





The hypothalamic hormone arginine vasopressin (AVP) potentiates the stimulatory action of CRH on ACTH secretion from pituitary corticotropes, but the underlying mechanism is elusive. Using the perforated patch-clamp technique to monitor membrane potentials in mouse corticotropes, we found that AVP triggered a transient hyperpolarization that was followed by a sustained depolarization. The hyperpolarization was caused by intracellular Ca2+ release that in turn activated the small conductance Ca2+-activated K+ (SK) channels. The depolarization was due to the suppression of background TWIK-related K+ (TREK)-1 channels. Direct activation of protein kinase C (PKC) reduced the TREK-1 current, whereas PKC inhibition attenuated the AVP-mediated reduction of the TREK-1 current, implicating the involvement of PKC. The addition of CRH (which stimulates the protein kinase A pathway) in the presence of AVP, or vice versa, resulted in further suppression of the TREK-1 current. In corticotropes with buffered cytosolic Ca2+ concentration ([Ca2+]i), AVP evoked a sustained depolarization, and the coapplication of AVP and CRH caused a larger depolarization than that evoked by AVP or CRH alone. In cells with minimal perturbation of [Ca2+]i and background TREK-1 channels, CRH evoked a sustained depolarization that was superimposed with action potentials, and the subsequent coapplication of AVP and CRH triggered a transient hyperpolarization that was followed by a larger depolarization. In summary, AVP and CRH have additive effects on the suppression of the TREK-1 current, resulting in a more robust depolarization in corticotropes. We suggest that this mechanism contributes to the potentiating action of AVP on CRH-evoked ACTH secretion.
During stress, neurons in the hypothalamic paraventricular nucleus release CRH and arginine vasopressin (AVP) into the portal circulation to stimulate ACTH secretion from corticotropes in the anterior pituitary gland. CRH acts on the CRH-1 receptors, which are coupled to the cAMP/protein kinase A (PKA) pathway (1, 2), to evoke a monophasic ACTH secretion that is dependent on voltage-gated Ca2+ channel (VGCC) activation (3). AVP, on the other hand, acts on VP-1b receptors, which are coupled to the phospholipase C pathway, which results in the generation of inositol 1,4,5-trisphosphate (IP3) and the activation of protein kinase C (PKC) (4). In contrast to CRH, AVP evokes a “spike and plateau” pattern of ACTH secretion; the “spike phase” is attributed to intracellular Ca2+ release, whereas the “plateau phase” involves PKC and VGCC activation (4, 5). Consistent with a key role of intracellular Ca2+ release in the spike phase of the AVP-induced ACTH secretion, we have shown previously that in rat corticotropes, the generation of IP3 during AVP stimulation triggers Ca2+ release from the intracellular store, and the resultant transient rise in [Ca2+]i triggers exocytosis (6, 7). We also demonstrated that the AVP-evoked intracellular Ca2+ release leads to the activation of the small conductance Ca2+-activated K+ (SK) channels, resulting in a transient hyperpolarization (6). However, it is unclear how AVP signaling is linked to the activation of VGCCs in corticotropes.
In comparison to CRH, AVP is a weaker ACTH secretagogue (1). Nevertheless, the action of AVP is essential for a full ACTH response when animals are challenged by a variety of acute stressors (4). For example, the elevation in plasma ACTH induced by acute restraint or forced swimming stress was attenuated in the mutant VP-1b receptor knockout mice and in wild-type mice that were pretreated with a VP-1b receptor antagonist (8). The important role of AVP in the acute stress response is largely mediated via its potentiating action on the CRH-evoked ACTH secretion (9). The mechanism underlying the potentiating action of AVP is not well understood, but the involvement of PKC has been implicated. Previous studies in rat pituitary cells have shown that the direct activation of PKC with phorbol ester stimulated ACTH secretion (10), and PKC depletion abolished the potentiating action of AVP (10, 11). AVP did not affect the cellular level of cAMP, but the CRH-induced accumulation of cAMP in pituitary cells was found to be enhanced by AVP via a PKC-dependent inhibition of phosphodiesterase (12, 13). These findings raised the possibility that the potentiating effect of AVP involved an increase in the CRH-induced cAMP accumulation. However, phorbol ester was able to potentiate ACTH secretion even in the presence of a high concentration of cAMP analog (14), suggesting that mechanisms other than an increase in the CRH-induced cellular cAMP accumulation may contribute to the potentiating action of AVP.
In a previous study, we reported that the resting membrane potential of corticotropes is determined by the activities of background TWIK-related K+ (TREK)-1 channels (15). We have also shown that CRH, acting via the cAMP/PKA–dependent pathway, suppresses the background TREK-1 current, resulting in a sustained depolarization (15, 16). The sustained depolarization activates VGCCs, leading to a persistent [Ca2+]i elevation (16, 17) and the triggering of exocytosis (7). Because TREK-1 channels can be phosphorylated by PKA as well as by PKC (18), we hypothesized that the potentiating action of AVP involves a suppression of the TREK-1 current. In the current study, we tested this hypothesis by examining the effect of AVP on the TREK-1 current and the membrane potential of corticotropes isolated from proopiomelanocortin (POMC)-enhanced green fluorescent protein (eGFP) transgenic mice. We found that in corticotropes with minimal disturbance to the cytosol (ie, recordings with the perforated patch-clamp technique), AVP caused a transient hyperpolarization that was followed by a sustained depolarization. The hyperpolarization was due to the activation of the SK channels during intracellular Ca2+ release, whereas the sustained depolarization was due to PKC-dependent suppression of the TREK-1 channels. In the presence of a maximal stimulatory concentration of CRH (10 nM), AVP further suppressed the TREK-1 current, resulting in an even more robust depolarization. Our findings indicate that a PKC-dependent suppression of the TREK-1 current is an important mechanism that contributes to both the plateau phase of the AVP-evoked ACTH secretion and the potentiating action of AVP on the CRH-evoked ACTH secretion.
Materials and Methods
Cell culture
POMC-eGFP transgenic mice were generated as described previously (19). To identify mice that express POMC-eGFP from the mixed litter, the pituitary gland was removed from mice euthanized in accordance with protocols approved by the animal care and use committee at the University of Alberta and standards of the Canadian Council on Animal Care. Because the intermediate lobe of the pituitary gland is composed of mostly melanotropes that express α-MSH, a POMC gene product, the intermediate lobe of the pituitary gland in the POMC-eGFP mouse exhibits intense green fluorescence when viewed under a dissecting microscope. The anterior lobes of the pituitary glands were dissected from these mice and dissociated enzymatically as described previously (15). Dissociated pituitary cells were plated on polylysine (0.1 mg/ml; Sigma-Aldrich Canada)–coated glass coverslips and maintained under standard culture conditions in DMEM supplemented with 10% (vol/vol) horse serum, 50 U/ml penicillin G, and 50 mg/ml streptomycin (all from Sigma-Aldrich) for 1 to 3 days. Individual corticotropes were identified from the heterogeneous pituitary cell population by their GFP fluorescence (observed under an epifluorescence microscope with an excitation filter at 470–490 nm and an emission filter at 520–560 nm). Cell cultures were obtained from 18 male and 15 female POMC-eGFP mice.
Electrophysiological recording
In most experiments (except for those shown in Figures 1, B and C, and 8), the membrane potential or current from individual corticotropes was recorded with the whole-cell patch-clamp technique with an EPC-9 patch-clamp amplifier (HEKA Electronics). The standard bath solution contained 150 mM NaCl, 10 mM Hepes, 8 mM glucose, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4). For experiments using a Ca2+-free extracellular solution, Ca2+ was omitted from the standard bath solution, and 1 mM EGTA was added. Except for the experiments shown in Figures 1A, 4, and 7, the standard whole-cell pipette solution contained 85 mM potassium aspartate, 20 mM Hepes, 20 mM KCl, 1 mM MgCl2, 2 mM Na2ATP, 0.1 mM Na4GTP, 5 mM CaCl2, and 10 mM EGTA (pH 7.4) and had a calculated [Ca2+] of 68 nM (20). For the experiments shown in Figures 4 and 7, the amplitude of the TREK-1 current was augmented by intracellular acidification (21) with a whole-cell pipette solution that was buffered to pH 7.0. For experiments involving perforated patch-clamp recording of membrane potentials (Figures 1, B and C, and 8), the standard perforated patch-clamp pipette solution contained 120 mM potassium aspartate, 20 mM K-Hepes, 20 mM KCl, 1 mM MgCl2, 5 mM Na2ATP, 0.1 mM Na4GTP (pH 7.4), and 250 μg/ml of amphotericin B. All experiments were performed at room temperature (20–23°C). A −10 mV junction potential was corrected throughout. For the generation of current voltage (I-V) plots, individual cells were whole-cell voltage clamped at −70 mV and then stepped to different potentials (−80 to −20 mV in 10 mV increment) for 200 msec. The current amplitude at different potentials was averaged from the last 40 msec of the voltage step.
Influence of AVP on [Ca2+]i, current, and membrane potential of GFP-fluorescent mouse corticotropes
![A, Application of AVP (100 nM) stimulated a transient elevation of [Ca2+]i. The [Ca2+]i rise was accompanied by activation of an outward current. [Ca2+]i was recorded simultaneously with the membrane current from a corticotrope that was whole-cell voltage clamped at −70 mV. The whole-cell pipette solution (pHint = 7.4) contained 0.1 mM Indo-1. B, Application of AVP (100 nM) evoked a transient hyperpolarization that was followed by a sustained depolarization (indicated by the dashed line) and the firing of action potentials. The dotted line indicates the resting membrane potential. The cell was recorded with the perforated patch-clamp technique using a pipette solution that was buffered to pH 7.4. C, In the presence of apamin (0.5 μM), a blocker of SK channels, application of AVP (100 nM) evoked a sustained depolarization that was not preceded by any hyperpolarization. The cell was recorded with the perforated patch-clamp technique using a pipette solution that was buffered to pH 7.4.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/156/10/10.1210_en.2015-1293/4/m_zee9991581690001.jpeg?Expires=1750265925&Signature=csJfkQhouO6wVdz0fTOlpOQQqPEzCWubPvuIXHTIivY7xKe9iSu6o73X~j9zys8c8uz6uKeoXlQapOqnhYeeQWIdhR6okbuVkd26xtkQTjywDN3Mw~HrK7~KQCIHcRMl80t1K5v4e5qsg~q8nRdKBkgKiTaX~ieP-17xVB8BQOAEHZW~6fVhPWRyxrkj~RtQ3JYESRBVQnZZTPEY-cskLLJ1eIHv-k0uSbEzXlV33c8rsVIia0j5p9J5EklnQH3Rc3nbr5EUVM5l3ot0uQX6JoxSyO3lMmy0ULwJe3hD9tkZcDFSVmGwp7okgXsvm6shTTXHbJy6D7i5DXyxD7NeVQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A, Application of AVP (100 nM) stimulated a transient elevation of [Ca2+]i. The [Ca2+]i rise was accompanied by activation of an outward current. [Ca2+]i was recorded simultaneously with the membrane current from a corticotrope that was whole-cell voltage clamped at −70 mV. The whole-cell pipette solution (pHint = 7.4) contained 0.1 mM Indo-1. B, Application of AVP (100 nM) evoked a transient hyperpolarization that was followed by a sustained depolarization (indicated by the dashed line) and the firing of action potentials. The dotted line indicates the resting membrane potential. The cell was recorded with the perforated patch-clamp technique using a pipette solution that was buffered to pH 7.4. C, In the presence of apamin (0.5 μM), a blocker of SK channels, application of AVP (100 nM) evoked a sustained depolarization that was not preceded by any hyperpolarization. The cell was recorded with the perforated patch-clamp technique using a pipette solution that was buffered to pH 7.4.
In corticotropes with minimal perturbation of the basal TREK-1 current and [Ca2+]i, the application of AVP in the continued presence of CRH triggered a biphasic pattern of membrane potential change

A, Application of CRH (10 nM) evoked a sustained depolarization (indicated by the dashed line) that was superimposed with the firing of action potentials. The addition of AVP (100 nM) in the continued presence of CRH caused a transient hyperpolarization, which was followed by a larger sustained depolarization (indicated by the solid line). B, AVP further increases the amplitude of the sustained depolarization evoked by CRH. The sustained depolarization evoked by CRH was 15 ± 6 mV and increased to 28 ± 4 mV after the coapplication of AVP and CRH (n = 6). In the experiment shown here, individual corticotropes were recorded with the perforated patch-clamp technique using a pipette solution that was buffered to the physiological pH of 7.4. *, P < .05.
AVP suppresses the TREK-1 current enhanced by intracellular acidification
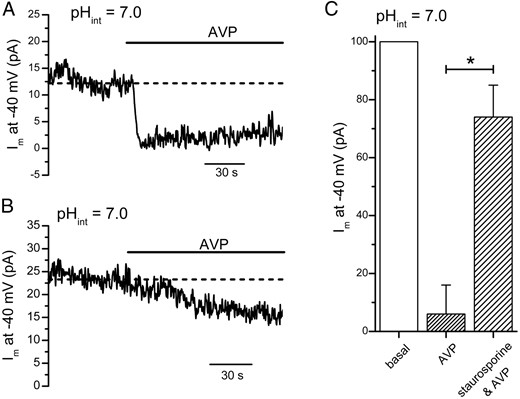
A, Intracellular acidification–enhanced TREK-1 current at −40 mV was reduced by AVP (100 nM). B, Pretreatment with staurosporine attenuated the AVP-mediated reduction of the intracellular acidification–enhanced TREK-1 current. C, In control cells, AVP (100 nM) reduced the intracellular acidification–enhanced TREK-1 current at −40 mV to 6 ± 10% of its basal value (n = 5). In cells pretreated with staurosporine (1 μM), 74 ± 11% of the TREK-1 current remained in the presence of AVP (n = 5). In the experiment shown here, individual cells were whole-cell voltage clamped at −40 mV with a pipette solution that contained 10 mM EGTA and 5 mM CaCl2 (pHint = 7.0). *, P < .05.
In corticotropes with buffered [Ca2+]i and intracellular acidification–enhanced TREK-1 current, AVP evoked a sustained depolarization and potentiated the CRH-induced depolarization

A, AVP (100 nM) evoked a sustained depolarization (indicated by the dashed line). Note that AVP did not trigger any hyperpolarization when the cytosol contained Ca2+ buffer. B, AVP potentiated the CRH-induced depolarization. Application of CRH (10 nM) evoked a sustained depolarization (indicated by the dashed line) that was superimposed with action potential firings. The addition of AVP (100 nM) increased the amplitude of the sustained depolarization (indicated by the solid line). C, Amplitude of the sustained depolarization (ΔVm) evoked by AVP alone, by CRH alone, or by the coapplication of CRH and AVP. AVP depolarized the membrane potential by 11 ± 2 mV (n = 13). In cells that were challenged with CRH and then a combination of CRH and AVP, the sustained depolarization evoked by CRH was 24 ± 3 mV and increased to 33 ± 3 mV after the combined application of AVP and CRH. In the experiments shown here, individual corticotropes were recorded with a whole-cell pipette solution that contained 10 mM EGTA and 5 mM CaCl2 with pH buffered to 7.0. *, P < .05.
Measurement of [Ca2+]i
[Ca2+]i was measured with Indo-1 fluorometry. Individual corticotropes were recorded with a whole-cell pipette solution that contained 120 mM potassium aspartate, 20 mM Hepes, 20 mM KCl, 1 mM MgCl2, 2 mM Na2ATP, 0.1 mM Na4GTP, and 0.1 mM Indo-1 (pH 7.4). Details of the instrumentation and procedures for [Ca2+]i measurement with Indo-1 were described previously (22).
Chemicals
Indo-1 K+ salt was from TEFlabs. AVP and CRH (Peptide International) were dissolved in 0.1 M acetic acid, lyophilized, and kept at −20°C. Phorbol 12-myristate 13-acetate (PMA) and staurosporine were kept as stock solutions in dimethylsulfoxide (DMSO) at −20°C. Apamin, arachidonic acid (sodium salt) (AA), PMA, staurosporine, and other chemicals were obtained from Sigma-Aldrich.
Statistical analysis
Origin (version 8; OriginLab Corp) was used for plotting and statistical procedures. When two populations of cells were compared, the two-sample Student t test was used. Any difference of P < .05 was considered statistically significant and was marked with an asterisk in the figures. All values shown are means ± SEM.
Results
AVP triggered a transient hyperpolarization that was followed by a sustained depolarization
In rat corticotropes, we have shown previously that AVP triggers Ca2+ release from the IP3-sensitive stores, and the rise in [Ca2+]i in turn activates the SK channels, resulting in a transient hyperpolarization (6). However, a recent study in mouse corticotropes reported that AVP increased only the frequency of spontaneous action potentials (23). Therefore, we examined the action of AVP on mouse corticotropes by simultaneously monitoring [Ca2+]i and the membrane current when the cell was whole-cell voltage clamped at −70 mV. To understand the mechanisms underlying the stimulatory action of AVP on corticotropes, we used 100 nM AVP, which has been shown to have a maximal stimulatory effect on ACTH secretion (1). As shown in the example in Figure 1A, the application of AVP (100 nM) evoked a transient rise in [Ca2+]i that gradually declined to the basal level even in the continued presence of AVP. The rise in [Ca2+]i was accompanied by an increase in outward current which diminished when [Ca2+]i declined near the basal level (Figure 1A). On average, the peak amplitude of the AVP-induced rise in [Ca2+]i was 1.0 ± 0.2 μM, and the peak amplitude of the accompanying outward current at −70 mV was 6.8 ± 1.9 pA. In 3 of the 12 cells examined, the amplitude of the AVP-induced [Ca2+]i rise was < 0.36 μM, and the Ca2+ signal was not accompanied by any transient outward current. Because the cells were voltage clamped at −70 mV, a potential at which VGCCs were not activated, the AVP-induced Ca2+ signal observed here arose primarily from intracellular Ca2+ release. Consistent with this notion, AVP evoked a comparable [Ca2+]i rise in cells bathed in a Ca2+-free extracellular solution (1.1 ± 0.2 μM; n = 5). The transient outward current was abolished by apamin (0.5 μM; n = 4), indicating that it was mediated via the SK channels.
To examine the influence of AVP on the membrane potential of mouse corticotropes, we used the perforated patch-clamp recording technique to minimize perturbation of the cytosol. In the 13 corticotropes examined, the resting membrane potential was −58 ± 4 mV. In 10 of the 13 cells, there were some fluctuations in the resting membrane potential under the basal condition, and the amplitude of the membrane potential fluctuation was between 5 and 10 mV. In the remaining 3 cells, the potential fluctuation was larger than 10 mV. Although a few spontaneous action potentials could be observed under the basal condition, we did not detect any high-frequency firing of spontaneous action potentials as reported by Duncan et al (23). As shown in Figure 1B, the application of AVP evoked a transient hyperpolarization. The hyperpolarization was followed by a sustained depolarization that was superimposed with robust action potential firings. On average, AVP hyperpolarized the membrane potential by 24 ± 6 mV, and the hyperpolarization lasted ∼20 to 40 seconds (n = 4). The mean amplitude of the sustained depolarization evoked by AVP in the same cells was 13 ± 3 mV (n = 4). For cells exposed to apamin (0.5 μM), AVP evoked a sustained depolarization, which was not preceded by any transient hyperpolarization (n = 4; Figure 1C). This finding confirms that the AVP-induced transient hyperpolarization is due to the activation of SK channels during intracellular Ca2+ release. In the 4 cells exposed to apamin, AVP evoked a larger sustained depolarization (30 mV ± 3 mV) compared with that for the control cells (13 ± 3 mV; n = 4). This result suggests that in control cells, the VGCC-mediated extracellular Ca2+ entry during the AVP-evoked depolarization activated SK channels that in turn limited the amplitude of the AVP-evoked depolarization. Because the resting membrane potential of corticotropes is primarily determined by the activities of the background TREK-1 channels (15), we examined whether the AVP-evoked depolarization involves a suppression of the TREK-1 current.
AVP reduced the TREK-1 current in corticotropes via a PKC-dependent pathway
A major challenge in studying the TREK-1 current in mouse corticotropes is the small amplitude of the basal TREK-1 current. As shown in our previous study (15), the amplitude of the basal TREK-1 current in mouse corticotropes at −40 mV is only ∼1.5 pA. In the same study, we found that AA suppresses the delayed rectifier K+ current but dramatically increases the amplitude of the basal TREK-1 current (15). Therefore, in the current study, we used AA to isolate and potentiate the TREK-1 current in mouse corticotropes. To minimize the activation of SK current during the AVP-induced intracellular Ca2+ release, we attenuated the AVP-induced [Ca2+]i rise by dialyzing individual corticotropes with a whole-cell pipette solution that contained 10 mM EGTA and 5 mM CaCl2. Figure 2A shows the current recorded from a corticotrope that was whole-cell voltage clamped at −70 mV. Application of AA (10 μM) induced the gradual development of an outward current, reflecting the potentiation of the basal TREK-1 current. When the amplitude of the TREK-1 current reached a peak, the cell was exposed to AVP (100 nM) in the continuous presence of AA. As shown in Figure 2A, AVP caused a partial reduction of the AA-enhanced TREK-1 current. The I-V plot in Figure 2B shows that before the application of AA, there was little current at potentials between −90 and −30 mV. Application of AA caused a dramatic increase in the outward current amplitude, and AVP reduced the AA-enhanced TREK-1 current at this entire range of potentials (Figure 2B). We have shown previously that the AA-enhanced TREK-1 current is largely abolished by pimozide, a diphenylbutylpiperidine antipsychotic (15). Therefore, we examined whether the inhibitory action of AVP can be occluded by pimozide. As shown in the example in Figure 2C and in 3 other cells, the application of pimozide (10 μM) essentially abolished the AA-enhanced TREK-1 current at −70 mV, and a subsequent application of AVP did not cause any additional reduction of the current. This finding confirms that the primary target of AVP is the TREK-1 current. To compare the inhibitory action of AVP on the AA-enhanced TREK-1 current among different corticotropes, we considered the current at −70 mV in the presence of AA (10 μM) in an individual cell to be 100% and then normalized the current amplitude after AVP application in the same cell accordingly. In the 47 corticotropes examined, application of AVP (100 nM) reduced the AA-enhanced TREK-1 current at −70 mV by 26 ± 2% (Figure 2D). In separate experiments, we examined the action of CRH under the same experimental conditions and found that CRH (10 nM), which has a maximal stimulatory effect on ACTH secretion (1), reduced the AA-enhanced TREK-1 current at −70 mV by 38 ± 6% (n = 18; Figure 2D). Thus, when CRH or AVP was applied at a concentration that had a maximal stimulatory effect on ACTH secretion, CRH caused a larger reduction of the TREK-1 current than AVP.
AVP reduces the AA-enhanced TREK-1 current in corticotropes
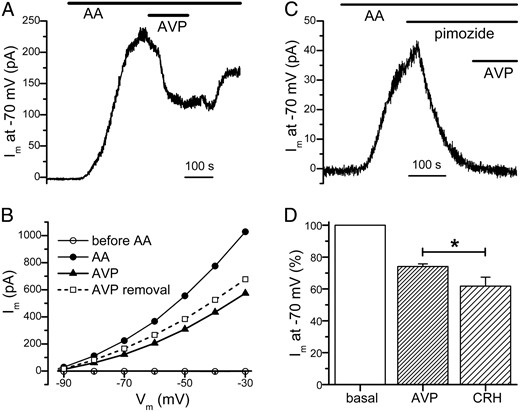
A, Application of AA (10 μM) enhanced the TREK-1 current, which was subsequently reduced by AVP (100 nM). B, Inhibitory action of AVP on the TREK-1 current could be observed at a wide range of potentials (same cell as in A). The I-V plots were generated when the cell was bathed sequentially in the standard bath solution (before AA), in AA, in AVP + AA, and after AVP removal (in the continued presence of AA). C, TREK-1 channel inhibitor pimozide occluded the action of AVP. Application of pimozide (10 μM) abolished the TREK-1 current enhanced by AA (10 μM). The subsequent addition of AVP (100 nM) failed to affect the holding current at −70 mV. D, In comparison to CRH, AVP had a weaker inhibitory effect on the TREK-1 current. The current of an individual cell at −70 mV in the presence of AA (10 μM) was considered to be 100%, and the current amplitude after AVP or CRH application in the same cell was normalized accordingly. The AA-enhanced TREK-1 current at −70 mV was reduced to 74 ± 2% of its basal value (n = 47) by AVP but to 62 ± 6% of its basal value (n = 18) by CRH. In the experiment shown here, individual cells were whole-cell voltage clamped at −70 mV with a pipette solution that contained 10 mM EGTA and 5 mM CaCl2 (pHint = 7.4). *, P < .05.
Because the stimulation of VP-1b receptors leads to PKC activation (4) and the direct activation of PKC was reported to suppress the mouse TREK-1 channels that were expressed in HEK293 cells (18), we hypothesized that the inhibitory action of AVP on the TREK-1 current is due to PKC activation. To test this hypothesis, we examined whether the direct activation of PKC with PMA, a phorbol ester, can suppress the TREK-1 current in corticotropes. Figure 3A shows that the application of PMA (1 μM) caused a robust suppression of the AA-enhanced TREK-1 current at −70 mV. As shown in the I-V plot of the same cell, the inhibitory action of PMA on the AA-enhanced TREK-1 current could be observed at the entire range of potentials (Figure 3B). In the 10 cells examined, PMA reduced the AA-enhanced TREK-1 current at −70 mV by 50 ± 5% (Figure 3C; cf 26 ± 2% by AVP in Figure 2D). In comparison to AVP, direct activation of PKC caused a more robust inhibition of the AA-enhanced TREK-1 current in corticotropes. We further examined whether staurosporine, a PKC inhibitor, can prevent the inhibitory effect of AVP on the TREK-1 current. In this set of experiments, cells were pretreated with staurosporine (1 μM) for 30 minutes before being challenged with AVP. Because the staurosporine stock solution contained DMSO, we treated some cells with the same concentration of DMSO for controls. In the 15 corticotropes that were pretreated with DMSO, AVP (100 nM) reduced the AA-enhanced TREK-1 current by 28 ± 5% (Figure 3C), similar to the inhibitory action of AVP observed in the untreated cells (26 ± 2%; Figure 2D). In the 14 cells pretreated with staurosporine, AVP reduced the AA-enhanced TREK-1 current by 14 ± 2%, which is significantly less than the inhibition observed in cells pretreated with DMSO (Figure 3C). These findings suggest that PKC has an important role in the AVP-mediated suppression of the TREK-1 current in corticotropes.
AVP suppresses the TREK-1 current via a PKC-dependent mechanism

A, Direct activation of PKC with the phorbol ester PMA (1 μM) caused a robust suppression of the AA-enhanced TREK-1 current. B, PMA reduced the AA-enhanced TREK-1 current at a wide range of potentials (same cell as in A). C, AVP-mediated reduction of the TREK-1 current was attenuated by staurosporine, a PKC inhibitor. On average, PMA reduced the AA-enhanced TREK-1 current at −70 mV to 50 ± 5% of its basal value (n = 10). In cells pretreated with DMSO, AVP (100 nM) reduced the current to 72 ± 5% of its basal value (n = 15). In cells pretreated with staurosporine (1 μM), 86 ± 2% of the AA-enhanced TREK-1 current remained in the presence of AVP (n = 14). In the experiment shown here, individual cells were whole-cell voltage clamped at −70 mV with a pipette solution that contained 10 mM EGTA and 5 mM CaCl2 (pHint = 7.4). *, P < .05.
In the experiments described in Figures 2 and 3, we examined the effect of AVP on the TREK-1 current that was enhanced by AA. In a previous study (15), we showed that the background TREK-1 current in mouse corticotropes can also be enhanced by lowering the pH of the whole-cell pipette solution (pHint) from 7.4 to 7.0. Therefore, we examined whether AVP can suppress the TREK-1 current that was enhanced by intracellular acidification. Because the amplitude of the TREK-1 current enhanced by intracellular acidification is several fold smaller than the AA-enhanced current (15), we voltage clamped the cells at −40 mV. Under this experimental condition, the average amplitude of the TREK-1 current at −40 mV was 30 ± 13 pA (n = 10). As shown in the example in Figure 4A, the application of AVP (100 nM) resulted in a robust suppression of the outward current at −40 mV. In 5 corticotropes examined, AVP reduced the holding current at −40 mV by 94 ± 10% (Figure 4C). Figure 4B shows that pretreatment with staurosporine (1 μM) largely occluded the inhibitory action of AVP on the intracellular acidification–enhanced TREK-1 current. In 5 corticotropes pretreated with staurosporine, AVP reduced the holding current at −40 mV by 26 ± 11% (Figure 4C). These findings further confirm that the TREK-1 current is a primary target of AVP and that PKC activation plays a dominant role in the inhibitory action of AVP.
AVP and CRH have an additive effect on the inhibition of the TREK-1 current
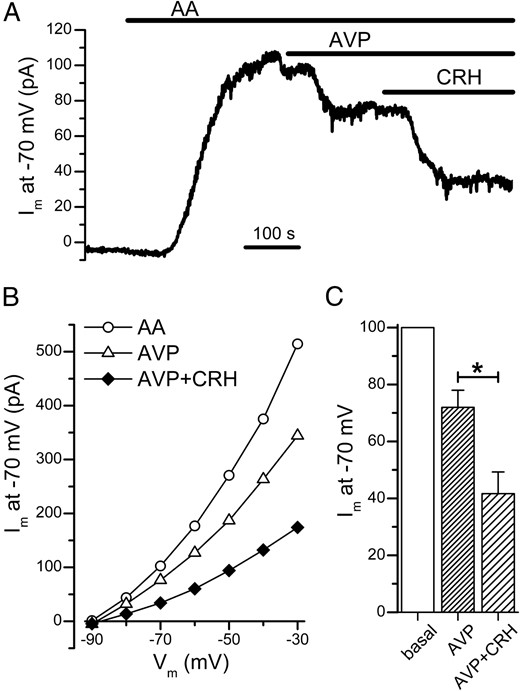
Our results above show that the TREK-1 channel is a common target for AVP and CRH. However, it is unclear whether the inhibitory actions of these two ACTH secretagogues on the TREK-1 current are additive. To address this, we first examined whether CRH can further suppress the TREK-1 current in the presence of AVP. In the example shown in Figure 5A, when AVP (100 nM) reduced the AA-enhanced TREK-1 current to a steady level, the subsequent addition of CRH (10 nM) in the continued presence of AVP further reduced the TREK-1 current. As shown in the I-V plot of the same cell, the addition of CRH in the continued presence of AVP further suppressed the AA-enhanced TREK-1 current at a wide range of potentials (Figure 5B). In the 8 cells examined with this protocol, AVP reduced the AA-enhanced TREK-1 current at −70 mV by 28 ± 6%, and the total current reduction following a subsequent challenge with CRH (in the continued presence of AVP) was 58 ± 8% (Figure 5C). Although AVP is less effective than CRH in the inhibition of the TREK-1 current (Figure 2D), we found that even in the presence of a maximal concentration of CRH (10 nM), AVP can further suppress the TREK-1 current. As shown in the example in Figure 6, A and B, when the AA-enhanced TREK-1 current decreased to a steady level in the presence of CRH (10 nM), the subsequent addition of AVP (100 nM; in the continued presence of CRH) further reduced the TREK-1 current. In the 12 cells examined with this protocol, CRH reduced the AA-enhanced TREK-1 current by 33 ± 6% and the total current reduction after the addition of AVP (in the continued presence of CRH) was 62 ± 6% (Figure 6C). These findings indicate that the inhibitory actions of AVP and CRH on the TREK-1 current are additive.
In the presence of AVP, CRH can further suppress the AA-enhanced TREK-1 current
A, After the AA-enhanced TREK-1 current at −70 mV was reduced to a steady level by AVP (100 nM), the addition of CRH (10 nM) further decreased the current. B, In the continued presence of AVP, CRH further suppressed the TREK-1 current at a wide range of potentials (same cell as in A). C, AVP reduced the amplitude of the AA-enhanced TREK-1 current at −70 mV to 72 ± 6% of its basal value, and the subsequent coapplication of CRH and AVP decreased the current to 42 ± 8% (n = 8). In the experiment shown here, individual cells were whole-cell voltage clamped at −70 mV with a pipette solution that contained 10 mM EGTA and 5 mM CaCl2 (pHint = 7.4). *, P < .05.
In the presence of CRH, AVP further suppresses the AA-enhanced TREK-1 current

A, After the AA-enhanced TREK-1 current at −70 mV was reduced to a steady level by CRH (10 nM), the addition of AVP (100 nM) further decreased the current. B, In the continued presence of CRH, AVP further suppressed the TREK-1 current at a wide range of potentials (same cell as in A). C, CRH reduced the amplitude of the AA-enhanced TREK-1 current at −70 mV to 67 ± 6% of its basal value, and the subsequent coapplication of AVP and CRH decreased the current to 37 ± 6% (n = 12). In the experiment shown here, individual cells were whole-cell voltage clamped at −70 mV with a pipette solution that contained 10 mM EGTA and 5 mM CaCl2 (pHint = 7.4). *, P < .05.
In corticotropes with buffered [Ca2+]i and intracellular acidification–enhanced TREK-1 current, AVP evoked a sustained depolarization and potentiated the CRH-induced depolarization
Next, we examined the impact of the AVP-mediated reduction of the TREK-1 current on membrane potential. To separate the effect of the TREK-1 current on membrane potential from that mediated by the SK current that was transiently activated by the AVP-evoked [Ca2+]i rise (Figure 1), we buffered [Ca2+]i using a whole-cell pipette solution that contained 10 mM EGTA and 5 mM CaCl2. To better observe the influence of the TREK-1 current in the AVP-mediated change in membrane potential, we enhanced the background TREK-1 current in corticotropes by lowering the pH of the whole-cell pipette solution from 7.4 to 7.0. Under these experimental conditions, the mean resting membrane potential of corticotropes was −72 ± 2 mV (n = 13; cf −58 ± 4 mV at pHint of 7.4), and the application of AVP (100 nM) did not cause any hyperpolarization but evoked a sustained depolarization (Figure 7A). In the 13 cells examined, AVP depolarized the membrane potential by 11 ± 2 mV (Figure 7C). We then examined whether AVP can further depolarize the membrane potential in the presence of CRH. In the example shown in Figure 7B, CRH (10 nM) evoked a sustained depolarization, which was superimposed with the firing of action potentials. The addition of AVP (100 nM) in the continued presence of CRH resulted in a further increase in the amplitude of the sustained depolarization. In the 14 cells examined with the same protocol as in Figure 7B, the sustained depolarization evoked by CRH alone was 24 ± 3 mV and increased to 33 ± 3 mV after the coapplication of AVP and CRH (Figure 7C). The larger depolarization evoked by the coapplication of AVP and CRH is consistent with the additive effect of CRH and AVP on the suppression of the AA-enhanced TREK-1 current (Figures 5 and 6). This result also shows that whereas AVP reduces ∼94% of the intracellular acidification–enhanced TREK-1 current at −40 mV (Figure 4C), the subsequent coapplication of AVP and CRH can further suppress the TREK-1 current and result in a more robust depolarization. Figure 7C also shows that the amplitude of the depolarization evoked by AVP was ∼2-fold smaller than that evoked by CRH. This result is consistent with the smaller inhibitory effect of AVP on the AA-enhanced TREK-1 current than that of CRH (26 vs 38%; Figure 2D).
Impact of AVP on the CRH-evoked depolarization in corticotropes under physiological conditions
An intriguing question about the interaction between AVP and CRH signaling on corticotropes is whether the hyperpolarization induced by AVP opposes the depolarization evoked by CRH. To examine how AVP affects the CRH-evoked depolarization under physiological conditions, we minimized the perturbation of [Ca2+]i and the basal TREK-1 current by recording the membrane potential in the perforated patch-clamp mode with a pipette solution that was buffered to physiological pH (pHint = 7.4; same recording condition as in Figure 1B). The example in Figure 8A shows that the application of CRH (10 nM) evoked a sustained depolarization that was superimposed with robust action potential firing. When the cell was challenged with AVP (100 nM) in the continued presence of CRH, the membrane potential hyperpolarized transiently to a level more negative than the resting membrane potential and then depolarized to a sustained level that was more positive than the depolarization evoked by CRH alone. In the 6 cells examined with this protocol, the sustained depolarization evoked by CRH alone was 15 ± 6 mV and increased to 28 ± 4 mV after the coapplication of AVP and CRH (Figure 8B). Therefore, we speculate that under physiological conditions, the simultaneous exposure of AVP and CRH should result in a transient phase of hyperpolarization that is dominated by the AVP-mediated hyperpolarization and then a sustained phase of depolarization that is mediated by both AVP and CRH.
Discussion
Influence of AVP on the membrane potential of mouse corticotropes
In our recordings with the perforated patch-clamp technique, the resting membrane potential of mouse corticotropes was ∼−58 mV, which is comparable to ∼−54 mV reported by Duncan et al (23) under similar experimental conditions. Although we found some fluctuations (5–10 mV) in the resting membrane potential, most of the corticotropes did not exhibit any spontaneous firing of action potentials, consistent with a low level of basal ACTH release (1, 5). We showed that AVP (100 nM) evoked a biphasic change in the membrane potential (Figure 1B). The first phase is a transient hyperpolarization that is due to the activation of SK channels during Ca2+ release from the intracellular stores (Figure 1A). In support of this, the transient hyperpolarization was abolished by the SK channel inhibitor, apamin (Figure 1C). This result is similar to our previous study in rat corticotropes (6) and is consistent with the dominant role of intracellular Ca2+ release during the spike phase of the AVP-evoked ACTH secretion (5). Our finding contrasts with a recent report that AVP at 2 nM did not evoke any hyperpolarization but increased the frequency of action potential firing in mouse corticotropes (23). As described earlier, we found that, in some corticotropes (3 of 12 cells), when the amplitude of the Ca2+ signal evoked by 100 nM AVP was small (<0.36 μM), the AVP-induced [Ca2+]i rise was not accompanied by the activation of SK current. Therefore, a possible explanation for the absence of AVP-mediated hyperpolarization in the study by Duncan et al (23) is that the lower concentration of AVP (2 nM) at room temperature may be less efficient in triggering intracellular Ca2+ release. This may result in a smaller AVP-mediated [Ca2+]i rise that is insufficient for the activation of SK channels.
The second phase of the AVP-mediated change in membrane potential is a sustained depolarization, which has not been described previously. As shown in Figure 1B, after the termination of the hyperpolarization, the cell depolarized to a plateau potential that was superimposed with action potential firings. In our previous studies, we have shown that a VGCC-mediated increase in [Ca2+]i is triggered when individual corticotropes were depolarized to potentials near −40 mV (16, 17). Thus, the sustained depolarization and action potential firings observed during the second phase (ie, after the transient hyperpolarization) of the AVP challenge should lead to VGCC-mediated extracellular Ca2+ entry and a rise in [Ca2+]i. The literature has shown that extracellular Ca2+ entry via VGCCs has a major role in the plateau phase of the AVP-evoked ACTH secretion (5), although the relationship between AVP signaling and VGCC activation is not understood. Therefore, our finding that AVP evokes depolarization and action potential firings after the termination of intracellular Ca2+ release provides an important link between AVP signaling and VGCC activation in corticotropes.
Sustained depolarization evoked by AVP was mediated via a PKC-dependent suppression of the TREK-1 current
Consistent with the dominant role of the TREK-1 current in setting the resting membrane potential of corticotropes, we found that the sustained depolarization evoked by AVP is due to the suppression of the TREK-1 current. In cells with [Ca2+]i buffered at ∼68 nM (see Materials and Methods) and an enhanced TREK-1 current (via the lowering of pHint to 7.0), AVP caused a sustained depolarization (Figure 7). When the TREK-1 current was enhanced by AA or intracellular acidification, AVP reduced the TREK-1 current (Figures 2 and 4). Consistent with the ability of PKC to phosphorylate TREK-1 channels (18), the PKC activator PMA reduced the AA-enhanced TREK-1 current in corticotropes (Figure 3). In cells treated with the PKC inhibitor staurosporine, the inhibitory effect of AVP on the TREK-1 current (enhanced by AA or intracellular acidification) was significantly attenuated (Figures 3C and 4C), suggesting an important role of PKC in the AVP-mediated suppression of the TREK-1 current. Note that in the presence of staurosporine, AVP could still cause a small reduction in the TREK-1 current (Figures 3C and 4C), suggesting that some PKC-independent mechanisms may also contribute to the AVP response. In bovine adrenal zona fasciculata cells, angiotensin II was reported to suppress the TREK-1 current via a Ca2+-dependent mechanism; it was postulated that the stimulation of intracellular Ca2+ release by angiotensin II generated a locally high [Ca2+] in proximity to some TREK-1 channels, resulting in a Ca2+-dependent inhibition of TREK-1 channels (24). This possibility cannot be ruled out here because we used 10 mM EGTA and 5 mM CaCl2 in our whole-cell pipette solution when the effect of AVP on the TREK-1 current was examined, and the buffering strength might not be sufficient to prevent the generation of Ca2+ microdomains. In addition, many other PKC-independent mechanisms have been proposed to inhibit TREK-1 channels, including phosphatidylinositol 4,5-bisphosphate depletion (25), a direct action of diacylglycerol (26), and a novel mechanism that requires ATP hydrolysis but is independent of phospholipase C and its downstream effectors (27). Thus, it is possible that one or more of these mechanisms may contribute to the PKC-independent component of the AVP effect. Overall, our finding suggests that PKC has an important role in the AVP-mediated suppression of the TREK-1 current and thus the AVP-mediated sustained depolarization. The important role of PKC is consistent with a previous study showing that PKC down-regulation attenuated the plateau phase of the AVP-evoked ACTH secretion (10).
Additive effect of AVP and CRH on the TREK-1 current contributes to the potentiating action of AVP on the stimulatory effect of CRH
It has been shown previously that the stimulatory action of AVP on ACTH secretion reaches a maximum at 100 nM AVP, whereas the CRH-evoked ACTH secretion reaches a maximum at 10 nM CRH (1). Here, we found that the inhibitory action on the AA-enhanced TREK-1 current at −70 mV induced by 100 nM AVP is significantly smaller than that mediated by 10 nM CRH (26% vs 38%; Figure 2D). We have also shown that in cells with intracellular acidification–enhanced TREK-1 current (Figure 7C), the sustained depolarization evoked by CRH (10 nM) is ∼2-fold larger than that evoked by AVP (100 nM). These results suggest that the degree of inhibition of the TREK-1 current in corticotropes correlates with the amplitude of the depolarization. Note that AVP appeared to have a larger inhibitory effect on the intracellular acidification–enhanced TREK-1 current at −40 mV (∼94%; Figure 4C) compared with the AA-enhanced TREK-1 current at −70 mV (∼28%; Figure 2D). One possible explanation is that the intracellular acidification–enhanced TREK-1 current is more susceptible to the inhibitory action of ACTH secretagogues. Consistent with this notion, we have shown in our previous study that CRH essentially abolished the intracellular acidification–enhanced TREK-1 current at −40 mV (15) but only reduced the AA-enhanced TREK-1 current at −70 mV by ∼38% (Figure 2D). It is also possible that the inhibition of the TREK-1 current by ACTH secretagogues is more robust at positive potentials. In the current study, CRH reduced the AA-enhanced TREK-1 current at −70 mV by ∼38% (Figure 2D), which is smaller than the inhibition of the AA-enhanced TREK-1 current at −40 mV (∼76%) shown in our previous study (15).
Our results in Figures 5 and 6 show that the suppression of the AA-enhanced TREK-1 current at −70 mV by maximal concentrations of AVP and CRH are additive. In cells first challenged with AVP (100 nM), the remaining AA-enhanced TREK-1 current was further suppressed by the addition of CRH (Figure 5). Conversely, in cells that were first challenged with CRH, the subsequent addition of AVP further reduced the TREK-1 current (Figure 6). On average, the coapplication of AVP and CRH reduced the AA-enhanced TREK-1 current at −70 mV by ∼60% (Figures 5C and 6C), similar to the 64% obtained by the summation of the inhibitory effect mediated separately by AVP (26%) and CRH (38%; Figure 2C). As described above, the inhibitory effect of AVP on the TREK-1 current is primarily mediated via PKC (Figures 3 and 4). We have also shown previously that CRH reduces the TREK-1 current via the cAMP/PKA pathway (15). Therefore, the additive effect of AVP and CRH on the suppression of the TREK-1 current is probably due to the independent actions of PKC and PKA on the TREK-1 channels. Consistent with this theory, the TREK-1 channels contain both PKA- and PKC-dependent phosphorylation sites and the PKC-mediated suppressing action of the TREK-1 channels is not attenuated by PKA inhibition (18).
The additional suppression of the TREK-1 current by AVP, even in the presence of a maximal concentration of CRH, results in a more robust depolarization. In cells with buffered [Ca2+]i and intracellular acidification–enhanced TREK-1 current, AVP depolarized the cells by ∼11 mV (Figure 7, A and C). Under the same experimental condition, the CRH-evoked plateau depolarization was ∼24 mV, and the addition of AVP increased the amplitude of the plateau depolarization by ∼10 mV (Figure 7, B and C). Thus, in cells with intracellular acidification–enhanced TREK-1 current, the effects of AVP and CRH on membrane potential are additive. In cells recorded under physiological conditions (without any added cytosolic Ca2+ buffer or enhancement of the basal TREK-1 current), the CRH-evoked plateau depolarization was slightly smaller (∼15 mV; Figure 8B), probably due to some activation of SK channels during the CRH-mediated [Ca2+]i rise. Under this condition, the addition of AVP caused a transient hyperpolarization (due to SK channel activation during intracellular Ca2+ release) and then further depolarized the CRH-evoked plateau depolarization by ∼13 mV (Figure 8B). This finding shows that, under physiological conditions, the AVP- and CRH-evoked depolarizations are additive. The additive effect of AVP and CRH on depolarization is expected to be translated into a synergistic action on ACTH secretion. We have shown previously that, at potentials between −40 and +10 mV, the activation of VGCCs in corticotropes increases steeply with depolarization (16). For example, the amplitude of the Ca2+ current in rat corticotropes evoked by a depolarization to −10 mV was ∼2-fold larger than the current evoked at −20 mV (16). Thus, the additional depolarization (∼15 mV) by AVP in the continued presence of CRH may increase the total VGCC-mediated Ca2+ entry by ∼2-fold. Moreover, we have shown previously that exocytosis in corticotropes is steeply dependent on [Ca2+] such that there is a third-power relationship between [Ca2+] and exocytosis (7). Therefore, a 15-mV additional depolarization by AVP in the presence of CRH may increase the CRH-evoked ACTH secretion by several fold.
Significance
Our results suggest that AVP potentiates the CRH-evoked ACTH secretion via two mechanisms: a transient rise of [Ca2+]i due to intracellular Ca2+ release and a larger VGCC-mediated extracellular Ca2+ entry. The latter mechanism is due to a PKC-dependent suppression of the TREK-1 current that is additive to the inhibition mediated by CRH via the PKA pathway. Importantly, we found that the TREK-1 channel is a common target of CRH and AVP. Thus, an increase in the opening of the TREK-1 channels will oppose the stimulatory effect of CRH and AVP on the electrical excitability of corticotropes and will, in turn, reduce the stress-induced ACTH release and the ensuing secretion of adrenal corticosteroids. Conversely, drugs that inhibit the TREK-1 channels are anticipated to increase the secretion of ACTH and corticosteroids. Note that the antidepressant fluoxetine is also an inhibitor of the TREK-1 channels, and we have shown previously that the CRH-evoked depolarization in corticotropes can be mimicked by fluoxetine (15). Consistent with the stimulatory action of fluoxetine on corticotropes, acute administration of fluoxetine has been shown to increase the plasma levels of ACTH and corticosteroids in rats (28) and in mice (29). Because hypothalamic-pituitary-adrenal axis hyperactivity is implicated in the pathophysiology of some types of depression, such as melancholic depression, fluoxetine treatment may exacerbate the elevated plasma cortisol levels in these patients, which may contribute in part to the poor response to fluoxetine treatment in depressive patients with hypothalamic-pituitary-adrenal hyperactivity (30).
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AA
arachidonic acid
- AVP
arginine vasopressin
- [Ca2+]i
cytosolic Ca2+ concentration
- DMSO
dimethyl sulfoxide
- eGFP
enhanced green fluorescent protein
- IP3
inositol 1,4,5-trisphosphate
- I-V
current-voltage
- PKA
protein kinase A
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- POMC
proopiomelanocortin
- SK
small conductance Ca2+-activated K+
- TREK
TWIK-related K+
- VGCC
voltage-gated Ca2+ channel.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}