





Abstract
There is a strong and ever-growing body of evidence regarding the use of pharmacogenomics to inform cardiovascular pharmacology. However, there is no common position taken by international cardiovascular societies to unite diverse availability, interpretation, and application of such data, nor is there recognition of the challenges of variation in clinical practice between countries within Europe. Aside from the considerable barriers to implementing pharmacogenomic testing and the complexities of clinically actioning results, there are differences in the availability of resources and expertise internationally within Europe. Diverse legal and ethical approaches to genomic testing and clinical therapeutic application also require serious thought. As direct-to-consumer genomic testing becomes more common, it can be anticipated that data may be brought in by patients themselves, which will require critical assessment by the clinical cardiovascular prescriber. In a modern, pluralistic and multi-ethnic Europe, self-identified race/ethnicity may not be concordant with genetically detected ancestry and thus may not accurately convey polymorphism prevalence. Given the broad relevance of pharmacogenomics to areas, such as thrombosis and coagulation, interventional cardiology, heart failure, arrhythmias, clinical trials, and policy/regulatory activity within cardiovascular medicine, as well as to genomic and pharmacology subspecialists, this position statement attempts to address these issues at a wide-ranging level.
Introduction
The completion of the human genome project in 2003 heralded an age of genomic promise.1,2 Advances in genomic knowledge and sequencing technology have provided vital tools for the implementation of personalized medicine; i.e. the ability to use genetic information to select the right pharmacological agent, to be used at the right dose, for the right person, and at the right time, thereby maximizing the efficacy of the intervention and minimizing adverse drug reactions (ADR).2
Pharmacogenomics (PGx) describes the role of genomic variation in drug response, which can relate to both pharmacokinetic and pharmacodynamic properties. Pharmacokinetics constitutes the effect of the body on the drug, categorized into effects on absorption, distribution, metabolism, and excretion of the drug. Pharmacodynamics reflects the effect of the drug on the body. Both pharmacokinetics and pharmacodynamics influence efficacy, effectiveness, and toxicity. Many drugs are metabolized in the liver by cytochrome P450 (CYP) enzymes. These enzymes are encoded by various genes named according to their family (e.g. ‘2’) and subfamily (e.g. ‘C’), based on the amino acid structure. A full code indicates a specific gene (e.g. CYP2C9). Other drug metabolizing enzymes, receptors, and transporters also play an important role in drug response. Indeed, whole-genome approaches, including sequencing, are likely to identify new pharmacogenes, including in therapeutic target genes, which determine drug efficacy and/or safety. It is inevitable that the number of pharmacogenes with evidence of clinical utility will continue to increase, which makes pharmacogenomics a dynamic area.
The cost of genome sequencing has decreased exponentially with the emergence of next-generation and third-generation sequencing technology, from 2.7 billion USD in 2003 to sequence the first human genome (which took 13 years) to <1000 USD to sequence a whole genome in 2019.3–5 In order to implement personalized medicine, three main tools to target discovery in PGx studies are used: genome-wide association studies (GWASs), candidate gene analyses, and next-generation sequencing (high throughput parallel sequencing).6 The first is an agnostic approach that locates a genome-wide signal to a locus, while the candidate gene approach starts from a gene hypothesized to be significant. This will often be a gene that encodes protein targets of the drug, or proteins that are involved in drug pharmacokinetics. When genetic loci or single-nucleotide polymorphisms (SNPs) of interest are identified, this should be replicated and may be followed by functional validation studies, although this is not always necessary, depending on the predictive accuracy of the variant. Evidence of clinical utility needs to be gathered which depends on the clinical phenotype and the gene-drug pair. The present rate of big data-based discovery, and the scale and complexity of genomic information interpretation and translation into useful therapeutic interventions present considerable challenges. The expectations of gold-standard evidence from randomized controlled trials (RCTs) for PGx may not be realistic (or indeed necessary) in PGx, as each trial would look at one or a few gene variant-drug pairs.7,8
Though PGx guided therapy has been endorsed by both regulators and international consortia, there is discordance in recommendations, and PGx has remained an area of specialist interest.9
Given the rapid advances which are occurring, there is a need for professional bodies and health systems to support prescribers in understanding, interpreting, and implementing evidence-based PGx in cardiovascular medicine. This is particularly timely as many prescribers begin to find themselves increasingly confronted with patients able to access PGx information through direct-to-consumer genetic testing.10 Moreover, prescribers may not have access to confirmatory testing within their place of work. Given the broad relevance of PGx to areas such as thrombosis and coagulation, interventional cardiology, heart failure, arrhythmias, clinical trials, and policy/regulatory activity within cardiovascular medicine, as well as to genomic and pharmacology subspecialists, this position statement attempts to address these issues at a wide-ranging level. The following sections address evidence for specific drug-gene pairs, as well as broad genomic approaches in cardiovascular medicine, and the potential role of genomics in clinical trials. The focus is on translational aspects most likely to be encountered by non-genomically trained practitioners in day-to-day practice.
Anticoagulants
Warfarin and other vitamin K antagonists
The coumarin derivatives, warfarin, acenocoumarol, and phenprocoumon, are used to prevent or treat thromboembolism. There are differences across the World and between European countries regarding which coumarin oral anticoagulant is preferred, but collectively they remain commonly prescribed, despite the rapidly increasing use of direct oral anticoagulants (DOACs).11,12 Coumarins inhibit vitamin K epoxide reductase complex subunit 1 (VKORC1), leading to hypofunctional clotting factors II, VII, IX, and X.13 Coumarin derivatives are all administered as a racemate. For warfarin, the S-warfarin enantiomer is preferentially metabolized by CYP2C9 and is ∼3–5x more potent than R-warfarin.14–16 Both acenocoumarol enantiomers are principally metabolized by CYP2C9 and, while S-acenocoumarol is more active, its short elimination half-life (1.8 h) means the majority of anticoagulant effect is through R-acenocoumarol.13 For phenprocoumon, both CYP2C9 and CYP3A4 are involved in its metabolism.17 Coumarin dose requirements to maintain an international normalized ratio (INR) between 2.0 and 3.0 are highly variable between individuals. For example, warfarin stable dose (WSD) ranges at least 25-fold (0.6–15.5 mg/day), and real-world registry data of patients anticoagulated for atrial fibrillation demonstrated a mean time in the therapeutic INR range (TTR) of 65% (±20%).18 This high inter-individual variation and narrow therapeutic index increase susceptibility to adverse events. Notably, 6–7% of patients prescribed warfarin were hospitalized due to bleeding over a mean follow-up of 425 days, with supra-therapeutic INRs increasing bleeding risk.19–21 Decreased TTR is also a predictor for increased ischaemic stroke, other thromboembolic events, major bleeding, and mortality.22,23
For warfarin, approximately ∼55–60% of variation in WSD can be principally explained by genetic variation in VKORC1 (∼25%) and CYP2C9 (∼15%), supplemented by CYP4F2*3 (∼1–7%) and clinical factors (e.g. age, body mass index, smoking, interacting drugs, collectively <20%)24–26 (Table 1).
Clinical trials testing genotype-guided warfarin dosing
| Gene | Variant(s) | Functional effect | Clinical effect |
|---|---|---|---|
| VKORC1 | −1639G>A (rs9923231) | Reduced VKORC1 transcription | Reduced warfarin requirments41 |
| CYP2C9 | *2, *3, *5, *6, *8, *11 | Decreased CYP2C9 metabolic activity | Reduced warfarin requirements42,43 |
| CYP4F2 | *3 (V433M) | Decreased hepatic CYP4F2 levels and reduced vitamin K hydroxylation | Higher warfarin requirments44 |
| Gene | Variant(s) | Functional effect | Clinical effect |
|---|---|---|---|
| VKORC1 | −1639G>A (rs9923231) | Reduced VKORC1 transcription | Reduced warfarin requirments41 |
| CYP2C9 | *2, *3, *5, *6, *8, *11 | Decreased CYP2C9 metabolic activity | Reduced warfarin requirements42,43 |
| CYP4F2 | *3 (V433M) | Decreased hepatic CYP4F2 levels and reduced vitamin K hydroxylation | Higher warfarin requirments44 |
Clinical trials testing genotype-guided warfarin dosing
| Gene | Variant(s) | Functional effect | Clinical effect |
|---|---|---|---|
| VKORC1 | −1639G>A (rs9923231) | Reduced VKORC1 transcription | Reduced warfarin requirments41 |
| CYP2C9 | *2, *3, *5, *6, *8, *11 | Decreased CYP2C9 metabolic activity | Reduced warfarin requirements42,43 |
| CYP4F2 | *3 (V433M) | Decreased hepatic CYP4F2 levels and reduced vitamin K hydroxylation | Higher warfarin requirments44 |
| Gene | Variant(s) | Functional effect | Clinical effect |
|---|---|---|---|
| VKORC1 | −1639G>A (rs9923231) | Reduced VKORC1 transcription | Reduced warfarin requirments41 |
| CYP2C9 | *2, *3, *5, *6, *8, *11 | Decreased CYP2C9 metabolic activity | Reduced warfarin requirements42,43 |
| CYP4F2 | *3 (V433M) | Decreased hepatic CYP4F2 levels and reduced vitamin K hydroxylation | Higher warfarin requirments44 |
At least 14 clinical trials testing genotype-guided warfarin dosing have been conducted, employing various algorithms that incorporate genetic (VKORC1, CYP2C9 ± CYP4F2) and clinical covariates.25,27,28 The largest European RCT of warfarin PGx was conducted by the EU-PACT collaboration and recruited patients starting warfarin for atrial fibrillation or venous thromboembolism (VTE).29 EU-PACT compared genotype-guided warfarin dosing on days 1–5 in 227 patients to standard warfarin loading in 228 patients, with all subsequent dosing in both arms according to routine practice, and found that the primary endpoint of 12-week TTR was significantly higher in the genotype-guided arm (67.4% vs. 60.3%, P < 0.001).29 There was no significant difference in bleeding or thromboembolic events. In EU-PACT, 99% of participants were Caucasian, and point-of-care (POC) genotyping was utilized.29 A subsequent UK-based real-world implementation study with POC genotyping similarly found genotype-guided dosing increased warfarin TTR by 7%.30 A recent meta-analysis incorporating EU-PACT data shows that genotype-guided warfarin dosing compares favourably to DOACs in cluster rank plots considering both thromboembolism and bleeding.31
In contrast, the US COAG trial did not find any significant difference between genotype-guided and clinically guided dosing algorithms (4-week TTR 45.2% vs. 45.4%, P = 0.91).32 An important distinction between the two trials is that 27% of patients recruited to COAG were African-American, but only the CYP2C9 alleles common in Caucasians, *2 and *3, were assessed. Genotype-guided African-American patients in COAG fared less well than those clinically dosed (4-week TTR 35.2% vs. 43.5%, P = 0.01), highlighting the importance of patient ethnicity in considering CYP2C9 variants.32 Another difference between the trials is that the US COAG trial comparator arm used a clinical dosing algorithm, whereas EU-PACT used standard care. However, clinical dosing algorithms have not been validated themselves vs. usual care and the comparator arm TTR was lower in COAG than in EU-PACT.
Genetics informatics trial was the largest and most recent RCT; it recruited patients undergoing elective hip or knee arthroplasty treated with perioperative warfarin and randomized them to genotype- (n = 808) or clinically guided (n = 789) dosing on days 1–11. Patients were genotyped for VKORC1 −1639G>A, CYP2C9*2, CYP2C9*3, and CYP4F2 V433M. The primary composite endpoint of INR ≥ 4, major bleeding, death (all within 30 days) plus VTE within 60 days, occurred in 10.8% and 14.7% of patients in the genotype and clinically guided groups, respectively (P = 0.02). This result was driven mainly by a decrease in INR ≥ 4 (P = 0.04) and borderline reduction in major bleeding (P = 0.06).27 Given the continued high real-world use of warfarin, these results are considered clinically relevant.
To aid implementation, there is supportive evidence for the cost-effectiveness of genotype-guided warfarin dosing, and multigene-drug testing will likely be even more advantageous.33,34 Clinical guidelines for genotype-guided warfarin dosing have been developed by both the Dutch Pharmacogenetics Working Group (DPWG) and Clinical Pharmacogenetics Implementation Consortium (CPIC).35,36
The EU-PACT collaboration also conducted RCTs of acenocoumarol and phenprocoumon PGx.37 Due to low enrolment, they were combined for analysis. Though the primary outcome of 12-week TTR in the 484 eligible participants did not differ between the genotype-guided and clinically guided dosing arms (61.6% vs. 60.2%, P = 0.52), follow-up analysis demonstrated that the acenocoumarol dosing algorithm, designed for a Dutch population, overestimated dose in Greek ancestry patients.37–39
On balance, we advocate genotype-guided dosing when commencing warfarin. To optimize the effectiveness of a genotype-informed strategy, genetic results should be available on the first day of warfarin dosing. This may require the use of point-of-care testing, or pre-emptive genotyping.
Although patient ethnicity is important in warfarin PGx, self-reported ethnicity may not always adequately represent genetic ancestry, for instance in admixed individuals. Genetic characterization to facilitate appropriate use of ethnicity-specific warfarin algorithms may be preferable, but arguably difficult to achieve in clinical practice.40 Therefore, rather than genotype for specific alleles in patients from different ethnic backgrounds, a better approach would be to genotype for all the relevant CYP2C9, VKORC1, and CYP4F2 alleles, irrespective of ethnicity/ancestry, and determine dose requirements using a ‘universal’ algorithm, which will require development.
Key clinical positions:
Prospective genotyping prior to warfarin initiation is advised where possible.
If genotypic information is already available, it should be used to guide warfarin initiation dosing.
Antiplatelets
Antiplatelet therapy is the backbone of treatment for atherothrombotic diseases, such as coronary artery disease and stroke. In acute coronary syndromes and patients undergoing percutaneous coronary intervention, dual antiplatelet therapy with aspirin and a P2Y12 receptor antagonist is usually recommended.45,46 P2Y12 receptor antagonists include clopidogrel, prasugrel, and ticagrelor. The more potent P2Y12 receptor antagonists, prasugrel, and ticagrelor, are usually preferred over clopidogrel in high-risk cases on the basis of improved cardiovascular outcomes, albeit with an increased bleeding risk.47 In stroke, single-antiplatelet therapy with clopidogrel alone is usually preferred for long-term secondary prevention.48 However, the effectiveness of antiplatelet therapy is limited by variability in patient response; particularly to clopidogrel, partly attributed to genetic variation.
Clopidogrel is a prodrug, transformed to its active metabolite in two sequential CYP2C19 dependent steps. Up to a third of patients have reduced enzymatic activity secondary to a loss of function variant in CYP2C19 (e.g. *2 or *3).49 This is associated with high on-treatment platelet reactivity and an increased risk of ischaemic events.50,51
Knowledge of an individual’s genetic profile can allow personalized antiplatelet strategies in order to optimize the balance of beneficial to adverse outcomes. However, genotypic variation does not always translate into adverse clinical outcomes as, unlike platelet function testing, genotyping does not provide a direct measure of response to therapy or an assessment of non-genetic factors on platelet function. Yet platelet function testing is limited by the need to perform testing while already on treatment and lacks standard reference values and thresholds. Given the large number of patients prescribed antiplatelet agents, use of PGx to personalize treatment could have a significant effect at a population level. A PGx polygenic risk score has also been proposed as an effective risk stratification tool, and merits further investigation in a prospective clinical setting.52
Clinical trials have used both phenotype-guided (platelet function testing while on treatment) and genotype-guided (CYP2C19 status assessed before/during treatment) approaches. Meta-analysis has shown phenotype-guided approaches in patients presenting with acute coronary syndrome or with coronary artery disease undergoing percutaneous coronary intervention are not superior to standard care.53 This is unsurprising given the limitations discussed. In contrast, meta-analysis of six randomized controlled trials using genotypic-based treatment, with 3764 patients undergoing percutaneous coronary intervention or with acute coronary syndromes, has shown a significant reduction in net adverse clinical events [hazard ratio (HR) 0.65; 95% confidence interval (CI) 0.45–0.95; P = 0.03], including reduced major adverse cardiovascular events (HR 0.59; 95% CI 0.43–0.82; P < 0.01) and reduced bleeding events (HR 0.75; 95% CI 0.61–0.93; P < 0.01).54 This meta-analysis pooled studies that use different strategies and different patient populations, including those with stable and acute coronary syndromes.
Notably, the large POPular Genetics RCT found that genotype-guided therapy in patients undergoing primary PCI was non-inferior to standard treatment with prasugrel or ticagrelor in terms of a combined primary outcome of thrombotic and bleeding events, and risk of bleeding was reduced.55 The recent TAILOR-PCI trial randomized 5302 PCI patients, 1849 of whom carried loss of function CYP2C19 alleles, to genotype-guided antiplatelet therapy vs. conventional therapy with clopidogrel.56 The composite cardiovascular primary endpoint was 4.0% in the genotype-guided and 5.9% in the non-genotype-guided cohort at 1-year follow-up (HR 0.66; 95% CI 0.43–1.02; P = 0.06).56 Unfortunately, the study was underpowered due to lower event rates than expected. However, a post hoc analysis found that the primary outcome was significantly reduced during the first 3 months after PCI (HR 0.21; 95% CI 0.08–0.54; P = 0.001), suggesting higher modifiable risk in the months immediately following stenting.56 This includes the highest risk period for early in-stent thrombosis, which is the first 30 days.57
This evidence base suggests that the genotype-guided approach could be superior to standard care in terms of both cardiovascular outcomes and reduced bleeding events, particularly in the months immediately following PCI. The availability of rapid diagnostic genetic tests means such strategies are a viable option in routine clinical practice. In other settings, such as stroke, there is still a lack of prospective large-scale RCT data. However, similar benefits might be anticipated, a hypothesis supported by systematic review and meta-analysis data.58,59
Clopidogrel PGx profiling for CYP2C19 remains only recommended in specific high-risk situations, such as patients with acute coronary syndrome undergoing percutaneous coronary intervention, or presenting with recurrent adverse events, and only if results are likely to change the treatment strategy. If patients are considered intermediate or poor metabolizers of clopidogrel, alternative antiplatelet agents are recommended.60 More large-scale trials in patients with coronary artery disease (e.g. GUARANTEE study) are ongoing and will inform this recommendation further. Additional prospective data from large-scale trials for other indications, such as stroke, are awaited before clear recommendations can be made (e.g. PLATELET study). Given the disease prevalence, even a small improvement with PGx use may translate to meaningful population-level health gains.
Key clinical positions:
Clopidogrel should be avoided in patients who are known to be intermediate or poor metabolisers from existing genotypic information.
Genotyping high-risk cardiovascular patients (either high risk of thrombosis or bleed) prior to prescribing clopidogrel should be considered where possible, particularly to prevent post-stent thrombosis.
Statins
Statins are the most commonly prescribed lipid-lowering medication for the treatment and prevention of cardiovascular disease (CVD).61,62 Meta-analyses show a consistent relationship between the extent of low-density lipoprotein cholesterol (LDL-C) reduction and CVD risk reduction, irrespective of statin choice, dose, or baseline risk.63–65 Statins are well-tolerated in most individuals but can rarely cause severe hepatotoxicity, and are associated with the development of diabetes mellitus.66 Statin-associated muscle symptoms (SAMS) are the most commonly reported side-effect and are a common reason for treatment discontinuation.67,68 SAMS range from mild myalgia without an elevation in creatine kinase to life-threatening rhabdomyolysis or autoimmune-necrotizing myositis.69,70 The underlying mechanism of muscle injury is increased systemic statin exposure with intracellular skeletal myocyte entry and disruption of muscle function.71
Statin disposition is dependent on multiple enzymes and transporters, with their relative importance varying between drugs. Solute carrier anion transporter family 1B1 (SLCO1B1) encodes organic anion transporting polypeptide 1B1 (OATP1B1) which is central to the hepatic uptake and consequent elimination of statins. A GWAS in patients with simvastatin-induced myopathy identified an increased risk of myopathy in heterozygous and particularly homozygous carriers of the reduced function SLCO1B1*5 variant, which significantly increases plasma concentrations of all statins, except fluvastatin (Table 2).72 In addition to this gene dose trend, a simvastatin dose trend was observed with the risk of myopathy increasing by 2.6 and 4.3 per copy of SLCO1B1*5 in patients on simvastatin 40 and 80 mg daily, respectively.72
Clinical trials testing genotype-guided warfarin dosing
| Statin | Increase in area under the curve in SLCO1B1*5 (loss-of-function) homozygotes87–91 |
|---|---|
| Simvastatin | 221% |
| Lovastatin | 186% |
| Pitavastatin | 208% |
| Atorvastatin | 145% |
| Pravastatin | 91% |
| Rosuvastatin | 65% |
| Fluvastatin | 19%, non-significant |
| Statin | Increase in area under the curve in SLCO1B1*5 (loss-of-function) homozygotes87–91 |
|---|---|
| Simvastatin | 221% |
| Lovastatin | 186% |
| Pitavastatin | 208% |
| Atorvastatin | 145% |
| Pravastatin | 91% |
| Rosuvastatin | 65% |
| Fluvastatin | 19%, non-significant |
Clinical trials testing genotype-guided warfarin dosing
| Statin | Increase in area under the curve in SLCO1B1*5 (loss-of-function) homozygotes87–91 |
|---|---|
| Simvastatin | 221% |
| Lovastatin | 186% |
| Pitavastatin | 208% |
| Atorvastatin | 145% |
| Pravastatin | 91% |
| Rosuvastatin | 65% |
| Fluvastatin | 19%, non-significant |
| Statin | Increase in area under the curve in SLCO1B1*5 (loss-of-function) homozygotes87–91 |
|---|---|
| Simvastatin | 221% |
| Lovastatin | 186% |
| Pitavastatin | 208% |
| Atorvastatin | 145% |
| Pravastatin | 91% |
| Rosuvastatin | 65% |
| Fluvastatin | 19%, non-significant |
The clinical association between simvastatin-induced myopathy and SLCO1B1*5 has been confirmed in a recent meta-analysis, which failed to identify any other genetic loci.73 There is ongoing uncertainty over the effect of SLCO1B1*5 on other statins as, in the example of atorvastatin, alternative anion transporters are partially responsible for hepatic uptake and its intrinsic myotoxicity may be less than compared to simvastatin.74,75 Interestingly, recent work has associated SLCO1B1*5 with rosuvastatin-induced myotoxicity in Han Chinese patients, despite it not previously being associated with myalgia in patients of European descent.76–78 This may be secondary to increased rosuvastatin exposure in individuals of Asian ancestry, which is partially explained by polymorphisms in ABCG2, an efflux transporter encoding breast cancer resistance protein (BCRP).79
Other factors, such as drug-drug interactions, exercise, neuromuscular disorders, mitochondrial impairment, mevalonate pathway perturbation, immune-mediated toxicity, muscle transcriptomics, and vitamin D deficiency can all contribute to myotoxicity.71 It is likely that SAMS often result from a combination of these and genetic factors.
There is little prospective data available to inform clinical decision making. Peyser et al.80 randomized 159 patients with prior statin myalgia to receive SLCO1B1 genotype informed statin therapy (GIST) vs. usual care. Rosuvastatin, pravastatin, or fluvastatin were recommended for SLCO1B1*5 carriers, whereas non-carriers were recommended to try any statin that they had not tried in the past. Over an 8-month follow-up, those who were randomized to GIST showed increased statin re-initiation and reduced LDL-C, but without an increase in self-reported medication adherence.80 Vassy et al.81 recently completed an RCT of 408 patients treated in primary care settings, to assess potential unintended harms of PGx testing. The findings were reassuring, showing that knowledge of SLCO1B1 genotype did not deter from appropriate initiation of statins in accordance with guidance, and did not result in inferior LDL-C at the 1 year endpoint.81
The minor allele frequency of rs4149056 (SLCO1B1*5) is approximately 1%, 8%, and 16% in African, Asian, and European populations, respectively,82 and so a substantial proportion of the population may be at risk. Both CPIC and DPWG, as well as the European summary of product characteristics, recommend that individuals carrying SLCO1B1*5 avoid high-dose simvastatin, but the suggested clinical approach differs.83,84 The CPIC guidelines recommend a lower simvastatin starting dose or an alternative statin in heterozygous carriers, alongside consideration of routine creatine kinase surveillance in those who are homozygous.85 In contrast, the DPWG recommends that homozygotes avoid simvastatin entirely.86 The DPWG guideline also recommends avoiding atorvastatin for individuals with other clinical risk factors for SAMS.86 The EU summary of product characteristic for simvastatin states ‘Where available, genotyping for the presence of the C allele should be considered as part of the benefit-risk assessment prior to prescribing 80 mg simvastatin for individual patients and high doses avoided in those found to carry the CC genotype.’
As the current evidence base is limited to specific drug-gene pairs and largely retrospective, PGx testing to assess the risk of SAMS has not yet transitioned into routine clinical care, and PGx testing while on statin therapy is not usually recommended.
Key clinical positions:
Avoid high-dose simvastatin (80 mg) and consider an alternative statin of equivalent LDL lowering efficacy in patients known to be homozygous for the SLCO1B1*5 reduced function variant.
Beta-blockers
Several beta-blockers, including metoprolol, carvedilol, and propranolol are metabolized by CYP2D6, a phase I drug-metabolizing enzyme implicated in the metabolism of 20–25% of clinically prescribed drugs,92–94 and encoded by the highly polymorphic gene, CYP2D6. Of these, metoprolol is most highly dependent on CYP2D6 for its elimination, with 70–80% of an oral dose undergoing CYP2D6-mediated biotransformation92 (Table 3). Other beta-blockers, such as atenolol and bisoprolol, undergo either no or negligible CYP2D6 metabolism.92,95
Clinical trials testing genotype-guided warfarin dosing
| CYP2D6 metabolizer phenotype | Fold difference in metoprolol exposure (compared with normal metabolizer)109 |
|---|---|
| Ultra-rapid | ∼0.4 |
| Intermediate | ∼2.5 |
| Poor | ∼4.9 |
| CYP2D6 metabolizer phenotype | Fold difference in metoprolol exposure (compared with normal metabolizer)109 |
|---|---|
| Ultra-rapid | ∼0.4 |
| Intermediate | ∼2.5 |
| Poor | ∼4.9 |
Clinical trials testing genotype-guided warfarin dosing
| CYP2D6 metabolizer phenotype | Fold difference in metoprolol exposure (compared with normal metabolizer)109 |
|---|---|
| Ultra-rapid | ∼0.4 |
| Intermediate | ∼2.5 |
| Poor | ∼4.9 |
| CYP2D6 metabolizer phenotype | Fold difference in metoprolol exposure (compared with normal metabolizer)109 |
|---|---|
| Ultra-rapid | ∼0.4 |
| Intermediate | ∼2.5 |
| Poor | ∼4.9 |
Several studies have investigated the clinical effects of CYP2D6 in patients taking metoprolol, mostly for heart failure (HF) or hypertension (HTN). The tolerated maintenance metoprolol dose in CYP2D6 poor metabolizers (PMs) relative to extensive (normal) metabolizers (EMs) is lower in some, but not all studies.96–101 A decreased heart rate (HR) has been frequently reported in intermediate metabolizers (IMs) and particularly PMs relative to EMs, and an increased incidence of (mostly asymptomatic) bradycardia (HR < 60 b.p.m.) has been reported in CYP2D6 PMs and occasionally IMs in some but not all studies.96,98,99,101–104 No association between CYP2D6 and systolic blood pressure in metoprolol users has been identified.96,98,101,103 However, at least three studies, including the two largest to date have linked CYP2D6 PM status to a ∼5 mmHg reduction in diastolic blood pressure relative to EMs, although this has not been replicated in all studies.96,98,99,101–103,105 Bar an early small case–control study, clinically apparent adverse events have not been associated with CYP2D6 PMs or IMs.98,99,101,102,105–107 One limitation of some of these studies is a reliance on a single, albeit common, loss-of-function variant, CYP2D6*4 (rs3892097) for assigning CYP2D6 predicted function.96,97,101
Due to the lower prevalence of CYP2D6 ultra-rapid metabolizers (UMs) compared to IM/PMs, less research has specifically focused on this group, although HR and blood pressure do not appear to be notably different from EMs with metoprolol.102,103 One study of 187 patients post myocardial infarction related CYP2D6 duplications to ventricular rhythm disturbances, although this group also had a higher proportion of ST-elevation myocardial infarction.104
The US Food & Drug Administration (FDA)-approved metoprolol product label states that CYP2D6 (genotype)-dependent metabolism seems to have little influence on the safety or tolerability of metoprolol.108 Nonetheless, the label also notes that strong CYP2D6 drug inhibitors are expected to mimic CYP2D6 PMs and recommends caution with co-administration of potent CYP2D6 inhibitors.108 This is one example of a trend in drug labelling that urges more (appropriate) caution for drug-drug interactions than for genetic polymorphisms with the same effect. The DPWG guidance for metoprolol-CYP2D6 recommends slower dose titration and reduced maximal doses in IMs and PMs in the event of symptomatic bradycardia or when a gradual reduction in heart rate is indicated, and suggests increasing metoprolol dose beyond the usual maximum or an alternative beta-blocker in UMs if effectiveness remains insufficient.35
On balance, if patients known to be a CYP2D6 PM or UM are started on a beta-blocker, avoiding metoprolol seems prudent. Similarly, CYP2D6 genotyping in patients that experience an ADR on metoprolol appears justifiable to minimize risk of future ADRs from other CYP2D6-metabolized drugs that the patient might subsequently be prescribed, and to refine the metoprolol-CYP2D6 evidence base.
Key clinical positions:
If genotype is known metoprolol should be avoided in CYP2D6 poor and ultrarapid metabolizers, and an appropriate substitute made (e.g. bisoprolol).
Prescribers should be aware that co-administration of potent CYP2D6 inhibiting drugs can lead to variable phenoconversion, the extent of which will be influenced by the underlying CYP2D6 genotype.
Hypertension
Primary hypertension (HTN) is highly prevalent in Europe and a leading cause of morbidity and mortality despite the existence of many pharmacologic therapies and lifestyle modification recommendations.110,111 There are now >1000 genetic loci linked with blood pressure regulation through large GWASs and meta-analyses, which supports a complex polygenic mechanism of partial heritability, with most common risk alleles exerting a small effect on blood pressure.112,113
Interestingly, one of the largest multi-ethnic meta-analysis of blood pressure traits to-date included a phenome-wide association study which demonstrated uniform distribution of clinical risk based on identified genetic loci across ethnic groups, including Caucasians, Hispanics, and persons of African descent.113 Though PGx trials have not provided an evidence base to support genotype-guided HTN pharmacotherapy, therapeutic recommendations remain stratified by self-reported ethnicity, with ‘black’ Europeans recommended a different treatment regime (extrapolated from African American population data from the USA).110
There is some evidence from the GenHAT study (a sub-study of ALLHAT), the largest RCT to explore gene panel risk prediction in stratified anti-hypertensive (anti-HTN) therapies that genetic panels can predict response to different anti-HTN therapies. However, this adequately powered study (N > 39 000) did not demonstrate clinically significant benefit to genetically stratified therapeutic choice.114 It also failed to demonstrate different clinical outcomes for use of the studied anti-HTN therapeutics in Caucasians or participants of African descent. This trial did not include beta-blockers, which are less routinely used to treat HTN.
Thus, current evidence does not support the routine use of any genomic targeted treatment or change to therapy for any variant reported for essential HTN (beta-blockers discussed separately above). There remains a need for further prospective clinical studies to either validate or discard the current practice of using self-reported ethnicity (black or white) to guide initial therapeutic choice. Prior assessment of evidence has concluded that this may be dubious.115
Key clinical positions:
No clinical action advised based on current evidence
Arrhythmias
The main arrhythmia-predisposing condition related to pharmacotherapy and amenable to genetically guided intervention is long QT.116
A prolonged QT interval is a well-known risk factor for ventricular arrhythmias [including torsade de pointes (TdP), a rare and unstable polymorphic ventricular tachycardia] and sudden cardiac death (SCD), and can be congenital or acquired.117–121 Acquired QT prolongation can be caused by cardiac disease, such as coronary heart disease and HF, but it is commonly caused by certain drugs [drug-induced long QT syndrome (diLQTS)].120,122 QT interval prolongation is one of the leading causes of therapeutic relabelling or withdrawal from the market. To date, >200 drugs are listed on CredibleMeds.org (a curated database maintained by AZCERT) as associated with QT prolongation and/or TdP.123
Susceptibility to acquired QT interval prolongation can be influenced by genetic variation; the heritability of QT interval duration in the general population (excluding congenital LQTS patients) is estimated to be ∼35%, and first-degree relatives of patients with congenital LQTS have a higher risk of drug-induced QT prolongation than non-related individuals.124–127 A large number of genes associated with QT interval duration have been identified by GWAS.128–130 The gene with the strongest signal related to QT interval duration is the nitric oxide synthase 1 adaptor protein gene (NOS1AP), located on chromosome 1.128–134 This gene also influences impulse propagation.135 Other key findings from GWAS included polymorphisms within genes known to be mutated in congenital LQTS, genes associated with intracellular calcium handling, as well as genes previously not known to influence cardiac repolarization.124,129,130 A polygenic risk score comprised of 61 variants previously associated with baseline QT interval at genome-wide level was also associated with drug-induced QT prolongation, and was a significant predictor of drug-induced TdP.136,137
Several other exploratory studies have sought to identify the underlying genetic architecture predisposing to diLQTS.136,138–140 One candidate gene study evaluated the genetic predisposition to diLQTS among 176 cases and 1044 controls and identified a nonsynonymous variant in KCNE1 (rs1805128, D85N) that conferred risk of diLQTS (odds ratio = 9.0, 95% CI 3.5–22.9).138 In an exome sequencing study of 65 diLQTS cases and 148 controls, a significant excess of rare variants in KCNE1 and ACN9 were identified. Moreover, 37% of diLQTS cases were also carriers of a rare variant in potassium channel genes compared with 21% of controls.140 These studies highlight the importance of individualized treatment and risk stratification in the setting of ADRs, but also the need for further work to translate this data to the clinical realm with thorough validation studies.
The most common mechanism by which drugs lead to acquired LQTS is blockade of the rapid component of the delayed rectifier potassium current, Ikr, encoded by KCNH2, also referred to as human ether-a-go-go-related gene.141 The risk of TdP seems to increase with higher concentrations of Ikr blocking drugs. Thus, genetic variants that alter drug metabolism and result in higher drug concentrations can predispose to diLQTS. The reduced repolarization reserve hypothesis proposes that patients remain asymptomatic until multiple hits (e.g. hypokalaemia, bradycardia, and/or exposure to a drug) unmask an extreme drug response phenotype (i.e. QT prolongation) that can lead to the development of fatal arrhythmias.117
Future prospects
Potentially dangerous effects of drugs may be masked when administered to a large number of patients. The risk may become evident only when genetic and environmental risk factors combine; for example, when an Ikr blocking drug is given to an individual with a silent mutation in one of the congenital LQTS genes in the setting of hypokalaemia.
Therefore, therapeutics that are both safe and efficacious for the majority of the population may be withdrawn due to unacceptable risk in a small minority of patients. With the current emphasis on avoiding QT prolongation in drug development, it is likely that drugs that would never result in TdP in a selected population are being abandoned due to perceived risk. The challenge lies in identifying which patients, in which settings, and with which drugs or combination of drugs (and at what doses) will develop diLQTS and avoiding the combination of factors that promote TdP.
Key clinical positions:
Medications known to prolong the QT interval should be avoided in those known to harbour long-QT-associated genetic variants.
Prospective genotyping in absence of phenotype or family history is not advised.
Chemotherapy-induced cardiomyopathy
Cardiomyopathy and HF resultant from chemotherapeutic myocyte toxicity [chemotherapy-induced cardiomyopathy (CCM)] is a feared side effect of several common chemotherapeutic agents, most prominently: doxorubicin, trastuzumab, paclitaxel, and 5-fluorouracil.142 The most studied amongst these is doxorubicin, with anthracycline-induced cardiotoxicity the subject of the sole existing PGx recommendation: The Canadian Pharmacogenomics Network for Drug Safety (CPNDS) recommends genetic testing for 3 SNPs (RARG rs2229774, SLC28A3 rs7853758, UGT1A6*4 rs17863783) prior to initiating doxorubicin or daunorubicin in children, endorsed by a moderate level B evidence-based support.143 Though these variants were each replicated in separate cohorts within a single published study, they have not yet been validated by further independent research. This recommendation was met with some consternation, as these SNPs lack functional validation as well as mechanistic plausibility (two are not expressed in cardiac tissue and don’t alter protein coding), and has not been incorporated into other guidelines or mainstream practice.144 A recent study from the USA found that a polymorphism present in both African and European ancestry populations was associated with higher risk of cardiomyopathy in childhood cancer survivors, with disproportionately higher risk to those of African ancestry (5.43- vs. 1.31-fold); however, this population included exposure to radiotherapy and anthracyclines.145
While PGx studies have not yet evolved to clinical utility in risk prediction within mainstream care, recent advances have localized additional risk to those cancer patients with TTN truncating variants (TTNtvs). The massive titin protein, coded for by TTN, is integral to sarcomere function, and implicated in familial forms of DCM.146,147 These genetic changes are hypothesized to create a stress cardiomyopathy phenotype that may also be expected in states such as pregnancy or alcohol excess.146
Nevertheless, the study that linked TTNtvs to CCM identified TTNtvs in only 7.5% of those with CCM, and not all the at-risk participants developed cardiomyopathy.146 However, this study sets the stage for the possibility of improved risk stratification which could prospectively investigate risk modification strategies for a genetically at-risk population. Work is urgently needed in this area to advance scientific gains to clinical care.
Key clinical positions:
No clinical action advised based on current evidence.
Clinical trials
Despite the growing worldwide burden of CVD, drug development in the field has been on a downward trend due to the numerous challenges associated with successfully bringing a new drug to the market. Compared to other therapeutic areas, developing new cardiovascular medicines is more expensive for many reasons including the requirement for trials with large sample sizes, new therapies usually add only incremental benefit to individuals on very effective polypharmacy, and increased late-phase failures. Therefore, to ensure the future of cardiovascular drug development, clinical trials need to evolve and embrace new advances to overcome these challenges.148
Pharmacogenomics could help address these difficulties by facilitating mechanism-based approaches for drug development in defined populations. This approach can be utilized at various stages in the drug development pathway.
Challenges for conducting PGx RCTs for drugs that are already licensed include each trial only being able to assess one or a few gene-drug pairs, and large numbers of participants needing to be recruited to include sufficient with the variant allele(s) of interest in the absence of accessible genomic data prior to recruitment.56 Moreover, there are specific concerns over the generalizability of PGx RCT evidence to ethnicities inadequately represented in such trials, as they may carry different gene variants relevant to the studied drug. Furthermore, there is a lack of consensus over the evidential threshold required for PGx biomarkers whose role is to guide dose/drug selection akin to prescribing adjustments based on renal/liver function tests.149
Phase II and III trials
Randomized controlled trials tend to require large patient numbers to show a clinical effect. However, there has been interested in improving patient selection to identify those who are most likely to benefit from the proposed treatment, which may potentially reduce the sample size requirements.
Post hoc subgroup analyses attempt to identify PGx markers from already completed RCTs. This has clear cost saving benefits, but studies risk being under-powered and results inconclusive.
Prospective study design takes adequate powering into consideration to produce more robust evidence. Examples of this design include the following150:
Enrichment design, where only patients who have the feature predicted to have clinical benefit are recruited or analysed. An example of this is the TAILOR-PCI trial discussed above, where the analysis was limited to those with CYP2C19 loss of function (*2, *3) variants.56
Adaptive trial design is categorized by adjusting trial features based on data amassed during the trial itself. An example of this is the GENETIC-AF trial.151
Biomarker based all-comers design which aims to investigate the interaction between treatment effect and biomarker status, more commonly employed in oncology.
Hybrid or combination biomarker trials, which are typically an enrichment study extending to include another technique such as adaptive design.
Successes, failures, and opportunities—lipid modulation trials
The discovery that mutations in PCSK9 were a rare cause of familial hypercholesterolaemia paved the way for the development of PCSK9 inhibitors for lipid management. Yet, cholesteryl ester transfer protein (CETP) inhibitors have proven that identification of a PGx marker does not guarantee successful drug development, and multiple CETP inhibitors have failed phase III trials.152 However, persistence with this therapeutic target is proving fruitful. The REVEAL trial, with the CETP inhibitor Anacetrapib showed a reduction in cardiovascular events.153 Additionally, the dal-GenE trial suggests improved cardiovascular outcomes when the CETP inhibitor dalcetrapib is used with a statin in the subset of patients with the AA genotype at rs1967309 in the ADCY9 gene.154 This further demonstrates the complexities of PGx application in trials as well as the need for robust research to ensure that potentially useful therapies are not wasted.
Pharmacogenomics has the potential to enrich the development of new therapies in cardiovascular medicine, resulting in a more efficient and cost saving drug development route. Significant challenges exist and well-designed trials are required to ensure we have robust evidence.
Real-world evidence to evaluate pharmacogenomics utility
Due to limitations in feasibility for RCT trials for each drug-gene pair, real-world evidence which uses the power of big data contained within electronic health records may prove useful. This approach would also be more likely to include more diverse populations, limiting concerns about external validity of data and health equality. An example of such an approach is the IGNITE study, which assessed outcomes for use of PGx-guided antiplatelet therapy following PCI (and found increased risk of cardiovascular events in those with a CYP2C19 loss-of-function allele who were prescribed clopidogrel).155
Key position:
Pharmacogenomics offers tools that can revolutionize drug design and clinical trials and make both more economically efficient. However, prospective designs should be optimized and post hoc subgroup analysis limitations recognized. Real-world evidence may fill important gaps.
Key paper positions
Thrombosis, ischaemic heart disease, and stroke: Evidence is strongest for pre-emptive genotyping prior to warfarin use. There is also enough evidence to advise genotype-guided antiplatelet therapy in selected high-risk CV patients.
Hypertension: Further research is needed to translate scientific gains to the bedside. Self-identified black/white racial stratification in HTN therapeutic algorithms is not supported by genetic evidence to-date.
Arrhythmias: Further research is needed to advance our understanding of genetic risk loci for drug-induced long QT and to trial a PGx approach in clinical practice. Known carriers of pathogenic LQT variants should not be given medication known to prolong the QTC.
Chemotherapy-induced cardiomyopathy: Further research is needed to translate scientific gains to the bedside.
Clinical trials: PGx has the potential to improve the success, efficacy, and cost profile of drug development and clinical trials but is in early stages of development.
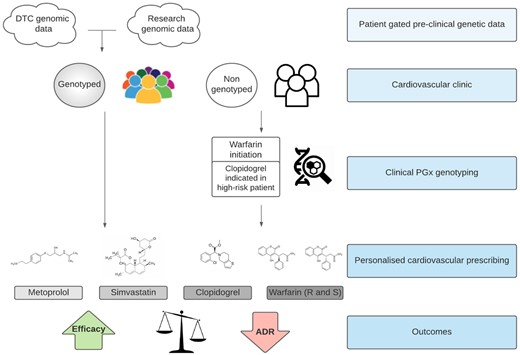
Summary graphic of ESC WG clinical PGx advice. This flow graphic illustrates the modes in which patients may have genetic testing, either prior to clinical encounter or indicated during a clinical encounter (where genotyping may be indicated prior to warfarin initiation or to guide antiplatelet therapy in high-risk patients). This information from diverse sources may then impact on the prescribing of metoprolol, simvastatin, clopidogrel, and warfarin, to increase therapeutic efficacy and decrease the probability of adverse drug reactions. ADR, adverse drug reactions; DTC, direct-to-consumer.
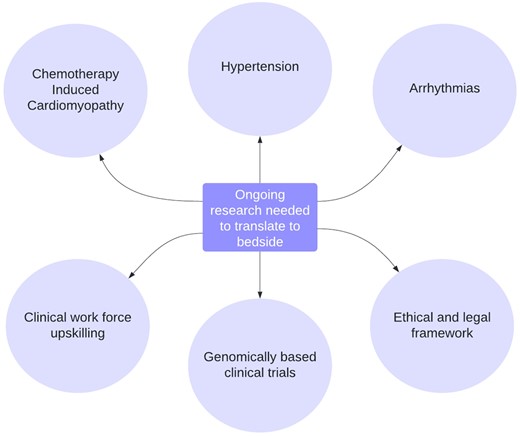
Areas with promise for PGx translation—key opportunities for research. Key opportunities for research to advance pharmacogenomics and facilitate translation to the bedside.
Acknowledgements
M.J.C. is an NIHR Senior Investigator and this work forms part of the research portfolio of the NIHR Biomedical Research Centre at Barts. M.P. is an Emeritus NIHR Senior Investigator, and wishes to thank the MRC Centre for Drug Safety Science and Wolfson Centre for Personalised Medicine for infrastructure support.
Conflict of interest: M.J.C. is seconded to Genomics England as a Chief Scientist and part of his salary is funded by Genomics England Ltd, a wholly owned Department of Health and Social Care Company. M.P. receives research funding from various organizations including the MRC, NIHR, EU Commission, HDR UK, and Health Education England. He has also received partnership funding for the following: MRC Clinical Pharmacology Training Scheme (co-funded by MRC and Roche, UCB, Eli Lilly, and Novartis); a PhD studentship jointly funded by EPSRC and Astra Zeneca; and grant funding from Vistagen Therapeutics. He has also unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb and UCB. He has developed an HLA genotyping panel with MC Diagnostics, but does not benefit financially from this. MP is also a member of the IMI Consortium ARDAT (www.ardat.org). None of the funding declared above has been directly used for the current review paper. S.A. receives Speaker fees and advisory board fees from: Boehringer-Ingelheim, Daiichi Sankyo, Bayer, Thermofisher. A.G.S. has received speaker honoraria and/or consulting fees from Sanofi, AbbVie, Novartis, Bayer, and Lilly. G.S. reports grants and personal fees from Vifor, grants and non-financial support from Boehringer Ingelheim, personal fees from Societa’ Prodotti Antibiotici, grants and personal fees from AstraZeneca, personal fees from Roche and Servier, grants from Novartis, personal fees from GENESIS, Cytokinetics, and Medtronic, grants from Boston Scientific, outside the submitted work. P.S. reports grants from Daiichi Sankyo, grants and personal fees from Boehringer-Ingelheim outside the submitted work.
References
Author notes
Emma Forton Magavern, Juan Carlos Kaski shared co-first authorship.
Munir Pirmohamed, Mark J. Caulfield shared co-last authorship.

{kind=link}
{kind=link}