





Abstract
Proprotein convertase subtilisin/kexin Type 9 (PCSK9) is now identified as an important and major player in hypercholesterolaemia and atherosclerosis pathophysiology. PCSK9, through promoting lysosomal degradation of hepatic low-density lipoprotein (LDL) receptor, can decrease the clearance of plasma LDLs, leading to hypercholesterolaemia and consequent atherosclerotic plaque formation. Hypercholesterolaemia has been found to promote systemic and vascular inflammation, which can cause atherosclerotic lesion formation and progression and subsequent incidence of cardiovascular disease. Recent studies have shown the involvement of PCSK9 in the inflammatory pathway of atherosclerosis. Although trials with PCSK9 inhibitors have not shown any alteration in plasma C-reactive protein levels, there is accumulating evidence showing lessened inflammatory response in the arterial wall that could attenuate atherosclerotic plaque development beyond the established LDL-lowering effect of PCSK9 inhibition. In this review, we represent mounting evidence indicating that PCSK9 can locally increase vascular inflammation and contribute to atherosclerotic plaque progression in patients with hypercholesterolaemia.
Introduction
Chronic inflammation plays an important role in the initiation and progression of atherosclerosis up to plaque rupture and erosion, resulting in atherosclerosis-related cardiovascular diseases (CVDs), such as myocardial infarction (MI) and stroke.1 Numerous genetic or acquired risk factors for atherosclerosis can damage and alter the normal homoeostatic functions of the endothelium, thereby inducing an inflammatory response.2
Among the CVD risk factors, hypercholesterolaemia is typically accompanied by vascular endothelium activation and macrophage-dominated inflammation in response to subendothelial cholesterol accumulation. The recognition and uptake by resident arterial macrophages of cholesterol-rich lipoproteins containing apolipoprotein B (apoB), mainly low-density lipoproteins (LDLs), causes a chronic inflammatory response that is generally localized but may have also systemic repercussions.3,4 Low-density lipoproteins infiltrate and are retained in the subendothelial layer via interactions between matrix proteoglycans and the LDL constituent apoB.5 Intimal-trapped LDL particles undergo oxidization through exposure to the products of oxidation such as reactive oxygen species (ROS) produced by macrophages, endothelial cells (ECs), and vascular smooth muscle cells (VSMCs). It is noteworthy that native LDLs have a much lower affinity for the scavenger receptors (SRs) than oxidized or chemically modified LDL. In this regard, oxidized LDLs (oxLDLs) act as a ligand for macrophage pattern recognition receptors including toll-like receptor-2 (TLR2), TLR4, and TLR6. Thus, oxLDLs can directly induce pro-inflammatory signalling pathways in resident macrophage of arterial lesions.6,7 Moreover, oxLDLs can be internalized by means of other macrophage pattern recognition receptors, the surfaces SRs, such as SR-A and CD36. Internalization of oxLDLs results in the production of lipid peroxides promoting the cellular accumulation of cholesterol esters, which in turn enhances TLR signalling and promotes foam cell formation.6,7
Accumulation of oxLDLs in the artery wall also contributes to cholesterol crystal generation. This accelerates atherogenesis and promotes cell injury and apoptosis within the lesion. The injured cells release endogenous materials that can provoke and intensify the inflammatory response. Uptake or formation of cholesterol crystals in macrophages has been found to activate the NLRP3 [Nucleotide-binding and Oligomerization Domain (NOD)-Like Receptor Protein 3] inflammasome. Activation is mediated by two pathways: one that mainly involves TLR4 activation, and a second that affects potassium ion efflux, lysosomal damage, or ROS production. The first pathway may occur because of TLR4-TLR6-CD36 signalling induction by oxLDLs. The second pathway can be a response to cholesterol crystals in lysosomes. This may occur through phagocytosis of extracellular cholesterol crystals or via CD36-mediated internalization of oxLDLs and free cholesterol release from the LDLs. Inflammasome activation can result in maturation and secretion of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18, which are principally expressed by activated macrophages, ECs, and VSMCs. The NLRP3 inflammasome, composed of the NOD-like receptor NLRP3, caspase 1 and the adaptor protein ASC, contributes to the cleavage of pro-IL-1β and pro-IL-18 precursors to their secretory and functional forms. NLRP3 has been found to be an important mediator of injury following myocardial ischaemia.8 Interestingly, it has been shown that neutralization of IL-1β with monoclonal antibodies may ameliorate atherosclerosis in animal models.9 Moreover, recent findings from the CANTOS trial also corroborated the benefits of canakinumab (a monoclonal antibody against IL-1β) in reducing cardiovascular (CV) outcomes in patients with MI and elevated high-sensitivity C-reactive protein (hs-CRP).10 Therefore, oxLDL-mediated enhancement of TLR activity can cause over-expression of chemokines and cytokines, augmentation of the inflammatory process and, once combined with the internalization or intracellular formation of cholesterol crystals, may result in the activation of inflammasomes and progression of inflamed plaque.
Over the last decade, proprotein convertase subtilisin/kexin Type 9 (PCSK9) has been the subject of increasing attention due to its pivotal role in cholesterol homoeostasis and atherosclerosis, mainly through the post-transcriptional regulation of hepatic LDL receptor (LDLR) and promoting its lysosomal degradation.11 Besides the hepatocyte LDLR, PCSK9 can regulate LDLR in other cells, including lymphocytes, macrophages, ECs, and VSMCs.12 Beyond cholesterol metabolism, other physiological processes are also mediated by PCSK9, including modulation of adipogenesis in visceral adipose tissue, regulation of immune response, modulation of neurogenesis during neuronal development, and interaction with very-LDL (VLDL) receptor (VLDLR) and the apoprotein E receptor 2 (Apo ER2).13 A possible role of PCSK9 in regulating genes affecting cellular proliferation, apoptosis and inflammation have also been suggested.14 In this review, we present evidence regarding the role of PCSK9 and PCSK9 inhibitors on atherosclerosis-related vascular inflammation.
Epidemiological evidence verifying the pro-inflammatory effect of PCSK9
The connection between PCSK9 and inflammation has been investigated in several epidemiological studies. These studies evaluated the association of PCSK9 with some key inflammatory markers including white blood cells (WBCs), fibrinogen, and hs-CRP (Table 1). White blood cell count (WBCC) is a traditional marker of inflammation. It has been shown to be significantly associated with early and advanced atherosclerosis in various populations with different levels of baseline risks, both asymptomatic and symptomatic patients with coronary artery disease (CAD).19 Furthermore, the present paradigm of the inflammatory nature of atherosclerosis strongly supports the relationship between WBCC and CAD.19 Plasma levels of PCSK9 were positively and independently associated with WBCC subsets, lymphocyte count and neutrophil count in univariate, and multivariate analyses in a single-centre study of CAD patients in China. These patients were not on lipid-lowering therapy. The molecular mechanisms underlying the association of the WBCC with the plasma PCSK9 levels are yet unknown.15
Correlation of plasma PCSK9 levels with inflammatory markers in patients with no lipid-lowering therapy
| Author/study (ref.) | Study design (sample size) | Inflammatory marker | Univariable analysis | Multivariable analysis | ||
|---|---|---|---|---|---|---|
| Coefficient (r) | P-value | Coefficient (β) | P-value | |||
| Li et al.15 | Single-centre cross-sectional study (251 stable CAD patients) | WBCC | 0.167 | 0.008 | 0.186a | <0.01 |
| Zhang et al.16 | Cross-sectional study (219 stable CAD patients) | Fibrinogen | 0.211 | 0.002 | 0.168b | 0.011 |
| hs-CRP | 0.153 | 0.023 | — | — | ||
| Gencer et al.17/ SPUM-ACS study | Multi-centre prospective cohort study (2168 ACS patients) | hs-CRP | 0.077 | 0.006 | — | — |
| Li et al.18 | Prospective study (552 CAD patients) | WBCC | 0.077 | 0.014 | — | — |
| Fibrinogen | 0.181 | <0.001 | — | — | ||
| hs-CRP | 0.101 | 0.003 | — | — | ||
| Author/study (ref.) | Study design (sample size) | Inflammatory marker | Univariable analysis | Multivariable analysis | ||
|---|---|---|---|---|---|---|
| Coefficient (r) | P-value | Coefficient (β) | P-value | |||
| Li et al.15 | Single-centre cross-sectional study (251 stable CAD patients) | WBCC | 0.167 | 0.008 | 0.186a | <0.01 |
| Zhang et al.16 | Cross-sectional study (219 stable CAD patients) | Fibrinogen | 0.211 | 0.002 | 0.168b | 0.011 |
| hs-CRP | 0.153 | 0.023 | — | — | ||
| Gencer et al.17/ SPUM-ACS study | Multi-centre prospective cohort study (2168 ACS patients) | hs-CRP | 0.077 | 0.006 | — | — |
| Li et al.18 | Prospective study (552 CAD patients) | WBCC | 0.077 | 0.014 | — | — |
| Fibrinogen | 0.181 | <0.001 | — | — | ||
| hs-CRP | 0.101 | 0.003 | — | — | ||
ACS, acute coronary syndrome; CAD, coronary artery disease; hs-CRP, high-sensitive C-reactive protein; SPUM-ACS, Special Program University Medicine Acute Coronary Syndromes and Inflammation; WBCC, white blood cell count.
Value shows correlation between PCSK9 and WBCC after adjustment for age, gender, body mass index, smoking, alcohol intake, hypertension, diabetes mellitus, familial history of CAD, and lipid parameters.
Value shows correlation between PCSK9 and fibrinogen after adjustment for age, gender, body mass index, smoking, systolic blood pressure, fasting blood glucose, familial history of CAD, lipid parameters, and CRP.
Correlation of plasma PCSK9 levels with inflammatory markers in patients with no lipid-lowering therapy
| Author/study (ref.) | Study design (sample size) | Inflammatory marker | Univariable analysis | Multivariable analysis | ||
|---|---|---|---|---|---|---|
| Coefficient (r) | P-value | Coefficient (β) | P-value | |||
| Li et al.15 | Single-centre cross-sectional study (251 stable CAD patients) | WBCC | 0.167 | 0.008 | 0.186a | <0.01 |
| Zhang et al.16 | Cross-sectional study (219 stable CAD patients) | Fibrinogen | 0.211 | 0.002 | 0.168b | 0.011 |
| hs-CRP | 0.153 | 0.023 | — | — | ||
| Gencer et al.17/ SPUM-ACS study | Multi-centre prospective cohort study (2168 ACS patients) | hs-CRP | 0.077 | 0.006 | — | — |
| Li et al.18 | Prospective study (552 CAD patients) | WBCC | 0.077 | 0.014 | — | — |
| Fibrinogen | 0.181 | <0.001 | — | — | ||
| hs-CRP | 0.101 | 0.003 | — | — | ||
| Author/study (ref.) | Study design (sample size) | Inflammatory marker | Univariable analysis | Multivariable analysis | ||
|---|---|---|---|---|---|---|
| Coefficient (r) | P-value | Coefficient (β) | P-value | |||
| Li et al.15 | Single-centre cross-sectional study (251 stable CAD patients) | WBCC | 0.167 | 0.008 | 0.186a | <0.01 |
| Zhang et al.16 | Cross-sectional study (219 stable CAD patients) | Fibrinogen | 0.211 | 0.002 | 0.168b | 0.011 |
| hs-CRP | 0.153 | 0.023 | — | — | ||
| Gencer et al.17/ SPUM-ACS study | Multi-centre prospective cohort study (2168 ACS patients) | hs-CRP | 0.077 | 0.006 | — | — |
| Li et al.18 | Prospective study (552 CAD patients) | WBCC | 0.077 | 0.014 | — | — |
| Fibrinogen | 0.181 | <0.001 | — | — | ||
| hs-CRP | 0.101 | 0.003 | — | — | ||
ACS, acute coronary syndrome; CAD, coronary artery disease; hs-CRP, high-sensitive C-reactive protein; SPUM-ACS, Special Program University Medicine Acute Coronary Syndromes and Inflammation; WBCC, white blood cell count.
Value shows correlation between PCSK9 and WBCC after adjustment for age, gender, body mass index, smoking, alcohol intake, hypertension, diabetes mellitus, familial history of CAD, and lipid parameters.
Value shows correlation between PCSK9 and fibrinogen after adjustment for age, gender, body mass index, smoking, systolic blood pressure, fasting blood glucose, familial history of CAD, lipid parameters, and CRP.
Fibrinogen, another important inflammatory marker, is a blood coagulation glycoprotein synthesized by the liver and is up-regulated in the inflammatory states.20 Fibrinogen has been known to accelerate the progression of atherosclerosis and consequent clinical CAD through stimulating plaque growth and/or inducing the adhesion of WBCs and platelets to the vascular wall.21 Moreover, elevated plasma levels of fibrinogen have been associated with increased CVD incidence, atherosclerosis severity, and CAD mortality.22 A cross-sectional study in patients with angiographically verified stable CAD showed that the circulating levels of PCSK9 were positively associated with those of fibrinogen. The association was found to be mechanistically independent of some potential confounders such as lipid profile indices and hs-CRP. The interaction between PCSK9 and fibrinogen was suggested to be one of the underlying mechanisms involved in the progression of atherosclerosis.16
C-reactive protein is an acute-phase mediator that is considered as a sensitive but non-specific biomarker of systemic inflammation.23 C-reactive protein can increase the uptake of LDLs into macrophages and lead to foam cell formation in atherosclerosis lesions24; therefore, it has been proposed as an atherogenic risk marker and risk factor.25 As determined by observational epidemiological studies, plasma levels of CRP show log-linear association with the risk of subsequent CAD,26 thereby are regarded as a powerful CV risk predictor.27 However, a causal association between CRP and CAD seems to be refuted by Mendelian randomization studies.28 PCSK9 levels were also found to be positively and significantly associated with hs-CRP levels in patients with stable CAD.16 A large prospective multi-centre cohort study of patients with acute coronary syndrome (ACS) showed that patients with higher levels of circulating PCSK9 have suffered higher degree of acute-phase inflammation as evaluated by hs-CRP levels.17 Likewise, a prospective case–control study in Chinese CAD patients who had no lipid-lowering therapy revealed that PCSK9 levels had positive and significant association with the incidence and severity of CAD. The effects of PCSK9 on CAD were found to be mediated by the elevated atherogenic lipids and inflammatory markers. Statistically, the elevated plasma levels of PCSK9 were associated with increased WBCC, fibrinogen, and hs-CRP in the studied CAD patients.18 Altogether, these findings show that PCSK9 levels have a plausible connection to inflammation, suggesting potential therapeutic effects of PCSK9 inhibition on atherosclerosis inflammation as well as incidence and severity of CAD. However, because a statistical association between PCSK9 and LDL-C levels has been inconsistently reported, how much of the PCSK9's impact on inflammation might be explained by its effect on LDL-C requires further investigation.
Sepsis as a model of the pro-inflammatory effects of PCSK9
The link between PCSK9 and inflammation has been supported in several experimental and human studies that evaluated the effects of differential gene expression of PCSK9 [PCSK9 knock-out or transgenic mice and humans carrying PCSK9 loss-of-function (LOF) or gain-of-function (GOF) mutations] on inflammatory responses during sepsis (Figure 1).
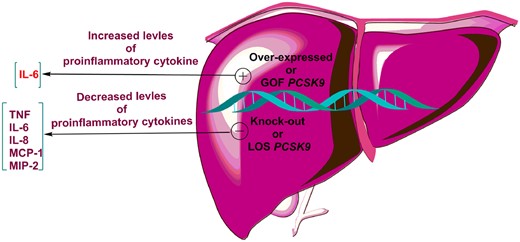
The effects of PCSK9 knock-out or transgenic mice and humans carrying PCSK9 loss-of-function or gain-of-function mutations on inflammatory cytokines. IL-6, interleukin 6; IL-8, interleukin 8; MCP, monocyte chemoattractant protein; PCSK9, proprotein convertase subtilisin/kexin type 9; TNF, tumour necrosis factor.
An in vivo study on lipopolysaccharide (LPS)-induced sepsis showed that there is a link between PCSK9 and inflammation. PCSK9 was found to intensify inflammation in mouse sepsis model; thus, overexpression of PCSK9 elevated the systemic release of inflammatory cytokine IL-6 and exacerbated lung and liver inflammation, while PCSK9 deficiency decreased circulating levels of IL-6 and ameliorated organ inflammation.29 Similar results were also reported in another in vivo investigation showing amelioration of LPS-induced inflammation and reduced serum levels of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), IL-6, macrophage chemotactic protein-1 (MCP-1), and macrophage inflammatory protein-2 (MIP-2) in PCSK9 knock-out mice.30 The in vivo findings are further supported by the results of a human study, in which septic shock patients carrying a PCSK9 LOF allele had significantly lower plasma levels of pro-inflammatory cytokines TNF-α, IL-6, IL-8, and MCP-1 compared with patients carrying a GOF allele.30 Overall, these results reveal a link between PCSK9 and inflammation, suggesting that PCSK9 can act as a pro-inflammatory mediator.
In the following sections, the relevance of PCSK9 in vascular inflammation and inflammatory cytokine production is illustrated to understand the mechanisms underlying the pro-inflammatory effect of PCSK9 in the development of atherosclerosis.
PCSK9, pro-inflammatory cytokines, and related signalling pathways
There have been numerous studies on the effect of PCSK9 on pro-inflammatory cytokines and vice versa (Figure 2). The TLR4/NF-κB is an important signalling pathway in the initiation and progression of atherosclerotic lesions through induction of vascular inflammatory responses.31 TLR4 can stimulate activation of NF-κB transcription factor that is responsible for the expression of numerous pro-inflammatory genes including TNF-α, IL-6, IL-1, and MCP-1.32–34 NF-κB, a master regulator of inflammatory response, is a redox-sensitive transcription factor activated by a wide range of stimuli including oxLDLs, ROS, TLRs, cytokines, and bacterial products such as LPS.
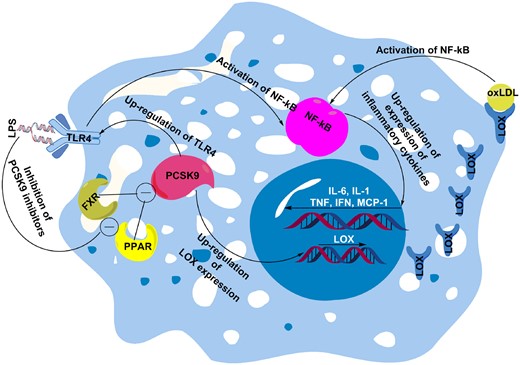
PCSK9 can mediate oxLDL-induced inflammation through enhancing expression of LOX, which can increase uptake of oxLDLs that induce inflammation via activation of NF-kB. PCSK9 can also increase expression of TLR4, which activates NF-kB. Overall, activated NF-kB up-regulates expression of inflammatory cytokines, such as IL-6, IL-1, TNF-α, IFN-γ, and MCP-1. Besides oxLDLs, lipopolysaccharide also known to enhance PCSK9-mediated inflammation through inhibition of PCSK9 inhibitors farnesoid X receptor and PPAR transcription factors. FXR, farnesoid X receptor; IFN-γ, interferon gamma; IL-1, interleukin 1; IL-6, interleukin 6; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein 1; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; oxLDL, oxidized low-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin Type 9; PPAR, peroxisome proliferator-activated receptors; TLR4, toll-like receptor 4; TNF-α, tumour necrosis factor alpha.
PCSK9 has been found to enhance the production of pro-inflammatory cytokines that play an important role in atherosclerosis plaque inflammation. Of note, the TLR4/NF-κB signalling pathway has been found to be one of the main affected signalling pathways mediating the PCSK9-induced increase in pro-inflammatory cytokines.35
OxLDL, as a key NF-κB activator, has been found to enhance the expression of PCSK9 by inducing the secretion of IL-1α, IL-6, and TNF-α in ECs and macrophages.36,37 Conversely, it was found that down-regulation of PCSK9 was able to diminish oxLDL-stimulated inflammatory responses of macrophages; thus, pro-inflammatory cytokines including IL-1α, IL-6, and TNF-α were decreased. PCSK9 inhibition in macrophages was found to decrease pro-inflammatory cytokines by reducing the degradation of IκB-α and nuclear translocation of NF-κB.36 Involvement of TLR4/NF-κB signalling in the PCSK9-mediated inflammatory pathway was also identified in an in vitro study on oxLDL-stimulated RAW264.7 macrophages. In this study, up- and down-regulation of PCSK9 expression could increase and decrease, respectively, oxLDL-induced secretion of the pro-inflammatory cytokines TNF-α, IL-1β, and MCP-1. This effect was linked to modulation of oxLDL-induced TLR4 expression and to p-IkB expression, IkB degradation, and NF-κB nuclear translocation. It was concluded that PCSK9 can elevate pro-inflammatory cytokines most likely through binding to the C-terminal domain of TLR4, leading to up-regulation of TLR4 expression and activation of the TLR4/NF-κB signalling pathway.35
The effect of PCSK9 on TLR4/NF-κB-mediated inflammation has also been described with LPS, which is able to cause inflammation through induction of PCSK9 expression.38 LPS is a well-known activator of TLR4 and can trigger NF-κB signalling pathway mainly through inducing TLR4, leading to systemic inflammation.39 In the LPS-induced mouse inflammation model decreased hepatic LDLR expression and increased plasma LDL-C were noted as a result of increased PCSK9 expression. In mechanism, LPS was suggested to increase PCSK9 expression possibly through down-regulation of PCSK9 inhibitors farnesoid X receptor and PPAR-α transcription factors.38 These findings show that LPS, as an inflammatory agent, can activate the TLR4/NF-κB pathway and induces PCSK9 expression, thus suggesting that the pro-inflammatory effects of PCSK9 might be mediated at least in part by the activation of the TLR4/NF-κB pathway.
Local effect of PCSK9 on the inflamed atheroma
The most evident mechanism connecting PCSK9 to the inflamed atheroma is the PCSK9-induced disturbance of lipid metabolism and eventual hypercholesterolaemia. Recent in vitro and in vivo investigations, as summarized below, have made it clear that pro-atherosclerotic impact of PCSK9 is not entirely due to its effect on the serum levels of apoB-containing lipoproteins but PCSK9 itself shows a primary role in atherosclerotic plaque formation independent of systemic lipid changes.
Although liver, kidney, and small intestine are the major sources of PCSK9 biosynthesis, VSMCs and, with a lesser extent, ECs have been found to express and secrete functional PCSK9 which can locally regulate LDLR expression on the adjacent cells, such as monocytes and arterial macrophages.40 In a Pcsk9−/− mouse model of vascular injury, absence of PCSK9 was associated with decreased neointimal formation and atherosclerotic plaques with low VSMC and collagen content.41 It was also shown that Pcsk9−/−SMCs have a reduced proliferation rate and a decreased migratory capacity.41 In aortic regions with low-shear stress, such as arch branching and iliac bifurcation area, PCSK9 expression was increased in VSMCs at the sites of inflammation, while higher fluid shear stress gently decreased the expression of PCSK9.42 In the cells exposed to various stress stimuli, such as shear stress, ROS are generated as by-products of the metabolism of molecular oxygen at the mitochondrial level. From a mechanistic standpoint, modulatory effect of fluid shear stress on PCSK9 expression was found to be mediated by NADPH oxidase-dependent ROS production in human VSMCs and ECs as well as in mouse aorta.42 Consistently, a bidirectional crosstalk between ROS production and PCSK9 expression has been reported that is mediated by NADPH oxidase system in aortic tissues during inflammatory states, which modulates deposition of native and oxLDLs in atherosclerosis-prone regions.43 The major receptor for oxLDLs on the aortic ECs and SMCs is lectin-like oxLDL receptor-1 (LOX-1) that is involved in the association between PCSK9, inflammation, and atherosclerosis. Lectin-like oxLDL receptor-1 is activated in numerous inflammatory diseases including atherosclerosis.37,44 A crosstalk between PCSK9 and LOX-1 has been shown in inflammatory microenvironments. In a positive-feedback loop, PCSK9 and LOX-1 can induce expression of each other.45 This interaction between PCSK9 and LOX-1 was shown to be regulated by mitochondrial ROS (mtROS) and NF-κB.42,46 Vascular smooth muscle cell-derived PCSK9 has been found to induce mitochondrial DNA damage and the resulting fragments could induce PCSK9/LOX-1 expression mediated by mtROS.46,47 Both LOX-1 activation and PCSK9 expression were also shown to elevate oxLDL uptake and increase the expression of vascular cell adhesion molecule-1, a regulator of immune surveillance and inflammation in VSMCs.47 Overall, in low-shear stress conditions such as during inflammatory states, PCSK9 could intensify atherosclerotic lesion inflammation through inflammation-activated self-reinforcing crosstalk of ROS/NF-κB/LOX-1/oxLDL axis in VSMCs.
PCSK9-stimulated plaque inflammation, independent of plasma lipid levels, is further supported by studies of transgenic mice over-expressing human (h) PCSK9 on the background of apoE−/− or LDLR−/−.14 The over-expressed PCSK9 was associated with enlarged aortic lesion size in apoE−/− mice but not in LDLR−/−mice, while no significant alterations in plasma atherogenic lipids were observed in both murine models. Furthermore, the atherosclerotic plaque in the absence of apoE was enriched with hPCSK9 and Ly6Chi (inflammatory) monocytes.48,49 These investigations established that PCSK9 promotes inflamed atheroma directly through enhancing the infiltration of inflammatory monocytes into the vascular wall through PCSK9-LDLR interaction in the plaque, and hence atherogenic effect of PCSK9 is not exclusively related to alterations in serum lipids.
Further evidence on the local effect of PCSK9 on the inflamed plaque comes from an experimental study conducted in chimeric mice transplanted with hPCSK9 transgenic marrow on apoE−/− or LDLR−/− background. This model permits investigation of the direct impact of PCSK9 in the atheromatous plaque without confounding by serum atherogenic lipoproteins. Non-circulating PCSK9 expressed in marrow-derived macrophages within atheroma could locally and directly enhance vascular inflammation through altering lesion composition independent of changes in lesion size and serum cholesterol levels.50 Although no change was observed in lipid levels or lesion size, macrophage-derived PCSK9 accumulated in the atheromatous plaques of chimeric mice and changed lesion composition through LDLR-dependent elevation of Ly6Chi monocyte infiltration.50 Moreover, this effect was associated with increased number of inflammatory monocytes in the spleen of chimeric mice as well as modulated expression of both pro- and anti-inflammatory cytokines in an LDLR-dependent fashion.50
The nexus between PCSK9 and monocyte-mediated plaque inflammation is supported by an in vitro study showing that local production of PCSK9 by VSMCs could suppress chemokine receptor Type-2 (CCR2)-dependent chemotaxis of monocytes trapped in plaque microenvironment, thus promoting their persistence in the atherosclerotic environment. It was suggested that PCSK9 reduces intracellular cholesterol via reducing LDLR expression in plaque trapped-monocytes, an effect that leads to decreased CCR2 expression and subsequent monocyte migration.51
To sum up, pro-inflammatory leucocytes have a pivotal role in atherosclerosis progression and can modulate atheroma lesion composition without a significant impact on cholesterol levels and lesion size.52 Likewise, composition of lesion rather than its size determines plaque rupture and thrombosis.53 Therefore, it can be concluded that hPCSK9 can directly and cholesterol-independently contribute to atherosclerosis inflammation by changing plaque composition and increasing infiltration and differentiation of inflammatory Ly6Chi monocytes in the plaque. This supports the notion that PCSK9 inhibitors may improve CV outcomes through both lipid-dependent and lipid-independent pathways.
PCSK9 inhibitors and vascular inflammation
Vaccination is a new strategy for PCSK9 inhibition that is actively pursued.54–56 Inhibition of circulating PCSK9 via active immunization by PCSK9 AT04A vaccine was tested in high-fat diet-fed APOE*3Leiden.CETP mice, a translational mouse model for hyperlipidaemia and atherosclerosis.57 The vaccine decreased serum levels of cholesterol (by 53%), systemic and vascular inflammation, as well as development of atherosclerosis.57 PCSK9 inhibition by AT04A also decreased the plasma levels of chemotactic inflammatory cytokines including M-CSF-1 and vascular endothelial growth factor A (VEGF-A) that are known activators of ECs and stimulators of monocyte/macrophage migration. Decreased plasma levels of M-CSF-1 and VEGF-A led to reduced expression of the intracellular adhesion molecule-1 (ICAM-1) in ECs, which in turn decreased monocyte/macrophage recruitment to the vascular wall.57 Plaque number and size were also decreased in AT04A-vaccinated mice.57 Another in vivo study with APOE*3Leiden.CETP mice showed that alirocumab decreases systemic and vascular inflammation inhibiting atherosclerosis plaque development.58 In alirocumab-treated mice, the pro-inflammatory Ly6Chi monocytes, precursors of pro-inflammatory M1 macrophages,59 were decreased.58 Alirocumab also decreased the adhesion of monocytes to the vascular endothelium via reducing endothelial expression of ICAM-1.58 Other vascular inflammatory markers including abundance of T-cell in the aortic root area, macrophage necrotic content, and cholesterol clefts of the arterial plaques were found to be decreased in APOE*3Leiden.CETP mice treated with alirocumab.58
Cholesterol crystals in atherosclerotic lesions induce local and systemic inflammatory responses.60 Likewise, increased macrophage content, a large necrotic core, a thin collagen-poor fibrous cap, and the reduced VSMCs are key traits of a vulnerable plaque that is prone to rupture.61 To sum up, in addition to lipid-lowering effects, alirocumab was revealed to significantly ameliorate vascular inflammation and considerably improve lesion morphology and stability in a mouse model of hyperlipidaemia and atherosclerosis.58
These experimental results are further supported by a clinical trial evaluating the effect of PCSK9 inhibition on inflammation in familial hypercholesterolaemia (FH) patients. Monocytes of FH patients were found to have a pro-inflammatory phenotype showing cytoplasm lipid accumulation, increased CCR2 expression, and enhanced transendothelial migration capacity.62 An elevated level of plasma LDL-C was associated with increased lipid content of monocytes, intensifying monocyte migration through inducing CCR2 expression.62 In the same proof-of-concept study using alirocumab and evolocumab, it was found that PCSK9 inhibition reduced intracellular lipid accumulation in monocytes and decreased their CCR2 expression and migratory capacity.62 Furthermore, PCSK9 inhibition was also associated with reduced levels of TNF-α and increased production of anti-inflammatory cytokine IL-10, signifying diminished inflammatory responses.62 These findings were suggested to be a consequence of plasma LDL-lowering effect of PCSK9 inhibitors which led to decreased CCR2 expression and subsequently decreased migration capacity of monocytes towards endothelial MCP-1+ cells62 (Table 2).
Effects of PCSK9 inhibition on systemic and vascular inflammation
| Ref. | Model/ participants of study | PCSK9 inhibitor | Effects on systemic inflammation | Effect on vascular inflammation |
|---|---|---|---|---|
| 57 | High-fat diet APOE*3Leiden.CETP mice | Anti-PCSK9 AT04A vaccine | Decreased plasma levels of M-CSF-1 and VEGF-A |
|
| 58 | High-fat diet APOE*3Leiden.CETP mice | Alirocumab | Decreased pro-inflammatory Ly6Chi monocytes |
|
| 62 | FH patients | Alirocumab and evolocumab | Decreased intracellular lipid accumulation and CCR2 expression in inflammatory monocytes |
|
| 63 | Patients at high risk for or with established CHD | RG7652 anti-PCSK9 mAb | Could not significantly affect circulating levels of hs-CRP and pro-inflammatory cytokines IL-6 and TNF-α | |
| Ref. | Model/ participants of study | PCSK9 inhibitor | Effects on systemic inflammation | Effect on vascular inflammation |
|---|---|---|---|---|
| 57 | High-fat diet APOE*3Leiden.CETP mice | Anti-PCSK9 AT04A vaccine | Decreased plasma levels of M-CSF-1 and VEGF-A |
|
| 58 | High-fat diet APOE*3Leiden.CETP mice | Alirocumab | Decreased pro-inflammatory Ly6Chi monocytes |
|
| 62 | FH patients | Alirocumab and evolocumab | Decreased intracellular lipid accumulation and CCR2 expression in inflammatory monocytes |
|
| 63 | Patients at high risk for or with established CHD | RG7652 anti-PCSK9 mAb | Could not significantly affect circulating levels of hs-CRP and pro-inflammatory cytokines IL-6 and TNF-α | |
CCR2, C–C chemokine receptor Type 2; CRP, C-reactive protein; ECs, endothelial cells; FH, familial hypercholesterolaemia; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin 6; M-CSF-1, macrophage colony-stimulating factor 1; TNF-α, tumour necrosis factor alpha; VEGF-A, vascular endothelial growth factor A.
Effects of PCSK9 inhibition on systemic and vascular inflammation
| Ref. | Model/ participants of study | PCSK9 inhibitor | Effects on systemic inflammation | Effect on vascular inflammation |
|---|---|---|---|---|
| 57 | High-fat diet APOE*3Leiden.CETP mice | Anti-PCSK9 AT04A vaccine | Decreased plasma levels of M-CSF-1 and VEGF-A |
|
| 58 | High-fat diet APOE*3Leiden.CETP mice | Alirocumab | Decreased pro-inflammatory Ly6Chi monocytes |
|
| 62 | FH patients | Alirocumab and evolocumab | Decreased intracellular lipid accumulation and CCR2 expression in inflammatory monocytes |
|
| 63 | Patients at high risk for or with established CHD | RG7652 anti-PCSK9 mAb | Could not significantly affect circulating levels of hs-CRP and pro-inflammatory cytokines IL-6 and TNF-α | |
| Ref. | Model/ participants of study | PCSK9 inhibitor | Effects on systemic inflammation | Effect on vascular inflammation |
|---|---|---|---|---|
| 57 | High-fat diet APOE*3Leiden.CETP mice | Anti-PCSK9 AT04A vaccine | Decreased plasma levels of M-CSF-1 and VEGF-A |
|
| 58 | High-fat diet APOE*3Leiden.CETP mice | Alirocumab | Decreased pro-inflammatory Ly6Chi monocytes |
|
| 62 | FH patients | Alirocumab and evolocumab | Decreased intracellular lipid accumulation and CCR2 expression in inflammatory monocytes |
|
| 63 | Patients at high risk for or with established CHD | RG7652 anti-PCSK9 mAb | Could not significantly affect circulating levels of hs-CRP and pro-inflammatory cytokines IL-6 and TNF-α | |
CCR2, C–C chemokine receptor Type 2; CRP, C-reactive protein; ECs, endothelial cells; FH, familial hypercholesterolaemia; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin 6; M-CSF-1, macrophage colony-stimulating factor 1; TNF-α, tumour necrosis factor alpha; VEGF-A, vascular endothelial growth factor A.
Failure of PCSK9 inhibitors to affect high-sensitivity C-reactive protein levels
The results of EQUATOR study, a randomized, multicentre, double-blind, and placebo-controlled Phase 2 trial conducted in nine countries, showed that 24 weeks of treatment with RG7652, a PCSK9 mAb, did not significantly affect circulating levels of hs-CRP and pro-inflammatory cytokines IL-6 and TNF-α in patients at high risk for or with established CAD, while levels of LDL-C were markedly decreased (48–60%).63 Additionally, a post hoc analysis of the SPIRE CV outcomes trials of bococizumab revealed that, according to on-treatment levels of hs-CRP and LDL-C measured 14 weeks after drug initiation, the residual inflammatory risk persisted among patients treated with both statin and the PCSK9 inhibitor bococizumab.64 These reports can be supported by a meta-analysis of randomized controlled trials that failed to find a significant impact of PCSK9 mAbs on plasma hs-CRP levels.65 Therefore, the aforementioned reports show that, despite the positive association between hs-CRP, PCSK9 levels, and acute-phase inflammation in CAD patients,16,17 hs-CRP is not the response molecule to PCSK9 inhibitors.
Conclusion
The pro-inflammatory role of PCSK9 in atherosclerosis was supported by numerous experimental studies (Figure 3). Recent clinical studies have also shown that higher levels of plasma PCSK9 are independently associated with the major systemic inflammatory markers including WBCs, fibrinogen, and hs-CRP in patients with ACS and CAD. Association of PCSK9 with systemic and vascular inflammation has also been confirmed by experimental and clinical evidence showing the effects of differential gene expression of PCSK9 on the plasma levels of reference systemic inflammatory cytokines including IFN-γ, TNF-α, IL-6, IL-1β, and MCP-1. However, the clinical consequences of PCSK9-related inflammation are still underinvestigated and it is unclear how much of the CV risk due to PCSK9 elevation is contributed by inflammation beyond the known effect of PCSK9 on LDL-C. PCSK9 inhibition can ameliorate systemic and vascular inflammation while inhibiting the development of atherosclerosis and improving plaque morphology in animal models of atherosclerosis (Figure 3). In addition, clinical trials confirmed the local anti-inflammatory effect of alirocumab therapy in FH patients through decreasing the lipid content and migratory capacity of inflammatory monocytes. While randomized trials showing clinical benefit of PCSK9 inhibitors did not show a decrease in hs-CRP concentrations, it is possible that these inhibitors could attenuate low-grade inflammation. Thus, due to the recognized negative prognostic role of some inflammatory markers on CVD risk and the overall anti-inflammatory impact of PCSK9 inhibition, it is possible that part of the beneficial influence of PCSK9 inhibitors on CV morbidity and mortality in humans is related to their ability to attenuate low-grade systemic inflammation. Extant evidence suggests that the reduction of LDL-C and, consequently of oxLDLs, accounts for much of the anti-PCSK9 mAbs' anti-inflammatory effects and reduction of CV events. However, a direct local anti-inflammatory action of PCSK9 inhibitors, independent of LDL-C reduction, has been shown in animal models and merits further investigation. Analyses from long-term clinical trials will also be necessary to explore the relationship between PCSK9 inhibition, attenuation of inflammation, and reduction of CV morbidity and mortality. Finally, since peripheral arterial disease (PAD) is also accompanied by a considerable inflammatory burden66 and PCSK9 inhibition has been shown to reduce major CV events and major limb events in patients with PAD67 it is recommended to explore the impact of PCSK9 inhibition on inflammatory indices in PAD in future investigations and post hoc analyses of clinical trials.
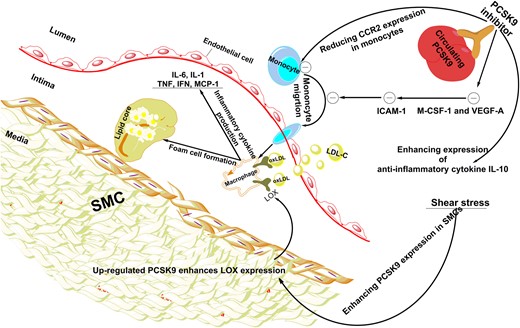
The effect of PCSK9 and PCSK9 inhibition on vascular inflammation. Shear stress results in the elevated expression of PCSK9 in SMCs. Up-regulated PCSK9 leads to the increased expression of LOX-1 in adjacent cells such as macrophages, which enhances oxLDL uptake and consequently promotes foam cell formation in the subendothelial layer. In addition, PCSK9 could enhance monocyte migration through elevating CCR2 expression. To sum up, over expression of PCSK9 causes vascular inflammation. On the other hand, PCSK9 inhibition could decrease main markers of vascular inflammation including M-CSF-1 and VEGF-A which leads to reduced ICAM-1 expression in endothelial cells and reduced infiltration of monocytes into the subendothelial layer. Furthermore, PCSK9 inhibition reduces CCR2 expression, which results in the inhibition of monocyte migration. Besides reduction of pro-inflammatory mediators, PCSK9 inhibitors could ameliorate vascular inflammation through elevating anti-inflammatory cytokines such as IL-10. CCR2, C–C chemokine receptor Type 2; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon gamma; IL-1, interleukin 1; IL-6, interleukin 6; IL-10, interleukin 10; LDL-C, low-density lipoprotein cholesterol; LOX-1, lectin-like oxidized LDL receptor-1; M-CSF-1, major histocompatibility complex; oxLDL, oxidized low-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin Type 9; SMCs, smooth muscle cells; TNF-α, tumour necrosis factor alpha; VEGF-A, vascular endothelial growth factor A.
Acknowledgements
We are thankful for the financial support from the National Institute for Medical Research Development (NIMAD), Tehran, Iran [grant no: 963401].
Conflict of interest: A.M.G. is a consultant for Kowa, a member of the Data Safety Monitoring Board for Ionis Pharmaceuticals and a board member of Esperion. M.B. has served on speaker’s bureau and as an advisory board member for Amgen, Sanofi-Aventis, and Lilly. And all other authors have no conflict of interest to declare.
References
Emerging Risk Factors Collaboration,
C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC),
Author notes
Amir Abbas Momtazi-Borojeni and Sarvenaz Sabouri-Rad authors contributed equally.

{kind=link}
{kind=link}
{kind=link}