





Abstract
Treatment of rat islets with the cytokine IL-1 results in the inhibition of mitochondrial function and insulin secretion, events that are mediated by β-cell expression of iNOS [inducible nitric oxide (NO) synthase] and production of NO. β-Cells recover from the inhibitory actions of NO, produced following 24 h incubation with IL-1, on islet oxidative metabolism and insulin secretion if iNOS enzymatic activity is inhibited and the islets are cultured (in the presence of IL-1 and iNOS inhibitors) for a brief period of 8 h. Islet recovery from cytokine- and NO-mediated damage is an active process that requires new gene expression, and NO itself is one activator of this recovery process. In this study, the mechanism by which NO stimulates islet recovery has been examined. Incubation of rat islets or RINm5F cells with the NO donor compound, sodium (Z)-1(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA-NO) for 1 h results in a 60% inhibition of mitochondrial aconitase activity. β-Cells completely recover aconitase activity if the cells are washed to remove the NO donor compound and incubated for an additional 5 h in the absence of DEA-NO. The recovery of mitochondrial aconitase activity correlates with a 4-fold increase in cyclic GMP accumulation and is prevented by the inhibition of guanylate cyclase. The recovery of aconitase activity also correlates with the activation of members of the MAPKs, p38, c-Jun N-terminal kinase (JNK) and ERK, and the activation p38 and JNK is attenuated by inhibition of guanylate cyclase. ERK and p38 do not appear to participate in the recovery process as selective inhibition of these kinases fails to prevent recovery of aconitase activity; however, transduction of β-cells with a dominant negative mutant JNK prevents β-cell recovery from NO-mediated damage. These findings support a role for guanylate cyclase and JNK in the recovery of β-cells from NO-mediated damage.
INSULIN-DEPENDENT DIABETES MELLITUS is characterized by islet inflammation followed by selective destruction of pancreatic islet β-cells (1, 2). Cytokines, released during islet inflammation, are believed to participate in β-cell damage during the development of autoimmune diabetes mellitus (3, 4). Incubation of isolated rat islets with the macrophage-derived cytokine IL-1 results in a time-dependent inhibition of glucose-stimulated insulin secretion, the oxidation of glucose to CO2, mitochondrial aconitase activity, and protein synthesis, as well as reduced cellular levels of ATP, and islet cell DNA damage (3, 5–8). Prolonged exposure of islets for 4 d with IL-1 results in islet degeneration (4, 9). The time-dependent inhibitory and damaging actions of IL-1 on islet function correlate with the time-dependent expression of iNOS [inducible nitric oxide (NO) synthase] and the production of NO (8, 10, 11). Inhibitors of NO synthase prevent the inhibitory actions of IL-1 on islet oxidative metabolism, glucose-stimulated insulin secretion, protein synthesis, islet degeneration, and the damaging actions of this cytokine on islet cell DNA integrity (5, 12–15). Consistent with a role for NO in mediating β-cell damage, islets isolated from iNOS knockout mice are resistant to the inhibitory actions of IL-1 and interferon (IFN)-γ on insulin secretion (16, 17). Pancreatic β-cells are the primary cellular source of iNOS in rat, mouse, and human islets following treatment with IL-1 alone, or IL-1 + IFN-γ (18–20). These findings support a primary role for β-cell production of NO as a mediator of the inhibitory and destructive actions of cytokine on β-cell function.
Although cytokines induce β-cell damage, the inhibitory actions of cytokines on β-cell function are reversible. Comens et al. (21) first showed that the inhibitory effects of a 15-h incubation of rat islets with IL-1 on glucose-stimulated insulin secretion can be reversed if the cytokine is removed by washing and the islets are allowed to culture for an additional 4 d in the absence of IL-1. This recovery of secretory function can be reduced from 4 d to 8 h by the inhibition of iNOS enzymatic activity (9). We have shown that the addition of iNOS inhibitors to rat islets pretreated for 18–24 h with IL-1 (without removing IL-1) results in a time-dependent recovery of insulin secretion that is complete within 8 h (9). The recovery of insulin secretion correlates with the simultaneous recovery of mitochondrial aconitase activity (9). Although islets maintain the ability to recover from the damaging actions of IL-1, this protective response is not absolute, as treatment of islets for 36 h or longer with IL-1 results in an irreversible inhibition of insulin secretion, aconitase activity, protein synthesis, and the commitment of islets to undergo morphological degeneration (14). The recovery of islets from the damaging actions of IL-1 appears to be an active process that requires new gene expression. We have shown that inhibitors of mRNA transcription prevent rat islet recovery from IL-1-induced damage as well as the recovery of human islet aconitase activity following IL-1 + IFN-γ-induced damage (9, 14). One stimulus for the activation of this recovery response in islets is NO. Treatment of rat or human islets with the NO donor sodium (Z)-1(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA-NO) for 1 h results in the inhibition of aconitase activity. If the NO donor is removed by washing and the islets are cultured for 5 additional h in the absence of DEA-NO, they completely recover aconitase activity (14, 22). Importantly, inhibitors of mRNA transcription prevent the recovery of aconitase activity (14, 22). In addition, the recovery of aconitase activity is not due to increased expression of this Kreb cycle enzyme (Corbett, J. A., and A. L. Scarim, unpublished observation). These findings suggest that NO is one stimulus that activates an adaptive protective response in β-cells, and that this protective response is associated with the expression of potential recovery factors in β-cells.
In this study, the mechanisms by which NO activates the protective recovery response in β-cells has been examined. Using aconitase activity as an index of β-cell recovery, we show that selective inhibition of guanylate cyclase prevents β-cell recovery from NO-mediated damage. In addition, we show that NO stimulates the phosphorylation of JNK and p38 in a guanylate cyclase-dependent fashion, and that selective inhibition of JNK, but not p38 or ERK, completely prevents β-cell recovery from NO-mediated damage. These findings support a role for the activation of guanylate cyclase and JNK in the recovery of β-cells from NO-mediated damage.
Materials and Methods
Materials and animals
Rat insulinoma RINm5F cells were obtained from the Washington University Tissue Culture Support Center (St. Louis, MO). Male Sprague Dawley rats (250–300 g) were purchased from Harlan (Indianapolis, IN). Enhanced chemiluminescence reagents were purchase from Amersham Pharmacia Biotech (Arlington Heights, IL). Collagenase type XI was from Sigma Chemical Co. (St. Louis, MO); RPMI 1640, and CMRL-1066, penicillin, streptomycin were from Life Technologies, Inc. (Grand Island, NY). Fetal calf serum was obtained from Hyclone Laboratories, Inc. (Logan, UT). Sodium (Z)-1(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA-NO), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), and LY-83583 were obtained from Alexis (San Diego, CA). Horseradish peroxide-conjugated donkey antirabbit IgG was obtained from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). Rabbit antiserum specific for p-38, JNK, and ERK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), the active forms of p-38 and JNK were obtained from Promega (Madison, WI), and the active form of ERK was from BioSource International (Camarillo, CA). Serine-73 phospo-specific c-Jun was from Cell Signaling Technology (Beverly, MA).
Islet isolation and culture
Islets were isolated from male Sprague Dawley rats (250–300 g) by collagenase digestion as described previously (23). Islets were dispersed into individual cells by treatment with 1 mg/ml trypsin in Ca2+ and Mg2+ free Hanks’ solution at 37 C for 3 min as described previously (24). Islets or dispersed islet cells were cultured overnight at 37 C in an atmosphere of 95% air and 5% CO2 in complete CMRL-1066 media (CMRL-1066 containing 2 mmol/liter l-glutamine, 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) before each experiment.
Intracellular cGMP assay
RINm5F cells (400,000 cells/400 μl complete CMRL-1066) were treated with the NO donor compound, DEA-NO with or without LY-83583, ODQ, 3-isobutyl-1-methylxanthine or (IBMX), culture media were removed and RINm5F cell cGMP levels were determined by competitive enzyme-immunoassay according to the manufacturer’s specifications (Amersham Pharmacia Biotech).
Western blot analysis
Following DEA-NO treatment, RINm5F cells were washed three times with PBS and lysed in sodium dodecyl sulfate (SDS) sample mix (0.25 m Tris-HCl, 20% β-mercaptoethanol, and 4% SDS). Proteins were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes under semi-dry conditions. Blots were blocked with TBST (20 mm Tris, 500 mm NaCl, and 0.1% Tween 20, pH 7.5) containing 5% nonfat dry milk overnight at 4 C and then incubated for 1.5 h at room temperature with either rabbit anti-active JNK (1:5000), rabbit anti-active p38 (1:2000), rabbit anti-active ERK (1:1000), or rabbit anti-phospo-specific c-Jun (serine-73; 1:1000). Horseradish peroxidase-conjugated donkey antirabbit secondary antibody (1:5000) was added and the blots were incubated for 40 min. The blots were washed and target proteins were detected by enhanced chemiluminescence (Amersham Pharmacia Biotech).
Aconitase activity
Dispersed rat islet cells or RINm5F cells were isolated by centrifugation and mitochondrial aconitase activity was determined using a modified version of a method previously developed for rat islet cells (9, 14). In brief, RINm5F cells or islet cells were prepared for aconitase activity measurements as described previously (9), and aconitase was assayed at 340 nm in a reaction containing 20 mm citrate, 0.2 mm nicotinamide adenine dinucleotide phosphate, 6 mm MnCl2, 50 mm Tris-Cl (pH 7.4), 0.6 U of isocitrate dehydrogenase, and 50 μl of islet-cell extract in a total volume of 200 μl using a Power Wave X-340 microtiter plate reader (BioTek Instruments, Winooski, VT) at room temperature. One unit of aconitase activity is defined as 1 pmol of nicotinamide adenine dinucleotide phosphate (reduced) formed per minute per microgram of protein.
Protein transduction
Full-length wild-type JNK and dnJNK (K55-R55) were subcloned into the pTAT expression vector provided by Steven Dowdy (Washington University School of Medicine, St. Louis, MO) (25, 26), and these vectors were transformed into the Escherichia coli strain BL21 cells. TAT-JNK and TAT-dominant negative (dn) JNK proteins were purified from BL21 cells as previously described by the Dowdy laboratory (25, 26). In brief, isolated cells were suspended in 10 ml of 8 m urea, 100 mm NaCl, 20 mm HEPES (pH 8.0), and sonicated on ice three times for 15 sec. The supernatant was clarified by centrifugation, applied on an nickel-nitrilotriacetic acid column (QIAGEN, Valencia, CA), and the fusion proteins were eluted with imidazole in a step-wise fashion (0.1, 0.25, 0.5, and 1.0 m). Fusion proteins, detected in the 0.1 m imidazole elution, were diluted 1:1 with 20 mm HEPES (pH 8.0), and further purified using a Mono-Q column equilibrated with 50 mm NaCl and 20 mm HEPES (pH 8.0). TAT-JNK and TAT-dnJNK fusion proteins were eluted with 1 m NaCl and desalted using PD-10 column equilibrated with PBS. Protein content was determined using the bincinchoninic acid protein assay kit (Pierce, Rockford, IL).
Statistical analysis
Statistical comparisons were made between groups using a one-way ANOVA. Significant differences between groups (P < 0.01) were determined using Bonferroni’s post hoc analysis.
Results
Inhibition of guanylate cyclase prevents recovery of aconitase activity
We have previously shown that islets and RINm5F cells possess the ability to recover from the damaging actions of NO and IL-1 on oxidative metabolism and secretory function, that the recovery process requires new gene expression, and that NO is one stimulus that activates the recovery process (9, 22). Because guanylate cyclase is a known cellular target activated by NO (27), we examined whether inhibition of guanylate cyclase affects the ability of rat islets or RINm5F cells to recover from NO-mediated damage. For these experiments, we evaluated the recovery of mitochondrial aconitase because: 1) aconitase is a known cellular target whose enzymatic activity is highly sensitive to NO; 2) the inhibitory actions of cytokines on insulin secretion are mediated, in part, by the inhibition of mitochondrial oxidation of glucose to CO2; and, 3) the kinetics of recovery of aconitase activity and insulin secretion following cytokine-mediated damage are nearly identical. Treatment of RINm5F cells (Fig. 1, A and B) or rat islets (Fig. 1C) for 1 h with DEA-NO results in a 60–90% inhibition of mitochondrial aconitase activity. Removal of DEA-NO by washing and continued culture of the islets or RINm5F cells for 5 h results in the near complete recovery of aconitase activity. Importantly, the inhibition of guanylate cyclase prevents the recovery of aconitase activity by both RINm5F cells and rat islets. The guanylate cyclase inhibitor LY-83583 at a concentration of 5 μm prevents the recovery of both RINm5F and islet mitochondrial aconitase activity, and a second guanylate cyclase inhibitor, ODQ, prevents the recovery of RINm5F cell aconitase activity at 500 μm. At concentrations of LY-83583 and ODQ that inhibit the recovery of aconitase activity, these guanylate cyclase inhibitors, in the absence of DEA-NO, do not affect RINm5F cells or rat islet cell mitochondrial aconitase activity. These findings support a role for guanylate cyclase activation in the recovery of islets and RINm5F cells from NO-mediated damage.
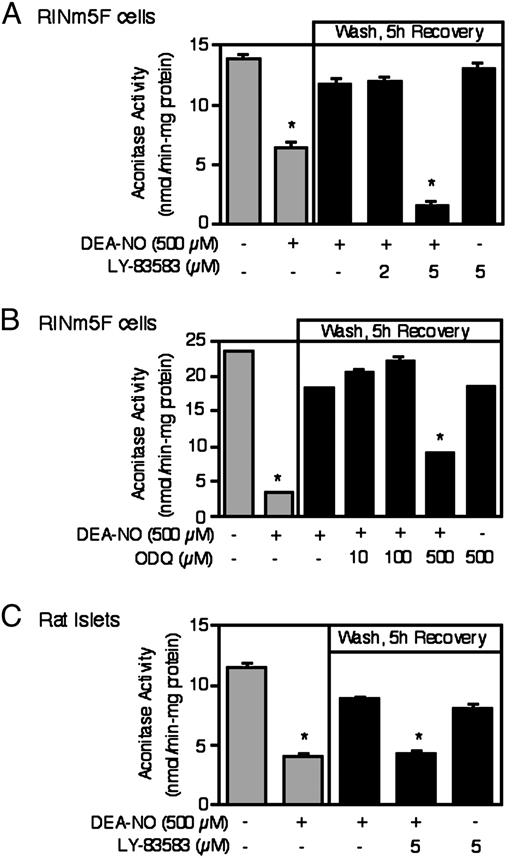
Inhibitors of guanylate cyclase prevent β-cell recovery from NO-mediated damage. A and B, RINm5F cells (1 × 106/ 2 ml of complete CRML-1066) or (C) rat islets (1000/3 ml complete CMRL-1066) were pretreated for 30 min with or without the indicated concentrations of the guanylate cyclase inhibitors LY-83583 or ODQ, DEA-NO (500 μm) was added and the cells or islets were cultured for an additional 1 h. DEA-NO was removed by washing and the cells or islets were allowed to recovery for an additional 5 h. Following the 1-h incubation with DEA-NO, or the 5-h recovery incubation, the cells or islets were isolated and mitochondrial aconitase activity was determined. Results are the average ± sem of three independent experiments. *, P < 0.01 vs. untreated control.
To confirm that DEA-NO stimulates guanylate cyclase activity, the effects of this NO donor compound on cGMP accumulation in RINm5F cells was examined. For these experiments, RINm5F cells were treated for 1 h with DEA-NO in the presence or absence of the phosphodiesterase inhibitor IBMX, the cells were isolated and cGMP levels determined. A 1-h incubation with DEA-NO results in a greater than 8-fold increase in the accumulation of cGMP in RINm5F cells (Fig. 2). Both LY-83583 and ODQ inhibit cGMP accumulation in a concentration-dependent manner with complete inhibition at 5 and 500 μm, respectively. In the absence of DEA-NO, treatment of RINm5F cells with IBMX for 1 h does not results in cGMP accumulation (absence of IBMX 58 ± 15 fmol/4 × 105 cells; presence of IBMX 35 ± 5 fmol/ 4 × 105 cells). Importantly, under conditions in which LY-83583 and ODQ completely prevent DEA-NO-induced cGMP accumulation, these guanylate cyclase inhibitors also prevent the recovery of mitochondrial aconitase activity in RINm5F cells and rat islets (Fig. 1, A–C). At concentrations of LY-83583 (2 μm) and ODQ (100 μm) that do not completely prevent DEA-NO-induced cGMP accumulation, these inhibitors also do not attenuate the recovery of aconitase activity in RINm5F cells and islets. These findings suggest that complete inhibition of guanylate cyclase activity is required to prevent the recovery of aconitase activity in RINm5F cells and rat islets.
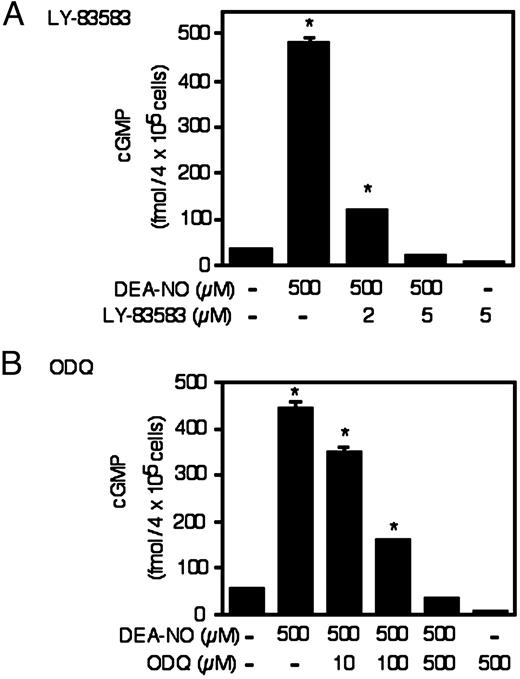
Effects of DEA-NO on cGMP accumulation in RINm5F cells. RINm5F cells (4 × 105/ 400 μl of complete CMRL-1066) pretreated for 30 min with or without the indicated concentrations of LY-83583 and ODQ were incubated for 1 h with 500 μm DEA-NO in the presence of the phosphodiesterase inhibitor IBMX (1 mm). The cells were isolated and cGMP levels determined. Results are the average ± sem of three independent experiments. *, P < 0.01 vs. untreated control.
Guanylate cyclase mediates DEA-NO-induced MAPK activation
Previous studies using fibroblasts have shown that NO stimulates the activation of the MAPK p38 (28). To determine if MAPKs are activated by NO in β-cells, the effects of DEA-NO on the phosphorylation of ERK, JNK, and p38 were examined by Western blot analysis using phosphospecific antibodies. Treatment of RINm5F cells for either 10 or 30 min with DEA-NO stimulates a greater than 10-fold increase in the levels of phosphorylated ERK (p42 and p44), and JNK-1 and JNK-2, and a 4- to 5-fold increase in the level of p38 phosphorylation (Fig. 3; fold stimulation determined by densitometry and normalized to loading controls, ERK, JNK, and p38, respectively). Treatment of RINm5F cells with inactivated DEA-NO does not stimulate MAPK phosphorylation (data not shown). To determine if guanylate cyclase participates in the activation of MAPKs by NO the effects of LY-83583 on ERK, p38, and JNK phosphorylation were examined. For this experiment, RINm5F cells were pretreated with or without LY-83583 for 30 min, DEA-NO was added and the cells were cultured for 10 or 30 additional min (Fig. 3). LY-83583, at a concentration that inhibits RINm5F cell recovery of aconitase and cGMP accumulation (5 μm), attenuates DEA-NO-stimulated phosphorylation of p38 by greater than 70% and JNK phosphorylation by greater than 90% following 10- and 30-min incubations. In a manner similar to LY-83583, ODQ (500 μm) also prevents DEA-NO stimulated JNK phosphorylation in RINm5F cells (data not shown). In contrast, LY-83583 does not attenuate DEA-NO-induced ERK phosphorylation. Alone, LY-83583 does not stimulate MAPK phosphorylation in RINm5F cells. These findings indicate that the activation of guanylate cyclase and accumulation of cGMP is required for the activation of p38 and JNK by NO, and activation of these MAPKs correlates with the ability of RINm5F cells to recover from NO-mediated inhibition of aconitase activity.
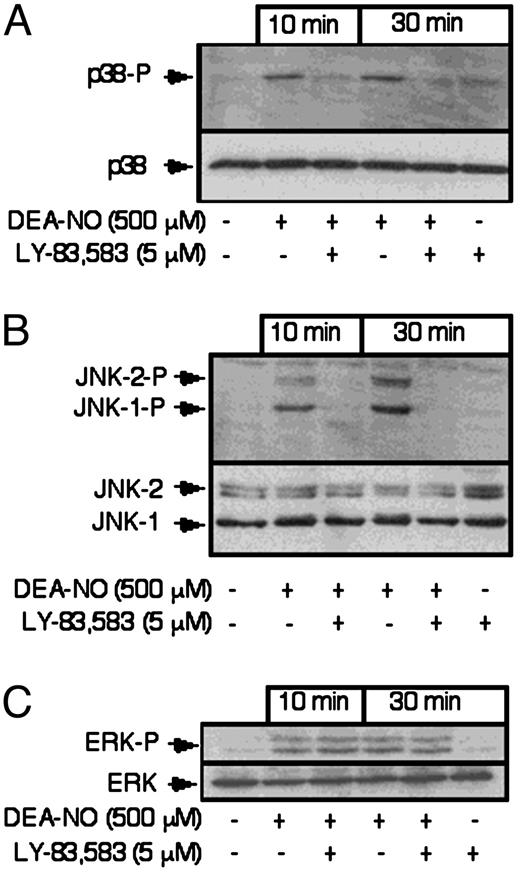
Effects of DEA-NO on p38, JNK, and ERK phosphorylation. RINm5F cells (4 × 105/ 400 μl of complete CMRL-1066), pretreated for 30 min with or without LY-83583, were incubated for 10 or 30 min with DEA-NO (500 μm). The cells were harvested, lysed in SDS sample mix, and the levels of phosphorylated p38, JNK, and ERK were determined by Western blot analysis using phospho-specific antiserum as described in Materials and Methods. The levels of p38, ERK (p42), and JNK were determined by Western blot analysis and are shown as loading controls. Results are representative of three independent experiments.
Effects of MAPK inhibition on the recovery of aconitase activity
Because NO activates MAPKs in a guanylate cyclase-dependent manner, we examined whether these kinases participate in the recovery of aconitase activity of RINm5F cells and rat islets. As shown in Fig. 4, selective inhibition of either p38 using SB-203580 or ERK using PD-98059 (an inhibitor of the upstream MAPK/ERK kinase, MEK) does not modulate the ability of RINm5F cells or rat islets to recover from the inhibitory actions of DEA-NO on mitochondrial aconitase activity. For these experiments, PD-98059 and SB-203580 were used at 10 μm, a concentration that inhibit RINm5F cell ERK and p38 activity by greater than 90% (data not shown). Increasing concentrations (up to 30 μm) also did not affect the ability of RINm5F cells to recover aconitase activity; however, at concentrations of 50 μm or greater both inhibitors induced RINm5F cell death (as determined by trypan blue exclusion and methylthiazoletetrazolium oxidation, data not shown). The lack of an inhibitory action of the ERK pathway inhibitor PD-98059 on the recovery of mitochondrial aconitase activity is consistent with the lack of an inhibitory action of guanylate cyclase inhibitors on DEA-NO-induced ERK phosphorylation (Fig. 3C). Although DEA-NO stimulates ERK and p38 phosphorylation, the activation of these MAPKs appears to correlate with but does not appear to participate in the recovery of RINm5F or rat islet cells from NO-mediated damage.
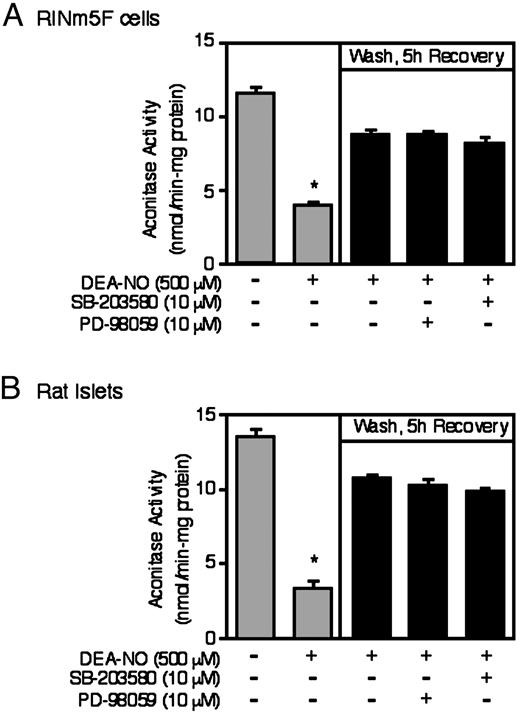
Effects of ERK and p38 inhibition on the recovery of aconitase activity. A, RINm5F cells (1 × 106/ 2 ml of complete CRML-1066); or B, rat islets (1000/3 ml complete CMRL-1066), pretreated for 30 min with or without the indicated concentrations of the ERK inhibitor PD-98059 or the p38 inhibitor SB-20358, were incubated for 1 h with DEA-NO. The cells or islets were washed to remove the NO donor compound, PD-98059 or SB-20358 were reapplied and the islets or RINm5F cells were allowed to recover for an additional 5 h. The RINm5F cells or islets were isolated and mitochondrial aconitase activity was determined following the 1-h incubation with DEA-NO or the 5-h recovery incubation as described in Materials and Methods. Results are the average ± sem of three independent experiments. *, P < 0.01 vs. untreated control.
Protein transduction was used to evaluate the role of JNK in the recovery of mitochondrial aconitase activity following NO-mediated damage. For these experiments, dnJNK and wild-type JNK were fused to the protein transduction domain of HIV-TAT (TAT), creating the TAT-dnJNK and TAT-JNK fusion proteins to be used for protein transduction. To confirm that TAT-dnJNK inhibits JNK activity, we evaluated the effects of TAT-dnJNK on IL-1-induced phosphorylation of the JNK substrate c-Jun, as previous studies have shown that IL-1 stimulates JNK activation in islets and RINm5F cells (29). As shown in Fig. 5A, a 30-min incubation of rat islets with IL-1 results in c-Jun phosphorylation, an effect that is prevented by TAT-dnJNK. We next evaluated the effects of dnJNK on the recovery of aconitase activity using RINm5F cells. For this experiment, RINm5F cells were pretreated for 30 min with either TAT, TAT-JNK, or TAT-dnJNK (30 μg/ml), DEA-NO was added and the cells were either cultured for 1 h, or cultured for 1 h, washed to remove the NO donor, and incubated for an additional 5 h in the presence of 30 μg/ml TAT, TAT-JNK, or TAT-dnJNK. The cells were then isolated and mitochondrial aconitase activity was determined. As shown in Fig. 5, RINm5F cells transduced with TAT-dnJNK fail to regain aconitase activity during the 5-h recovery incubation. In contrast, RINm5F cells transduced with the protein transduction domain of HIV-TAT (TAT) or TAT-JNK completely recover mitochondrial aconitase activity. In addition, RINm5F cells transduced with TAT-dnJNK appear to be more sensitive to NO-induced damage, as a 1-h incubation with DEA-NO inhibits aconitase activity by approximately 70% in cells transduced with TAT or TAT-JNK, whereas aconitase activity is reduced by approximately 90% in RINm5F cells transduced with TAT-dnJNK. These findings support a role for JNK in the recovery of mitochondrial aconitase activity of RINm5F cells.
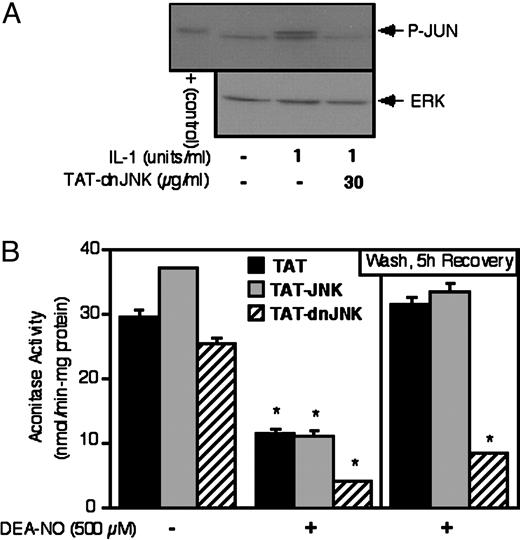
dnJNK prevents the recovery of RINm5F cell aconitase activity following NO-mediated damage. a) RINm5F cells (4 × 105/ 400 μl of complete CMRL-1066) were pretreated for 30 min with 30 μg/ml of TAT-dnJNK, IL-1 (1 U/ml) was added and the cells were cultured for 30 additional min. The cells were harvested and the Western blot analysis was used to examine c-Jun phosphorylation. Phosphorylated c-Jun is shown as a positive control. The blots were stripped and total ERK (p44) levels were determined as a protein loading control. B, RINm5F cells (3 × 106/ 2 ml of complete CMRL-1066), pretreated for 30 min with or without 30 μg/ml of TAT, TAT-JNK, or TAT-dnJNK, were treated for 1 h with DEA-NO. The NO donor was removed by washing, TAT, TAT-JNK, or TAT-dnJNK was reapplied, and the cells were cultured for an additional 5 h. Aconitase activity was determined following the 1-h incubation with DEA-NO, or following the 5-h recovery incubation. Results are the average ± sem of three independent experiments. *, P < 0.01 vs. untreated control.
Discussion
The molecular mechanisms by which cytokines mediate β-cell damage have been examined in detail. These studies have shown that cytokines such as IL-1 and IFN-γ stimulate iNOS expression and NO production by β-cells (5, 13). The production of NO by β-cells leads to an impairment of mitochondrial oxidation of glucose to CO2, reduced Kreb cycle activity (inhibition of aconitase), inhibition of electron transport, and reduced cellular levels of ATP (5, 13). Consistent with a requirement for increased cellular levels of ATP for closure of ATP-inhibited K+-channels and β-cell depolarization in the stimulus secretion coupling mechanism by which glucose induces insulin secretion (30), reduced cellular levels of ATP due to the inhibition of oxidative metabolism is one mechanism by which NO impairs insulin secretion by β-cells. NO, produced following cytokine treatment, or supplied exogenously by donor molecules also induces DNA damage, mediates islet morphological degeneration, and induces islet cell death (7, 13). Although the type of islet cell death (apoptosis or necrosis) has been debated (7, 13, 15), it is clear that NO participates in both the inhibition of islet oxidative metabolism and the loss of islet cell viability in response to cytokine treatment.
Although the mechanisms by which cytokines and NO mediate β-cell damage have been extensively studied, little is known concerning endogenous β-cell defense pathways that confer resistance to or stimulate the recovery from cytokine- or free radical-mediated damage. Comens et al. (21) first showed that β-cells have the ability to recover from the damaging actions of a 15-h incubation with IL-1, if the cytokine is removed by washing and the islets are allowed to incubate in cytokine-free media for extended periods of up to 4 d. We extended these original observations by showing that the addition of iNOS inhibitors to islets cultured for 18–24 h with IL-1, followed by continued culture in the presence of both the iNOS inhibitor and cytokine, results in a time-dependent recovery of islet secretory function that is complete within 8 h (9). The recovery of islet secretory function correlates with a simultaneous recovery of aconitase activity (9). Importantly, the recovery of β-cell function following cytokine-mediated damage appears to be an active response that is sensitive to inhibition by the transcriptional inhibitor actinomycin D (9, 14). NO is one stimulus that activates this recovery response in β-cells, as the inhibitory actions of NO-donor compounds on islet and RINm5F cell aconitase activity can be prevented by actinomycin D (22). Whereas β-cells possess the ability to recover from cytokine-mediated damage, this protective response is limited. Insulin secretion, oxidative metabolism, and protein synthetic capacity are irreversibly inhibited, and islets are commitment to morphological degeneration if they are cultured for 36 h or longer with IL-1 (14).
In the current study, the mechanisms by which NO activates the recovery response in β-cells have been examined using mitochondrial aconitase activity as an index of the recovery response. Importantly, islet recovery of secretory function correlates with the recovery of aconitase activity (9, 14). In this study, we show that inhibition of guanylate cyclase activity prevents the recovery of RINm5F cell and islet aconitase activity. Consistent with a role for guanylate cyclase activation in the recovery response to NO-mediated damage, a 1-h incubation of RINm5F cells with DEA-NO results in the accumulation of high levels of cGMP and that concentrations of the guanylate cyclase inhibitors LY-83583 and ODQ that prevent the recovery of aconitase activity, inhibit cGMP accumulation in response to the NO donor. DEA-NO also stimulates the phosphorylation of ERK, JNK, and p38 in RINm5F cells, and p38 and JNK phosphorylation in response to the NO donor is sensitive to inhibition by ODQ and LY 83583 under conditions in which these guanylate cyclase inhibitors prevent the recovery of aconitase activity. ERK and p38 do not appear to participate in the recovery response because: 1) DEA-NO-induced ERK phosphorylation is not sensitive to inhibition by the guanylate cyclase inhibitors; and 2) selective inhibition of the upstream ERK kinase, MEK, using PD-98059 or p38 using SB-203580 does not modulate the recovery response in RINm5F cells. In contrast, JNK appears to participate in the recovery response as DEA-NO stimulates JNK phosphorylation in a guanylate cyclase-dependent fashion, and transduction of dnJNK into RINm5F cells prevents the recovery of aconitase activity (schematic of proposed pathway is shown in Fig. 6). In addition to inhibiting the recovery response, RINm5F cells transduced with dnJNK appear to be significantly more sensitive to the inhibitory actions of NO on aconitase activity following a 1-h incubation. These findings suggest that NO may participate in the induction of a protective response in β-cells by activating guanylate cyclase, and the downstream activation of JNK. Because new gene expression is required for β-cell recovery from cytokine and NO-mediated damage, it is likely that genes whose transcription is regulated by JNK may participate in β-cell recovery from cytokine- and NO-induced damage.

Proposed mechanism for the activation of islet recovery from NO-mediated damage. Highlighted in bold are the signaling pathways activated by NO in RINm5F cells and rat islets that are believed to participate in islet recovery from NO-induced damage. Also shown are the sites of action of the selective inhibitors used in this study.
The recent success of islet transplantation as a method to treat brittle type 1 diabetics by the Edmonton group has renewed enthusiasm for islet transplantation as a treatment for type 1 diabetes (31). However, this approach suffers from several limitations that include a lifetime of immunosuppression, the limited availability of human islets, and the loss of islet mass following islet isolation. A number of research groups have explored strategies to confer β-cell resistance to inflammation and cytokine- mediated damage as methods to reduce immunosuppression and increase the availability of insulin-producing donor cells. The strategies that have been used to date include the development of insulin-secreting cell lines that express genes conferring cellular resistance to free radical or cytokine-mediated damage. These approaches include the expression of free radical scavenging enzymes such as MnSOD, the expression of dominant negative mutants of specific components of inflammatory signaling pathways, and the expression of antiapoptotic genes (32–38). Although these approaches have been successful in generating cell lines that are protected against cytokine- or free radical-mediated damage, a major challenge of this approach has been the development of cell lines that are responsive to glucose and that release insulin to levels similar in magnitude to the levels produced by primary β-cells. In an effort to overcome the difficulties associated with the use of cell lines, we have focused our efforts on elucidating the natural defense mechanisms that β-cells possess that confer resistance of these cells to cytokine- and free radical-mediated damage. In this report, we have identified one potential protective pathway in β-cells that is activated by NO and that allows β-cells to recover from NO-mediated damage. This pathway includes the activation of guanylate cyclase and the downstream activation of JNK. Currently, we are using array analysis to identify genes whose expression is activated by NO and whose expression is sensitive to inhibitors of guanylate cyclase and JNK in an effort to identify potential recovery genes that are expressed by β-cells. Future studies designed to identify small molecules that activate this recovery pathway in β-cells, and the identification of genes expressed endogenously in β-cells that protect β-cells from cytokine- and free radical-mediated damage are ongoing, as they may provide novel targets and strategies designed to reduce the immunosuppression requirements and loss of islet mass following transplantation. In support of this approach, we have identified hsp 70 as one gene whose expression is stimulated by NO in β-cells, and that when β-cells express hsp 70 following heat shock cytokine signaling is attenuated (22). In addition, peroxisome proliferator-activated receptor-γ agonists troglitazone and 15-deoxy-Δ (12, 14)-prostaglandin J2, have been identified as two small molecules that stimulate hsp 70 expression and attenuate cytokine signaling in β-cells (29, 39).
Acknowledgments
We thank Colleen Kelly Bratcher for expert technical assistance.
This work was supported by NIH Grants DK-52194 and AI-44458.
Abbreviations
- DEA-NO
Sodium (Z)-1(N,N-diethylamino) diazen-1-ium-1,2-diolate
- dn
dominant negative
- IBMX
3-isobutyl-1-methylxanthine
- iNOS
inducible NO synthase
- IFN-γ
interferon-γ
- JNK
c-Jun N-terminal kinase
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- SDS
sodium dodecyl sulfate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}