





Abstract
Determining the type of amyloid deposits is clinically important for choosing the specific therapies for cardiac amyloidosis.
A 78-year-old woman who had been experiencing fluid retention and dyspnoea on exertion for 6 months was referred to our hospital for the management of heart failure with left ventricular hypertrophy. Since 99mTc-hydroxymethylene diphosphonate scintigraphy showed mild cardiac uptake and significant elevation of serum free lambda chain (with a difference of 263 mg/L in free light chain), we suspected immunoglobulin light-chain amyloidosis (AL), and endomyocardial biopsy was performed. The deposit site within the myocardial tissue exhibited positive for Congo red staining and transthyretin immunostaining, however negative or non-specific for light-chain immunostaining including lambda and kappa staining. Genetic testing confirmed a mutation in V122I, variant-type transthyretin amyloidosis (ATTRv). Despite the administration of patisiran, her condition exhibited progressive deterioration. Additionally, she displayed macroglossia, an atypical manifestation in ATTRv amyloidosis. Further biopsies from tongue and abdominal wall fat culminated in a final diagnosis: the coexistence of ATTRv and AL (of the lambda type). Although treatment with melphalan and dexamethasone was started, she passed away 24 months after the initial visit. When the endomyocardial biopsy specimen underwent mass spectrometry as a post hoc analysis, both ATTR and AL amyloid were significantly detected.
Coexistence of ATTRv and AL within cardiac amyloidosis is extremely uncommon. In situations where incongruities arise between the amyloid type determined via immunohistochemistry findings and the amyloid type assumed based on other clinical findings, mass spectrometry should be considered.
The coexistence of the two amyloid types, specifically variant-type transthyretin and light chain, is extremely uncommon and therefore difficult to diagnose in accordance with the current guideline.
The amyloid type should be reconsidered, if the pathologic diagnosis is inconsistent with imaging findings or the clinical course.
Mass spectrometry is useful for the accurate classification of amyloidosis.
Introduction
Cardiac amyloidosis (CA) is a progressive disease caused by the deposition of amyloid fibrils in the myocardium, leading to heart failure. There are two major types of cardiac amyloidosis. Light-chain amyloidosis (AL) develops due to the deposition of amyloid derived from monoclonal immunoglobulin light chains, and the other is ATTR amyloidosis derived from transthyretin produced mainly in the liver. Furthermore, ATTR amyloidosis is divided into wild-type ATTR amyloidosis (ATTRwt) associated with aging and variant-type ATTR amyloidosis (ATTRv) caused by genetic mutations. The prognosis for both types is poor without treatment, but the development of treatment methods is progressing. Therefore, early determination of the amyloid type and start of treatment has become important.1 Diagnosis of amyloidosis is not easy, but in rare cases, the same patient may have pathologically different amyloid types,2 in which cases diagnosis is very difficult. Because the prevalence of ATTRwt and AL amyloidosis increases with age, several cases have been reported in which there is coexistence of two types of amyloidosis.3,4 However, this is an extremely rare case in which the coexistence of ATTRv and AL amyloidosis has been pathologically proved, and treatment initiated.
Summary figure
| 6 months before | The patient presented with symptoms of fluid retention and exertional dyspnoea. |
| 2 months before | Echocardiography showed hypertrophy and diastolic dysfunction. |
| Age 78 | She was referred to our hospital with suspected cardiac amyloidosis. 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) scintigraphy was negative and bone marrow biopsy led to the diagnosis of monoclonal gammopathy of undetermined significance (MGUS). |
| 3 months later | She was diagnosed as ATTRv because TTR was positive pathologically and genetic mutation (V122I) was revealed. |
| 5 months later | Although patisiran was initiated, congestion and pleural effusion got worse. |
| 10 months later | She was diagnosed as coexistence of ATTRv and AL with an additional biopsy from the tongue. Combination therapy with melphalan and dexamethasone was added for AL. |
| 20 months later | The treatment for AL was terminated due to worsening of heart failure |
| 24 months later | She died of heart failure. |
| 6 months before | The patient presented with symptoms of fluid retention and exertional dyspnoea. |
| 2 months before | Echocardiography showed hypertrophy and diastolic dysfunction. |
| Age 78 | She was referred to our hospital with suspected cardiac amyloidosis. 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) scintigraphy was negative and bone marrow biopsy led to the diagnosis of monoclonal gammopathy of undetermined significance (MGUS). |
| 3 months later | She was diagnosed as ATTRv because TTR was positive pathologically and genetic mutation (V122I) was revealed. |
| 5 months later | Although patisiran was initiated, congestion and pleural effusion got worse. |
| 10 months later | She was diagnosed as coexistence of ATTRv and AL with an additional biopsy from the tongue. Combination therapy with melphalan and dexamethasone was added for AL. |
| 20 months later | The treatment for AL was terminated due to worsening of heart failure |
| 24 months later | She died of heart failure. |
| 6 months before | The patient presented with symptoms of fluid retention and exertional dyspnoea. |
| 2 months before | Echocardiography showed hypertrophy and diastolic dysfunction. |
| Age 78 | She was referred to our hospital with suspected cardiac amyloidosis. 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) scintigraphy was negative and bone marrow biopsy led to the diagnosis of monoclonal gammopathy of undetermined significance (MGUS). |
| 3 months later | She was diagnosed as ATTRv because TTR was positive pathologically and genetic mutation (V122I) was revealed. |
| 5 months later | Although patisiran was initiated, congestion and pleural effusion got worse. |
| 10 months later | She was diagnosed as coexistence of ATTRv and AL with an additional biopsy from the tongue. Combination therapy with melphalan and dexamethasone was added for AL. |
| 20 months later | The treatment for AL was terminated due to worsening of heart failure |
| 24 months later | She died of heart failure. |
| 6 months before | The patient presented with symptoms of fluid retention and exertional dyspnoea. |
| 2 months before | Echocardiography showed hypertrophy and diastolic dysfunction. |
| Age 78 | She was referred to our hospital with suspected cardiac amyloidosis. 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) scintigraphy was negative and bone marrow biopsy led to the diagnosis of monoclonal gammopathy of undetermined significance (MGUS). |
| 3 months later | She was diagnosed as ATTRv because TTR was positive pathologically and genetic mutation (V122I) was revealed. |
| 5 months later | Although patisiran was initiated, congestion and pleural effusion got worse. |
| 10 months later | She was diagnosed as coexistence of ATTRv and AL with an additional biopsy from the tongue. Combination therapy with melphalan and dexamethasone was added for AL. |
| 20 months later | The treatment for AL was terminated due to worsening of heart failure |
| 24 months later | She died of heart failure. |
Case presentation
A 78-year-old woman was referred to our hospital due to concentric left ventricular hypertrophy identified on echocardiography, prompting suspicions of cardiac amyloidosis. Her medical history encompassed a stroke, ossification of the posterior longitudinal ligament, and lumbar spondylolisthesis. There was no family history of amyloidosis. Her medical therapy is azosemido at dose of 30 mg. During the physical examination, she presented with macroglossia, pronounced atrophy of the thenar muscles, peripheral pitting oedema, and manifestations of heart failure (classified as New York Heart Association functional grade III). Her blood pressure registered at 116/76 mmHg, with a pulse rate of 58 beats/min. A chest radiograph depicted mild cardiomegaly and signs of congestion, while electrocardiography unveiled a sinus rhythm accompanied by low voltage. Echocardiography revealed left ventricular wall thickening and brightness, coupled with a left ventricular ejection fraction of 67% and an E/A ratio of 2.3 (Figure 1A–C). Laboratory assessments disclosed elevated serum brain natriuretic peptide (BNP) levels, heightened serum high-sensitivity troponin T levels, and a diminished serum kappa/lambda ratio. Serum immunofixation electrophoresis unveiled the presence of IgG lambda monoclonal protein (Table 1). Bone marrow biopsy exhibited scattered plasma cells of 8.8%, which led to the diagnosis of monoclonal gammopathy of undetermined significance (MGUS). 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) scintigraphy confirmed mild cardiac uptake (Perugini score: grade 1). Cardiac magnetic resonance disclosed hypertrophy in the intertribal and interventricular septum, mid-wall late gadolinium enhancement and washout (dark blood pool) of the intracardiac contrast material (Figure 1D and E). Based on these findings, AL cardiac amyloid was suspected, leading to the undertaking of a myocardial biopsy. This biopsy unveiled amyloid deposits that exhibited Congo red staining and manifested apple-green birefringence under polarized light. On immunostaining, there was diffuse kappa and lambda staining in different areas from amyloid deposition. Therefore, this staining was deemed non-specific. On the other hand, the area with amyloid deposits exhibited positive transthyretin (TTR) staining (Figure 2), subsequent genetic testing of TTR disclosed a V122I mutation, leading to the diagnosis of ATTRv. Given the absence of indication of treatment for MGUS, patisiran was commenced for ATTRv. While serum transthyretin levels reduced and neurological deficits exhibited marginal improvement under treatment, the patient’s clinical trajectory deviated from the anticipated course of ATTRv, including poor response to diuretics, aggravated congestion and pleural effusion observed on chest radiography, and compounded by worsening dyspnoea and macroglossia (Figure 3). The need for a re-evaluation of the amyloidosis type arose, the decision was to conduct biopsies of the tongue and abdominal wall fat. Subsequent analysis illuminated the presence of both transthyretin and lambda staining within the areas exhibiting amyloid deposition (Figure 4). Consequently, the diagnosis of coexisting ATTRv and AL amyloidosis was made. As a response, combination therapy with melphalan (6 mg orally for 4 days every 3 weeks) and dexamethasone (20 mg orally on the same day) was added. The condition of heart failure showed little improvement with any treatment, and her appetite gradually decreased. Patisiran was administered until shortly before her passing, but the treatment for AL was terminated 10 months after diagnosis due to deterioration of performance status. She passed away 24 months after her initial visit to our hospital. When the initial endomyocardial biopsy specimen underwent mass spectrometry as a post hoc analysis, both ATTR and AL amyloid were significantly detected.
![(A) Chest radiography showed mild cardiomegaly and congestion. (B) electrocardiography showed sinus rhythm (heart rate: 58 beats/min) with low voltage. (C) Transthoracic echocardiogram (parasternal long-axis view) revealed increased left ventricular wall thickness and speckled myocardium. Transmitral Doppler was restrictive pattern (E wave = 94 cm/s, E′ = 2.8 cm/s, E/E′ = 23). (D) 99mTc-hydroxymethylene diphosphonate scintigraphy showed mild myocardial uptake [Perugini score: 1, heart to contralateral ratio (H/CL ratio): 1.2]. (E) Cardiac magnetic resonance showed hypertrophy in dual ventricular and atrial septum, mid-wall late gadolinium enhancement (arrows) and washout (dark blood pool) of the intracardiac contract material.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ehjcr/8/6/10.1093_ehjcr_ytae264/4/m_ytae264f1.jpeg?Expires=1747974069&Signature=aSKIpnTORoG5EnjTSPQsGxsY0DD91H~eJbfhNwCTuLz~qQFBQCdZsBcxOqoqJJYCvPA3M-N0lUC9d7rwfTO5hUOK250OsD2vc9z0C8Fim~xqd1R5WdaLEfuXLCauKv1UkvEOinXaoXjQHXriPige9rUjvjeRr6QYMnsH-V5r4j1ch3XZFXaI2QBE-10~UTcUQsyXix6a-ZRU5ZETMevOZSV57MZnrHV1QiBjREx48BbqtlSmhwtEausaZbuhyHwG~LOuED2jHLsGWuxL6vw6KsVjMvJW8GMZ0uKVYzWowXBOo2TFb9kwFXhz04tCfVKL0iAEs2OYOGwCQ4kmHJFWoQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Chest radiography showed mild cardiomegaly and congestion. (B) electrocardiography showed sinus rhythm (heart rate: 58 beats/min) with low voltage. (C) Transthoracic echocardiogram (parasternal long-axis view) revealed increased left ventricular wall thickness and speckled myocardium. Transmitral Doppler was restrictive pattern (E wave = 94 cm/s, E′ = 2.8 cm/s, E/E′ = 23). (D) 99mTc-hydroxymethylene diphosphonate scintigraphy showed mild myocardial uptake [Perugini score: 1, heart to contralateral ratio (H/CL ratio): 1.2]. (E) Cardiac magnetic resonance showed hypertrophy in dual ventricular and atrial septum, mid-wall late gadolinium enhancement (arrows) and washout (dark blood pool) of the intracardiac contract material.
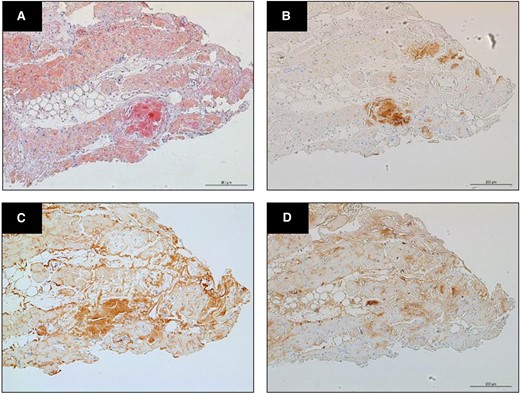
(A) Myocardial biopsy showed amyloid deposits staining with Congo red (×40 magnification). (B) On immunohistochemistry, staining was positive for anti-transthyretin antiserum. (C) Lambda and (D) kappa staining was diffusely stained in areas different from amyloid deposition, therefore these staining was considered to be non-specific.
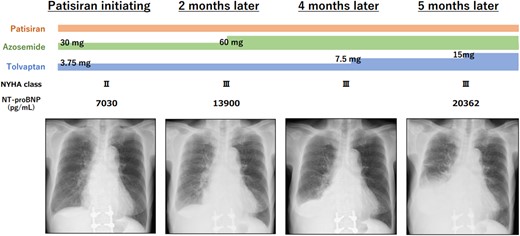
Patisiran (0.3 mg per kg of body weight intravenously once every 3 weeks) was initiated for ATTRv. Although her clinical course was atypical for ATTRv, including increased diuretics, increased congestion, and pleural effusion on chest radiography.
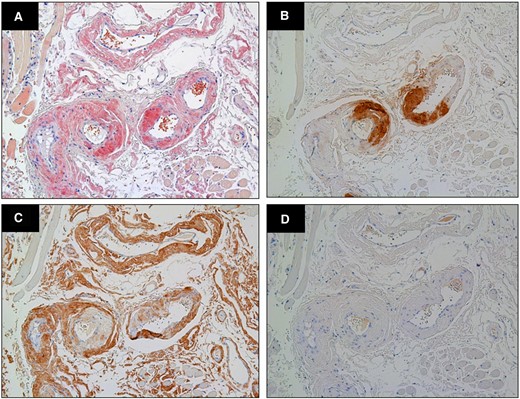
(A) Congo red stained biopsy of tongue showed amyloid deposits (×400 magnification). On immunohistochemistry showed intense transthyretin (B) and lambda light-chain (C) immunostaining, with negative kappa staining (D).
Laboratory data at diagnosis
| Investigations | Results | Normal ranges | |
|---|---|---|---|
| White blood cell count | 6.3 | ×103/μL | 3.3–8.6 |
| Haemoglobin | 13.2 | g/dL | 11.6–14.8 |
| Platelet count | 23.1 | ×104/μL | 15.8–34.8 |
| Creatinine | 0.94 | mg/dL | 0.46–0.79 |
| High sensitive troponin T | 0.051 | ng/mL | 0.000–0.014 |
| N-terminal pro-brain natriuretic peptide | 782.6 | pg/mL | <125 |
| IgG | 2858 | mg/dL | 861–1747 |
| Lambda light chain | 293.0 | mg/L | 5.7–26.3 |
| Kappa light chain | 30.5 | mg/L | 3.3–19.4 |
| Free light-chain ratio | 0.10 | 0.26–1.65 | |
| Serum immunofixation electrophoresis | IgG lambda M protein | ||
| Investigations | Results | Normal ranges | |
|---|---|---|---|
| White blood cell count | 6.3 | ×103/μL | 3.3–8.6 |
| Haemoglobin | 13.2 | g/dL | 11.6–14.8 |
| Platelet count | 23.1 | ×104/μL | 15.8–34.8 |
| Creatinine | 0.94 | mg/dL | 0.46–0.79 |
| High sensitive troponin T | 0.051 | ng/mL | 0.000–0.014 |
| N-terminal pro-brain natriuretic peptide | 782.6 | pg/mL | <125 |
| IgG | 2858 | mg/dL | 861–1747 |
| Lambda light chain | 293.0 | mg/L | 5.7–26.3 |
| Kappa light chain | 30.5 | mg/L | 3.3–19.4 |
| Free light-chain ratio | 0.10 | 0.26–1.65 | |
| Serum immunofixation electrophoresis | IgG lambda M protein | ||
Laboratory data at diagnosis
| Investigations | Results | Normal ranges | |
|---|---|---|---|
| White blood cell count | 6.3 | ×103/μL | 3.3–8.6 |
| Haemoglobin | 13.2 | g/dL | 11.6–14.8 |
| Platelet count | 23.1 | ×104/μL | 15.8–34.8 |
| Creatinine | 0.94 | mg/dL | 0.46–0.79 |
| High sensitive troponin T | 0.051 | ng/mL | 0.000–0.014 |
| N-terminal pro-brain natriuretic peptide | 782.6 | pg/mL | <125 |
| IgG | 2858 | mg/dL | 861–1747 |
| Lambda light chain | 293.0 | mg/L | 5.7–26.3 |
| Kappa light chain | 30.5 | mg/L | 3.3–19.4 |
| Free light-chain ratio | 0.10 | 0.26–1.65 | |
| Serum immunofixation electrophoresis | IgG lambda M protein | ||
| Investigations | Results | Normal ranges | |
|---|---|---|---|
| White blood cell count | 6.3 | ×103/μL | 3.3–8.6 |
| Haemoglobin | 13.2 | g/dL | 11.6–14.8 |
| Platelet count | 23.1 | ×104/μL | 15.8–34.8 |
| Creatinine | 0.94 | mg/dL | 0.46–0.79 |
| High sensitive troponin T | 0.051 | ng/mL | 0.000–0.014 |
| N-terminal pro-brain natriuretic peptide | 782.6 | pg/mL | <125 |
| IgG | 2858 | mg/dL | 861–1747 |
| Lambda light chain | 293.0 | mg/L | 5.7–26.3 |
| Kappa light chain | 30.5 | mg/L | 3.3–19.4 |
| Free light-chain ratio | 0.10 | 0.26–1.65 | |
| Serum immunofixation electrophoresis | IgG lambda M protein | ||
Discussion
To our knowledge, this report represents a rare case in which both ATTRv and AL amyloid depositions have been confirmed in the same pathological specimen and subsequently treated. In complicated and uncertain cases, it is essential to quickly utilize reliable tests, such as mass spectrometry and not only immunohistochemistry, to begin appropriate therapy as soon as possible.
Although two types of amyloidosis rarely coexist, some previous reports suggested the possibility that two types could coexist.2–4 The study conducted at the Mayo Clinic reported that out of 1094 cases of amyloidosis diagnosed via mass spectrometry, nine cases exhibited the presence of two distinct types of amyloidosis.2 All instances of coexistence were diagnosed ATTR amyloidosis as one of the amyloid types. Among these cases, six featured AL amyloidosis as the secondary type. Eight out of nine cases showed no mutations in the TTR gene and were consequently diagnosed as ATTRwt. Only one case unveiled an amino acid abnormality (V122l), leading to its diagnosis as ATTRv. Unlike our case, this instance saw the diagnosis of ATTRv many years after an initial AL diagnosis, and its clinical trajectory remains unclear. While the coexistence of two types of amyloidosis may be rare, it’s crucial for us to remain mindful of the potential for such coexistence, as it could entail distinct treatment strategies.2 This awareness is particularly important in the context of the evolving landscape of amyloidosis treatment.
We can deduce the type of CA based on the clinical course, serum immunofixation electrophoresis, or imaging. In most cases, the pathologically diagnosed CA type aligns with the assumed type derived from the clinical course. However, in our case, disparities emerged between the pathological diagnosis and other findings, including the clinical course, a physical examination, and 99mTc-HMDP scintigraphy results. According to ESC guidelines, when scintigraphy shows cardiac uptake and at least one of the monoclonal protein tests is abnormal, consideration should be given to ATTR with concomitant MGUS, AL amyloidosis, or the coexistence of AL and ATTR. In intricate cases like these, the definitive diagnosis hinges on pathological findings, overriding other clinical findings.1 Initially, our case was diagnosed with as ATTR concomitant MGUS based on immunohistochemistry findings of myocardial biopsy specimens according to the diagnostic flow chart. The initial discrepancy was that 99mTc-HMDP scintigraphy revealed only weak uptake, despite the case’s pathological diagnosis of ATTR amyloidosis. 99mTc-HMDP scintigraphy is high sensitivity for ATTR diagnosis,5 and the diagnostic efficacy for ATTR cardiac amyloidosis demonstrates a sensitivity of 58–99% and specificity of 79–100%.5,6 However, it can be negative in the initial stages of ATTR.
Furthermore, the clinical trajectory was swift despite the ATTR.7 In cases where the patients suffered from only ATTR amyloidosis, rapid advancement subsequent treatment initiation is atypical. Given the elevated serum troponin levels upon diagnosis and the rapid clinical course, it is improbable that this was at an early stage of the ATTRv. Because the type of amyloid influences how the organs are impaired,8 the contradiction in our case was resolved by performing biopsies from different organs.
Regarding the myocardial biopsy conducted during the initial diagnosis, the immunostaining of lambda was deemed non-specific due to incongruity between the immunostaining area and the Congo red staining area. The query persists: how did heart failure progress rapidly despite the absence of AL amyloid deposition in the myocardium? In retrospect, these depositions might have manifested as light chains deposition disease without the accrual of amyloid,9 suggesting that heart failure could also have been caused by the light chains. On the other hand, the reduced stainability of AL amyloid with Congo red staining, in comparison to TTR amyloid, as observed in the tongue biopsy (Figure 4), may have made it difficult to identify the Congo red staining areas of AL amyloid in the myocardial biopsy specimens. Previous reports also have stated that the use of immunoelectron microscopy and mass spectrometry has enabled the accurate classification of amyloidosis.10 In our case, mass spectrometry detected both ATTR and AL amyloid in the initial myocardial biopsy, leading to the interpretation of the pathophysiology.
Conclusion
Determining the type of amyloid and initiating treatment are of clinical importance. In cases where there are disparities between immunohistology findings and other clinical findings, it becomes essential to re-evaluate the amyloid type using mass spectrometry and immunoelectron microscopy.
Lead author biography

Dr Yuko Eda is a physician at Kitasato University School of Medicine. Her areas of interest include cardiac amyloid and heart failure.
Acknowledgements
The authors are grateful to Fuyuki Kametani, PhD of Institute for Biomedical Sciences, Shinshu University for performing a mass spectrometry. We also thank Masahide Yazaki, MD, PhD of Institute for Biomedical Sciences, Shinshu University for extracting amyloid from the specimen by laser microdissection.
Consent: The authors confirm that written consents for submission and publication of this case report including images and associated text have been obtained from the patients in line with COPE guidance.
Funding: None declared.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Author notes
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments