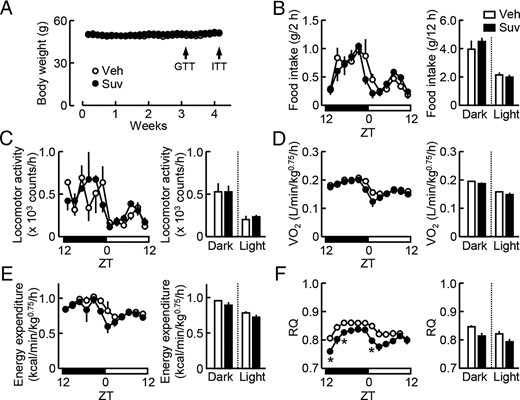
Influence of the daily awake-phase administration of suvorexant on the regulation of the energy balance in db/db mice. Suvorexant (closed symbols, Suv, 30 mg/kg) or vehicle (open symbols, Veh) was administered by p.o. at ZT12 for 4 weeks to db/db mice (males, 11 wk old). Daily changes in the energy balance were analyzed using a metabolism measuring system. A, Time course of body weight gain. B, Food intake. Left, Daily changes in the meal size every 2 hours. Right, Total amount during the light and dark phases. C–F, Locomotor activity (C), O2 consumption (D), energy expenditure (E), and RQ (F). Left, Daily changes in the respective parameters expressed as average levels during 2-hour measurements. Right, Average levels during the light and dark phases. Data were the mean ± SEM (n = 3–4). *, P < .05, determined by the t test. GTT, glucose tolerance test. ITT, insulin tolerance test.
Effects of suvorexant on glucose metabolism in db/db mice
In order to investigate whether suvorexant affects glucose metabolism, db/db mice were acutely or chronically treated with suvorexant (30 mg/kg) at ZT0. On day 1, blood glucose elevations during the glucose tolerance test were not significantly different between the suvorexant- and vehicle-treated groups (Figure 5A). However, after 3 weeks of its daily administration, the peak levels of blood glucose at 60 minutes during the glucose tolerance test were significantly reduced by suvorexant (Figure 5B). Basal serum insulin and glucagon levels under 6-hour fasting conditions were not affected by the 2-week administration of suvorexant (data not shown). Furthermore, serum insulin levels after the loading of glucose (1 g/kg) were not affected by suvorexant (Figure 5C). Suvorexant had no effect on glucose levels during the insulin tolerance test (Figure 5D). We also tested the effects of suvorexant at different doses. The daily administration of suvorexant at 50 mg/kg, but not at 10 mg/kg, for 2 weeks reduced elevations in glucose levels during the glucose tolerance test (Figure 5, E and F). These results indicate that the chronic administration of suvorexant at more than 30 mg/kg improved glucose metabolism in db/db mice.
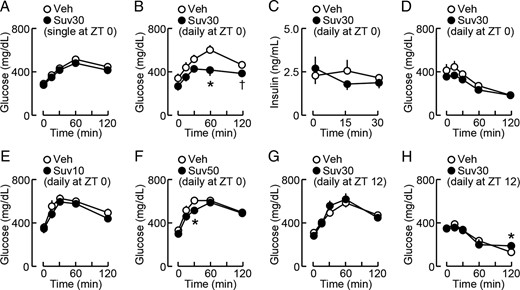
Pharmacological profiles of effects of suvorexant on glucose metabolism in db/db mice. Suvorexant (closed circles; Suv10, 10 mg/kg; Suv30, 30 mg/kg; Suv50, 50 mg/kg) or vehicle (open circles, Veh) was administered by p.o. once or daily at the indicated time of day to db/db mice (males, 11 wk old), and the glucose tolerance test (GTT) and insulin tolerance test (ITT) were then performed at ZT6. A, GTT after the single administration of suvorexant (30 mg/kg, ZT0) on day 1; n = 12. B, GTT after the daily administration of suvorexant (30 mg/kg, ZT0) for 3 weeks; n = 10–11. C, Changes in serum insulin levels during GTT conducted after the daily administration of suvorexant (30 mg/kg, ZT0) for 6 weeks; n = 6. D, ITT after the daily administration of suvorexant (30 mg/kg, ZT0) for 4 weeks; n = 4–5. E and F, GTT after the daily ZT0 administration of suvorexant at 10 mg/kg (E) or 50 mg/kg (F) for 2 weeks; n = 6. G, GTT after the daily administration of suvorexant (30 mg/kg, ZT12) for 3 weeks; n = 4–5. H, ITT after the daily administration of suvorexant (30 mg/kg, ZT12) for 4 weeks; n = 4–5. Data were the mean ± SEM. *, P < .05 and †, P < .1 vs vehicle.
We then examined the effects of suvorexant administered at different times of day. After 3–4 weeks of the daily administration of suvorexant (30 mg/kg) at ZT12, blood glucose levels during the glucose tolerance test (Figure 5G) or insulin tolerance test (Figure 5H) in the light phase were not significantly different between the suvorexant- and vehicle-treated groups in db/db mice. The suvorexant-treated group showed higher glucose levels at 120 minutes in the insulin tolerance test (Figure 5H). Thus, the effects of suvorexant on glucose metabolism appear to depend on the treatment time of day.
In order to elucidate the mechanisms underlying the improvements observed in glucose metabolism, insulin signaling in insulin-targeted tissues was analyzed after 3 weeks of the daily administration of suvorexant (50 mg/kg, ZT0) in db/db mice. In the liver, insulin-induced increases in the phosphorylation of Akt were not significantly different between the suvorexant- and vehicle-treated groups (Figure 6A). Similar results were obtained in skeletal muscle (Figure 6B) and epididymal adipose tissues (Figure 6C). These results indicate that suvorexant improved glucose tolerance independently of the effects of insulin.
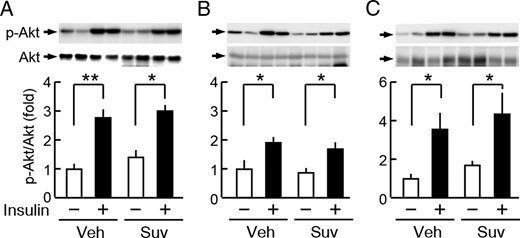
Suvorexant did not affect insulin signaling in the peripheral tissues of db/db mice. db/db mice were treated daily with suvorexant (0 or 50 mg/kg) at ZT0 for 3 weeks. A–C, Insulin-induced phosphorylation of Akt in the liver (A), skeletal muscle (B), and epididymal adipose tissues (C). In this analysis, mice were treated with suvorexant (Suv) or vehicle (Veh), fasted for 16 hours, and then iv injected with insulin (4.0 U/kg, 15 min) or saline. Values represent fold changes in the phosphorylation level (p-Akt/Akt) from the saline-injected controls in mice treated with vehicle. Data were the mean ± SEM (n = 3–5). *, P < .05 and **, P < .01. p, phosphorylated.
Consistent with the above results showing suvorexant-induced reductions in energy expenditure (Figure 3), the more prolonged daily administration (>5 wk) of suvorexant (30 mg/kg) at ZT0 increased the body weight of db/db mice (Figure 7A). The mRNA levels of uncoupling protein 1 (Ucp1) in brown adipose tissue (Figure 7B), but not in epididymal adipose tissues (Figure 7C), were decreased by the administration of suvorexant. In epididymal adipose tissues, the number of smaller sized adipocytes (such as 103.6–3.7 μm2) was decreased, whereas that of larger sized ones (such as 104.1–4.4 μm2) was slightly increased by the suvorexant treatment (Figure 7, D and E). These changes appeared to be due to increase in sleep time and the associated chronic reduction in energy expenditure in the light phase. Nevertheless, the expression levels of proinflammatory markers, including monocyte chemoattractant protein 1 (MCP-1), Tnf-α, and IL-6 mRNAs, were not altered under this condition (Figure 7F). Moreover, no drug effects were observed on liver morphology (Supplemental Figure 4A), hepatic triglyceride content (Supplemental Figure 4B), or the mRNA levels of Tnf-α and IL-6 in the liver of db/db mice (Supplemental Figure 4C). Serum nonesterified fatty acid levels were also unaltered (Supplemental Figure 4D). These results indicate that lipid metabolism and inflammation in peripheral tissues were not affected, even after the prolonged administration of suvorexant.
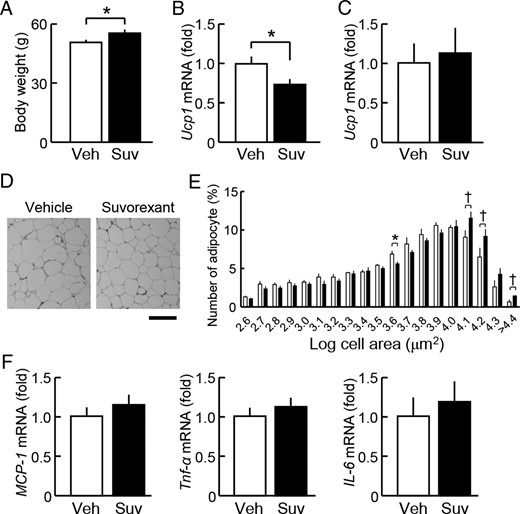
Influence of the prolonged treatment with suvorexant on the energy balance in db/db mice. Suvorexant (closed columns, Suv, 30 mg/kg) or vehicle (open columns, Veh) was administered by p.o. at ZT0 for 6 weeks to db/db mice (males, 11 wk old). Adipose tissues were dissected at ZT6 under the 6-hour fasting condition. A, Body weight. B and C, mRNA levels of Ucp1 in brown (B) and white (epididymal) adipose tissues (C). D, Typical hematoxylin and eosin staining images of epididymal adipose tissue sections from vehicle-treated (left) and suvorexant-treated db/db mice (right). Scale bar, 200 μm. E, Distribution histogram of the adipocyte size. The areas of adipocytes were measured (magnification, ×4) in 4 slices of epididymal adipose tissues from each mouse and averaged. F, mRNA levels of MCP-1, Tnf-α, and IL-6 in epididymal adipose tissues. Data were the mean ± SEM (n = 10–11). *, P < .05 and †, P < .1.
We then examined the influences of suvorexant on the regulation of hepatic glucose production. When suvorexant (30 mg/kg) was administered daily at ZT0 for 6 weeks, the mRNA expression levels of the hepatic gluconeogenic factors, phosphoenolpyruvate carboxykinase (Pepck) and peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α), were significantly reduced, whereas that of glucose-6-phosphatase (G6Pase) was slightly reduced in the liver of db/db mice (Figure 8A). Moreover, the phosphorylation levels of CREB, a major promotor of gluconeogenic genes (26), were reduced by this drug treatment (Figure 8B), whereas those of FoxO1, another gluconeogenic gene promotor regulated by insulin, were not affected (data not shown). Under this condition, neither serum corticosterone levels (Figure 8C), serum glucagon levels (Figure 8D), nor hepatic cAMP levels (Figure 8E) were affected by the suvorexant treatment. The mRNA levels of D-box-binding protein (Dbp), a clock-controlled gene, were reduced by suvorexant in the liver at ZT6 under 6-hour fasting conditions; however, no change was detected in the other clock or clock-controlled genes examined: Clock, Bmal1, Period 1, Period 2, Cry1, Rev-erbα, and Nampt (Supplemental Figure 5). Hepatic glycogen content in db/db mice was not altered by daily administration of suvorexant (30 mg/kg, ZT0) for 2 weeks (data not shown). Thus, the daily resting-phase administration of suvorexant appeared to suppress the expression of genes involved in hepatic gluconeogenesis, at least independently of glucagon and corticosterone, although direct measurement of gluconeogenesis was not conducted.
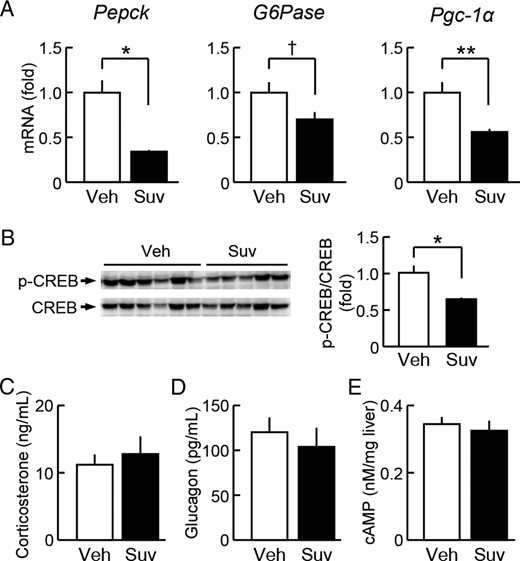
Influence of the daily resting-phase administration of suvorexant on hepatic glucose regulation in db/db mice. Suvorexant (closed columns, Suv, 30 mg/kg) or vehicle (open columns, Veh) was administered by p.o. at ZT0 for 6 weeks to db/db mice (males, 11 wk old). Liver tissues and blood samples were then obtained at ZT6 under 6-hour fasting conditions. A, mRNA levels of Pepck, G6Pase, and Pgc-1α in the liver. B, Phosphorylation levels of CREB in the liver. C and D, Serum levels of corticosterone (C) and glucagon (D). E, cAMP levels in the liver. Data were the mean ± SEM; n = 10–11. *, P < .05; **, P < .01; and †, P < .1 vs vehicle.
Discussion
Circadian rhythms in glucose metabolism are known to be perturbed in type 2 diabetic patients (27). Improvements in deranged circadian rhythms by time-restricted feeding or the administration of melatonin have prevented metabolic disorders in individuals with diet-induced obesity (28, 29). Accumulating evidence also indicates that the maintenance of sleep homeostasis is crucial for normal glucose homeostasis (6); however, it remains unknown whether the amelioration of sleep disturbances may help to improve metabolic disorders, including diabetes. In the present study, we found that the daily resting-phase administration of suvorexant, an antiinsomnia drug, improved sleep disturbances and glucose metabolism in type 2 diabetic db/db mice. The suvorexant-induced amelioration of sleep disturbances preceded improvements in glucose metabolism. Thus, the results of the present study indicate that timed suvorexant treatments are a valuable chronopharmacological intervention to block the pathological link between sleep disturbances and type 2 diabetes.
Suvorexant is one of the DORAs that promote REM and non-REM sleep in normal C57BL/6 mice (30, 31). In diabetic db/db mice, the wakefulness time in the resting phase was found to be abnormally increased. Suvorexant exhibited the ability to increase REM and non-REM sleep times and decrease the awake time in db/db mice when administered at the beginning of the resting phase. Drug efficacy was less potent in db/db than in db/m+ mice, and this may have been due to the reduced expression of orexin (ie, the target ligand of suvorexant) by hyperglycemia in the diabetic state (32). Alternatively, because leptin is known to inhibit the activity of orexin neurons in the hypothalamus (15), lack of the leptin action may cause disinhibition of the orexinergic system in db/db mice in some circumstances, thereby affecting the effect of suvorexant. We anticipate that although the orexinergic system is suppressed to some extent under type 2 diabetic conditions, the nontimely actions of orexin in the resting phase cause abnormal awakening. This is the first study to provide evidence for the promotion of sleep by suvorexant, even in the diabetic state.
Suvorexant readily passes through the blood-brain barrier; therefore, drug concentrations in the brain and plasma after its iv administration are almost identical (33). In the present study, we orally administered suvorexant at a dose of 30 mg/kg to db/db and db/m+ mice in order to promote sleep, and this was based on a previous study using normal C57BL/6 mice (31). In addition, we observed that suvorexant (30 mg/kg, p.o.) blocked the effect of icv-injected orexin A on blood glucose levels in C57BL/6J mice. This dose was higher than the clinical dose (≤20 mg) used in the treatment of patients with insomnia (21); however, relatively higher doses of suvorexant are known to be required to promote sleep in rodents than in dogs or humans because of its higher clearance (33). Suvorexant (30 mg/kg, ZT0) acutely promoted sleep, but did not significantly affect the total awake/sleep time in the next 12-hour dark phase in db/db and db/m+ mice, indicating that “next-day somnolence” was not caused by this treatment. These results were consistent with previous findings obtained in normal rats (33) and humans (21). Therefore, we consider the dose of suvorexant used in the present study to be appropriate for evaluating pharmacological profiles in mice.
The present study also provided a novel key result: the daily resting-phase administration of suvorexant improved not only sleep but also glucose metabolism in db/db mice. The improvement of glucose tolerance was observed under the 16-hour fasting condition in which the glucose counter-regulatory responses, including hepatic gluconeogenesis, are known to be promoted (Figure 5B) but not under the 4-hour fasting condition (data not shown). Moreover, this improvement was associated with reductions in the mRNA levels of Pepck and Pgc-1α and the phosphorylation of CREB in the liver of db/db mice, all of which contribute to enhancing hepatic gluconeogenesis. Although these transcriptional changes may not necessarily reflect the functional change, reductions in hepatic gluconeogenesis appear to explain the mechanism underlying the improving effects of suvorexant on glucose metabolism. We consider suvorexant to suppress hepatic gluconeogenesis via neural rather than hormonal pathways because no alterations in serum insulin, glucagon, or corticosterone levels were detected. In addition, systemic insulin sensitivity and peripheral insulin signaling were not altered by the drug treatment. These results appeared to be independent of the dose of insulin used (4 U/kg), because the insulin tolerance test with 2-U/kg insulin also showed no difference in blood glucose levels between the suvorexant (50 mg/kg, ZT0 for 2 wk)- and vehicle-treated db/db mice (data not shown). The sympathetic nervous system contributes to increases in hepatic glucose production (34). Basal sympathetic tone is abnormally increased in db/db mice (11) and type 2 diabetic patients (35). The blockade of OX2R in the rostral ventrolateral medulla, a central region controlling sympathetic nerve activity, has been reported to reduce blood pressure in spontaneously hypertensive rats (36). Therefore, we suggest that suvorexant centrally suppresses sympathetic efferent signals to the liver, thereby preventing sustained hepatic gluconeogenesis in db/db mice.
On the other hand, we and others previously reported that the hypothalamic orexin system plays crucial roles in the regulation of energy and glucose metabolism (20, 37). An orexin deficiency was found to induce narcolepsy in association with obesity in humans (38), and orexin-deficient mice developed severe obesity under high-fat diet conditions (25, 39). Moreover, an orexin deficiency caused insulin resistance profoundly in the liver under pathophysiological conditions, such as aging, obesity, and depression in mice (24, 25, 40). In contrast, transgenic orexin overexpression prevented diet-induced obesity and improved insulin sensitivity in mice (41). These findings indicate that orexin is required to maintain energy and glucose homeostasis, whereas we showed that an orexin receptor antagonist exerted beneficial effects in the present study. However, these apparent discrepancies may be reconciled by taking into account daily changes in the effects of orexin. Orexin levels in the cerebrospinal fluid increase daily during the awake phase and decrease during the resting phase (16). The administration of orexin A in the awake phase according to this rhythm has been shown to improve the daily regulation of hepatic gluconeogenesis, thereby reducing hyperglycemia in the resting phase in db/db mice, whereas no improvement was produced by its resting-phase administration (24). In contrast, the resting-phase, but not awake-phase administration of suvorexant improved glucose tolerance in the resting phase in db/db mice. Therefore, we suggest that the timely activation and inhibition of the orexin system amplifies the daily effects of orexin, thereby improving the sleep/wake cycle, autonomic nerve balance, and metabolic functions.
The effects of short-term blockade of orexin receptors on the sleep/wake state are different from those of chronic orexin deficiency. Partial OX2R occupancy (>65%) by DORAs is known to be sufficient to promote sleep (16), and suvorexant rapidly increased both the REM and non-REM sleep episode durations in normal mice (31). In contrast, orexin-deficient mice, an animal model of narcolepsy, have fragmented sleep and wakefulness because of the behavioral state instability (42). The unstable sleep/wake state is considered to be caused by chronic compensatory changes in the wake-promoting process, such as nontimely activation of noradrenergic neurons in the brain stem in orexin neuron-ablated mice (43). Therefore, we consider that the observed effects of suvorexant in the present study are independent of narcolepsy-like behavioral changes.
The limitation of the present study was that we only used 1 diabetic mouse model (ie, db/db mice) to examine the effects of suvorexant. Furthermore, the effects of suvorexant on glucose metabolism were relatively modest. The diabetic spontaneous mutant db/db mice are leptin resistant due to lack of the main leptin receptor isoform Ob-Rb that mediates the central actions of leptin, and they develop severe obesity and hyperphagia, thereby secondarily leading to metabolic disorders, such as hyperglycemia and insulin resistance; however, leptin receptor deficiency is not a crucial contributor to the etiology of type 2 diabetes in humans (9). Thus, it may be difficult to predict the impact of suvorexant in humans based on the results of the present study. However, because the non-REM and REM sleep periods are more consolidated in humans than in mice, we anticipate that suvorexant may cause similar or more profound effects in humans. Further studies are warranted in order to examine the impact of suvorexant on the pathological links between sleep disturbances and metabolic disorders in patients with type 2 diabetes.
In conclusion, we here found that the improvements in sleep caused by the daily resting-phase administration of suvorexant were associated with the better control of blood glucose in type 2 diabetic mice. In this process, gene expression associated with gluconeogenesis was suppressed. Abnormal increases in hepatic gluconeogenesis are the main cause of hyperglycemia during resting periods in type 2 diabetes (44), and sleep loss is one of the main factors increasing hepatic glucose production (4). Therefore, we suggest that the timed administration of suvorexant is a promising chronopharmacological intervention to treat metabolic diseases associated with sleep disturbances, including type 2 diabetes.
Acknowledgments
We thank T. Matsushima and T. Fujimori (University of Toyama, Toyama, Japan) for their technical assistance.
This work was supported by Grants-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (JSPS KAKENHI Grants 15K09380 and 15K15599) and by grants from the Foundation for Growth Science (T.Sas.), the Japan Diabetes Foundation (T.Sas.), and the Tamura Science and Technology Foundation (H.T.).
Disclosure Summary: The authors have nothing to disclose.
Appendix
See Table 1.
Antibody Table
| RRID | Name of Antibody | Peptide/Protein Target | Antigen Sequence (if Known) | Vendor | Catalog Number | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|---|
| AB_626658 | Akt1 (B-1) antibody | AKT1 | Santa Cruz Biotechnology | sc-5298 | Mouse; monoclonal | 1:1000 | |
| AB_329825 | Phospho-Akt (Ser473) antibody | Phospho-Akt (Ser473) | Cell Signaling Technology | 9271 | Rabbit; polyclonal | 1:1000 | |
| AB_10695751 | CREB (48H2) rabbit mAb | CREB | Cell Signaling Technology | 9197S | Rabbit; monoclonal | 1:1000 | |
| AB_10544696 | Phospho-CREB (Ser133) (D1G6) rabbit mAb antibody | Phospho-CREB (Ser133) | Cell Signaling Technology | 4276S | Rabbit; monoclonal | 1:1000 | |
| AB_2106495 | FoxO1 (C29H4) rabbit mAb | FoxO1 | Cell Signaling Technology | 2880 | Rabbit; monoclonal | 1:1000 | |
| AB_329831 | Phospho-FoxO1 (Ser256) antibody | Phospho-FoxO1 (Ser256) | Cell Signaling Technology | 9461 | Rabbit; polyclonal | 1:1000 |
| RRID | Name of Antibody | Peptide/Protein Target | Antigen Sequence (if Known) | Vendor | Catalog Number | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|---|
| AB_626658 | Akt1 (B-1) antibody | AKT1 | Santa Cruz Biotechnology | sc-5298 | Mouse; monoclonal | 1:1000 | |
| AB_329825 | Phospho-Akt (Ser473) antibody | Phospho-Akt (Ser473) | Cell Signaling Technology | 9271 | Rabbit; polyclonal | 1:1000 | |
| AB_10695751 | CREB (48H2) rabbit mAb | CREB | Cell Signaling Technology | 9197S | Rabbit; monoclonal | 1:1000 | |
| AB_10544696 | Phospho-CREB (Ser133) (D1G6) rabbit mAb antibody | Phospho-CREB (Ser133) | Cell Signaling Technology | 4276S | Rabbit; monoclonal | 1:1000 | |
| AB_2106495 | FoxO1 (C29H4) rabbit mAb | FoxO1 | Cell Signaling Technology | 2880 | Rabbit; monoclonal | 1:1000 | |
| AB_329831 | Phospho-FoxO1 (Ser256) antibody | Phospho-FoxO1 (Ser256) | Cell Signaling Technology | 9461 | Rabbit; polyclonal | 1:1000 |
Antibody Table
| RRID | Name of Antibody | Peptide/Protein Target | Antigen Sequence (if Known) | Vendor | Catalog Number | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|---|
| AB_626658 | Akt1 (B-1) antibody | AKT1 | Santa Cruz Biotechnology | sc-5298 | Mouse; monoclonal | 1:1000 | |
| AB_329825 | Phospho-Akt (Ser473) antibody | Phospho-Akt (Ser473) | Cell Signaling Technology | 9271 | Rabbit; polyclonal | 1:1000 | |
| AB_10695751 | CREB (48H2) rabbit mAb | CREB | Cell Signaling Technology | 9197S | Rabbit; monoclonal | 1:1000 | |
| AB_10544696 | Phospho-CREB (Ser133) (D1G6) rabbit mAb antibody | Phospho-CREB (Ser133) | Cell Signaling Technology | 4276S | Rabbit; monoclonal | 1:1000 | |
| AB_2106495 | FoxO1 (C29H4) rabbit mAb | FoxO1 | Cell Signaling Technology | 2880 | Rabbit; monoclonal | 1:1000 | |
| AB_329831 | Phospho-FoxO1 (Ser256) antibody | Phospho-FoxO1 (Ser256) | Cell Signaling Technology | 9461 | Rabbit; polyclonal | 1:1000 |
| RRID | Name of Antibody | Peptide/Protein Target | Antigen Sequence (if Known) | Vendor | Catalog Number | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|---|
| AB_626658 | Akt1 (B-1) antibody | AKT1 | Santa Cruz Biotechnology | sc-5298 | Mouse; monoclonal | 1:1000 | |
| AB_329825 | Phospho-Akt (Ser473) antibody | Phospho-Akt (Ser473) | Cell Signaling Technology | 9271 | Rabbit; polyclonal | 1:1000 | |
| AB_10695751 | CREB (48H2) rabbit mAb | CREB | Cell Signaling Technology | 9197S | Rabbit; monoclonal | 1:1000 | |
| AB_10544696 | Phospho-CREB (Ser133) (D1G6) rabbit mAb antibody | Phospho-CREB (Ser133) | Cell Signaling Technology | 4276S | Rabbit; monoclonal | 1:1000 | |
| AB_2106495 | FoxO1 (C29H4) rabbit mAb | FoxO1 | Cell Signaling Technology | 2880 | Rabbit; monoclonal | 1:1000 | |
| AB_329831 | Phospho-FoxO1 (Ser256) antibody | Phospho-FoxO1 (Ser256) | Cell Signaling Technology | 9461 | Rabbit; polyclonal | 1:1000 |
Abbreviations
- CREB
cAMP-response element-binding protein
- CV
coefficient of variation
- DORA
orexin receptor antagonist
- EEG
electroencephalogram
- EMG
electromyogram
- FoxO1
forkhead box protein O1
- icv
intracerebroventricular
- OX1R
orexin receptor-1
- Pepck
phosphoenolpyruvate carboxykinase
- Pgc-1α
peroxisome proliferator-activated receptor-γ coactivator-1α
- p.o.
oral gavage
- REM
rapid eye movement
- RQ
respiratory quotient
- ZT
zeitgeber time.
References
Author notes
H.T. and K.K. contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}