





Abstract
Amyloidosis is a systemic disorder of abnormal protein folding and deposition resulting in a range of symptoms including neuropathy, heart failure, renal disease, and dermatologic findings. The two most common types of amyloidosis that affect the heart are transthyretin (ATTR) amyloidosis and light chain (AL) amyloidosis, which vary in clinical presentation. Skin findings such as periorbital purpura are considered more specific for AL amyloidosis. However, there are rare cases of ATTR amyloidosis causing the same dermatologic findings.
A 69-year-old female presented for evaluation of amyloidosis after cardiac imaging done at the time of a recent atrial fibrillation ablation showed signs of infiltrative disease. On examination, she had periorbital purpura which she reportedly had for years without receiving a diagnosis, as well as macroglossia with teeth indentation. These exam findings, in addition to her transthoracic echocardiogram showing apical sparing, are typically considered characteristic of AL amyloidosis. Subsequent workup revealed the presence of hereditary ATTR (hATTR) amyloidosis with a heterozygous pathogenic variant in the TTR gene producing the p.Thr80Ala mutation.
Spontaneous periorbital purpura is thought to be pathognomonic for AL amyloidosis. However, we describe a case of hereditary ATTR amyloidosis with the Thr80Ala TTR genetic variant presenting initially with periorbital purpura, the first case documented in the literature to our knowledge.
Systemic amyloidosis can present in several ways, including subtle dermatologic manifestations like macroglossia and periorbital purpura.
While the light chain variant of amyloidosis has been classically associated with periorbital purpura, it is important to recognize that there are cases of hereditary amyloidosis causing these findings.
Multidisciplinary care is critical in the management of patients with systemic amyloidosis with the advantages of optimizing patient care, outcomes, and satisfaction.
Introduction
Cardiac amyloidosis is an infiltrative cardiomyopathy caused by aggregation of misfolded precursor proteins into insoluble amyloid fibrils. Transthyretin (ATTR) amyloidosis and light chain (AL) amyloidosis are known subtypes. ATTR amyloidosis is characterized by the presence of a mutation [hereditary ATTR (hATTR) amyloidosis] or absence of a mutation (wild-type ATTR amyloidosis).1 Endomyocardial biopsy (EMB) provides definitive diagnosis of cardiac amyloidosis.2 However, non-invasive diagnostic criteria have emerged and include the use of bone scintigraphy, in the absence of a monoclonal protein, for the diagnosis of ATTR amyloidosis.3
Dermatologic signs, such as periorbital purpura, were previously thought to be pathognomonic for AL amyloidosis, but we present a rare case of periorbital purpura as the first manifestation of hereditary ATTR amyloidosis.
Timeline
| Time | Event |
|---|---|
| May 2022 | Patient underwent second ablation for atrial fibrillation Cardiac magnetic resonance image raised concerns for possible infiltrative heart disease |
| Early June 2022 | Evaluated in the Multidisciplinary Amyloidosis Clinic Presented with periorbital purpura, macroglossia, and teeth indentation Serologic workup was negative for AL amyloidosis |
| Late June 2022 | Patient underwent right ventricular endomyocardial biopsy |
| July 2022 | Congo red-positive amyloid deposits were present on biopsy Liquid chromatography with tandem mass spectrometry (LC MS/MS) detected ATTR-type amyloid deposition Liquid chromatography with tandem mass spectrometry detected an amino acid sequence abnormality in the transthyretin protein (Thr80Ala or T80A) |
| August 2022 | Follow-up in the Multidisciplinary Amyloidosis Clinic Started therapy with transthyretin stabilizer (tafamidis) Nerve conduction studies demonstrated small fibre polyneuropathy |
| September 2022 | Started therapy with small-interfering RNA agent (patisiran) |
| October 2022 | Patient had worsening volume overload and diuretic doses were increased |
| January 2023 | Patient endorsed an improvement in symptoms with diuretics Patient was advised to continue tafamidis and patisiran |
| Time | Event |
|---|---|
| May 2022 | Patient underwent second ablation for atrial fibrillation Cardiac magnetic resonance image raised concerns for possible infiltrative heart disease |
| Early June 2022 | Evaluated in the Multidisciplinary Amyloidosis Clinic Presented with periorbital purpura, macroglossia, and teeth indentation Serologic workup was negative for AL amyloidosis |
| Late June 2022 | Patient underwent right ventricular endomyocardial biopsy |
| July 2022 | Congo red-positive amyloid deposits were present on biopsy Liquid chromatography with tandem mass spectrometry (LC MS/MS) detected ATTR-type amyloid deposition Liquid chromatography with tandem mass spectrometry detected an amino acid sequence abnormality in the transthyretin protein (Thr80Ala or T80A) |
| August 2022 | Follow-up in the Multidisciplinary Amyloidosis Clinic Started therapy with transthyretin stabilizer (tafamidis) Nerve conduction studies demonstrated small fibre polyneuropathy |
| September 2022 | Started therapy with small-interfering RNA agent (patisiran) |
| October 2022 | Patient had worsening volume overload and diuretic doses were increased |
| January 2023 | Patient endorsed an improvement in symptoms with diuretics Patient was advised to continue tafamidis and patisiran |
| Time | Event |
|---|---|
| May 2022 | Patient underwent second ablation for atrial fibrillation Cardiac magnetic resonance image raised concerns for possible infiltrative heart disease |
| Early June 2022 | Evaluated in the Multidisciplinary Amyloidosis Clinic Presented with periorbital purpura, macroglossia, and teeth indentation Serologic workup was negative for AL amyloidosis |
| Late June 2022 | Patient underwent right ventricular endomyocardial biopsy |
| July 2022 | Congo red-positive amyloid deposits were present on biopsy Liquid chromatography with tandem mass spectrometry (LC MS/MS) detected ATTR-type amyloid deposition Liquid chromatography with tandem mass spectrometry detected an amino acid sequence abnormality in the transthyretin protein (Thr80Ala or T80A) |
| August 2022 | Follow-up in the Multidisciplinary Amyloidosis Clinic Started therapy with transthyretin stabilizer (tafamidis) Nerve conduction studies demonstrated small fibre polyneuropathy |
| September 2022 | Started therapy with small-interfering RNA agent (patisiran) |
| October 2022 | Patient had worsening volume overload and diuretic doses were increased |
| January 2023 | Patient endorsed an improvement in symptoms with diuretics Patient was advised to continue tafamidis and patisiran |
| Time | Event |
|---|---|
| May 2022 | Patient underwent second ablation for atrial fibrillation Cardiac magnetic resonance image raised concerns for possible infiltrative heart disease |
| Early June 2022 | Evaluated in the Multidisciplinary Amyloidosis Clinic Presented with periorbital purpura, macroglossia, and teeth indentation Serologic workup was negative for AL amyloidosis |
| Late June 2022 | Patient underwent right ventricular endomyocardial biopsy |
| July 2022 | Congo red-positive amyloid deposits were present on biopsy Liquid chromatography with tandem mass spectrometry (LC MS/MS) detected ATTR-type amyloid deposition Liquid chromatography with tandem mass spectrometry detected an amino acid sequence abnormality in the transthyretin protein (Thr80Ala or T80A) |
| August 2022 | Follow-up in the Multidisciplinary Amyloidosis Clinic Started therapy with transthyretin stabilizer (tafamidis) Nerve conduction studies demonstrated small fibre polyneuropathy |
| September 2022 | Started therapy with small-interfering RNA agent (patisiran) |
| October 2022 | Patient had worsening volume overload and diuretic doses were increased |
| January 2023 | Patient endorsed an improvement in symptoms with diuretics Patient was advised to continue tafamidis and patisiran |
Case presentation
A 69-year-old female was referred to the outpatient cardiology clinic due to concern for cardiac amyloidosis. She had a medical history of paroxysmal atrial fibrillation requiring two prior catheter ablations and anticoagulation with apixaban. Transthoracic echocardiography (TTE) demonstrated increased biventricular wall thickness, and subsequent cardiac magnetic resonance imaging suggested an infiltrative process with diffuse subendocardial late gadolinium enhancement and abnormal nulling of the myocardial signal. Therefore, she was referred to our Cardiac Amyloidosis Clinic for further evaluation.
Her review of systems was notable for fatigue, shortness of breath, and lightheadedness for more than six months. She mentioned having bruising around her eyes for many years, and although she previously sought evaluation for this symptom, she was told that she had fragile blood vessels with no further investigation. She denied ocular trauma, carpal tunnel syndrome, neuropathy, lumbar stenosis, biceps tendon rupture, or inflammatory arthropathy. The patient denied tobacco use or recreational drug use but consumed one–two glasses of red wine per week. Family history was significant for pulmonary sarcoidosis, unspecified heart failure in her father, and trigeminal neuralgia in her mother. The patient reported being of British, French, and Irish ancestry.
On physical examination, the patient had macroglossia with teeth indentation and scalloping along the lateral margins of the tongue. Ocular exam demonstrated periorbital purpura (Figure 1). The jugular venous pressure was elevated, and she had an irregular rhythm with a normal heart rate. The remainder of her physical exam was unremarkable.
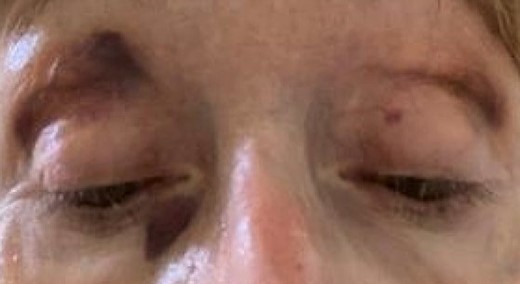
Bilateral periorbital purpura.
Initial serologic workup revealed a normal complete blood count, renal function, and hepatic function profile. Repeat TTE was performed with longitudinal strain imaging and demonstrated increased biventricular wall thickness, thickened atrioventricular valves, biatrial enlargement (Video 1), Doppler haemodynamics with E velocity of 1.2 m/sec (reference range 0.4–0.9 m/s) and medial e’ velocity of 3 cm/sec (reference range ≥ 8 cm/s), and abnormal global longitudinal strain of −10.2% with relative apical preservation (Figure 2). Given these findings, there was concern for cardiac amyloidosis.4 Haematologic testing revealed a normal serum-free AL ratio of 1.45 (reference range 0.26–1.65) without evidence of monoclonal paraprotein on serum or urine immunofixation. However, given her unusual presentation of spontaneous periorbital purpura, which is often associated with AL amyloidosis, EMB was recommended.
Transthoracic echocardiogram with a small left ventricular chamber, concentric hypertrophy, ejection fraction of 56%, and no regional wall abnormalities.
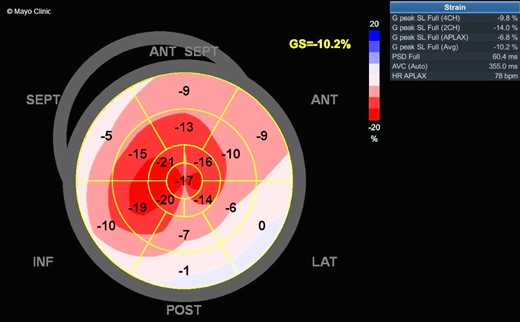
Myocardial strain imaging demonstrating abnormal global longitudinal strain of −10.2% with apical sparing.
Right ventricular EMB with Congo red staining revealed amyloid deposits within the myocardium and vessel walls. Low power images of the biopsy showed normal cardiac myofibres within the tissue, with amorphous extracellular deposits (Figure 3). On high power magnification, a pale-pink hyaline material was present that stained strongly with Congo red (Figure 3). Liquid chromatography with tandem mass spectrometry (LC MS/MS) detected a peptide profile consistent with ATTR amyloidosis and an amino acid sequence abnormality in the ATTR protein. Genetic testing confirmed a Thr80Ala (legacy nomenclature Thr60Ala, which omits the 20 amino acid peptide) mutation in the TTR gene.
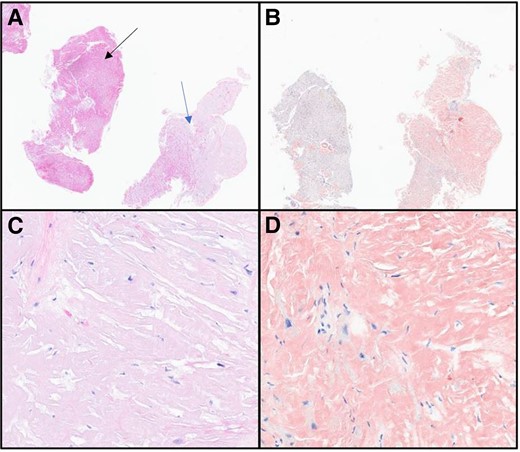
(A) Haematoxylin and eosin 2×. Tissue on the left (black arrow) showing normal myocardial muscle with pale, pink deposits within vessels and infiltration into the tissue on the right (blue arrow). (B) Congo red 2×. Congo red staining positive in the amyloid deposits. (C) Haematoxylin and eosin 40×. Pale-pink, glassy extracellular material present. No plasma cells identified. (D) Congo red 40×. Strong Congo red staining identified, confirming the diagnosis of amyloidosis.
After being diagnosed with hereditary ATTR amyloidosis, tafamidis therapy (61 mg capsule once daily) was started, which is currently the only therapy approved for cardiac amyloidosis by the Food and Drug Administration in the USA. Subsequent electromyography to assess for nerve involvement showed mild bilateral median mononeuropathies at the wrists consistent with bilateral carpal tunnel syndrome.2 Nerve conduction studies demonstrated decreased postganglionic sudomotor function in the hand and proximal leg, concerning for a small fibre neuropathy. Given the diagnosis of hereditary ATTR amyloidosis with cardiac involvement and newly diagnosed polyneuropathy, patisiran, a small-interfering RNA that targets TTR mRNA, was also initiated intravenously at a dose of 0.3 mg/kg every three weeks. The patient continues to be closely followed in the Cardiac Amyloidosis Clinic.
Discussion
Amyloidosis constitutes a group of disorders caused by abnormal deposition and aggregation of misfolded precursor proteins. Over 30 different protein types have been identified as amyloidogenic, but the two most frequently implicated in cardiac amyloidosis are ATTR and AL amyloid.5 Non-invasive imaging has emerged as a reliable mode of diagnosis. However, in circumstances where differentiation between AL and ATTR amyloidosis is challenging, a biopsy is critical. Early detection and accurate diagnosis enable prompt initiation of appropriate therapy, especially since AL amyloidosis requires chemotherapy while ATTR amyloidosis does not.
Light chain amyloidosis is often associated with skin manifestations such as nodular, indurated, or bullous lesions.6 Periorbital purpura is a common manifestation of amyloid deposition in superficial vessels, leading to vessel fragility and increased susceptibility to ecchymoses with minor trauma. Other soft tissue findings such as macroglossia with teeth indentation and scalloping, muscle pseudohypertrophy, enlarged glandular tissue, and submandibular infiltrations are also observed. Periorbital purpura is thought to be pathognomonic for AL amyloidosis.4 However, a few case reports describe the presence of periorbital purpura in hereditary ATTR amyloidosis. One case identified a patient with periorbital and scleral purpura who, upon genetic testing, was found to have a heterozygous Ser77Tyr mutation in the TTR gene consistent with a diagnosis of hATTR.7 Another case reported a Glu54Gly mutation in the TTR gene in a patient who had spontaneous periocular bruising.8 Spontaneous periorbital purpura has not been described with the Thr80Ala mutation to our knowledge.
There are over 120 pathogenic variants reported in the TTR gene, and these mutations have been shown to cluster in groups with shared geography or ethnicity.1 Hereditary ATTR amyloidosis has an autosomal dominant pattern of inheritance.8 The most common pathogenic TTR variant in the USA is the Val142Ile mutation, which occurs in 3.4% of African Americans.9 Thr80Ala is the second most common mutation in the USA, and this variant originated in the northern parts of the Republic of Ireland.8 Symptoms in this cohort of patients typically present between 50 and 60 years of age, with predominantly neurologic and cardiac phenotypes.1TTR genetic testing is recommended for all patients with ATTR amyloidosis, particularly given that this has implications for first-degree relatives. The role of concomitant use of stabilizers and silencer therapy for ATTR amyloidosis is still being investigated, as is the best way to monitor progression and response to therapy in ATTR cardiomyopathy, with further studies needed.
Recent advancements in treatment for ATTR amyloidosis include strategies for ATTR stabilization and silencing of ATTR production, although the latter is only approved for familial amyloid polyneuropathy.10 The stabilizer, tafamidis, is approved for ATTR cardiac amyloidosis in the USA. Our patient was initiated on both silencer (patisiran) and stabilizer therapies. Although there is limited evidence to support concomitant use of these classes at this time, ongoing trials such as HELIOS B (ClinicalTrials.gov Identifier: NCT04153149) and CardioTTRansform (ClinicalTrials.gov Identifier: NCT04136171), in which patients are on both therapies, may offer additional guidance.
Lead author biography
 Dr Nikita Jhawar is an internal medicine resident physician at Mayo Clinic in Jacksonville, FL, who aspires to pursue a fellowship in cardiology.
Dr Nikita Jhawar is an internal medicine resident physician at Mayo Clinic in Jacksonville, FL, who aspires to pursue a fellowship in cardiology.
Supplementary material
Supplementary material is available at European Heart Journal – Case Reports.
Slide sets: A fully edited slide set detailing this case and suitable for local presentation is available online as Supplementary data.
Consent: Written consent and verbal consent were obtained directly from the patient in accordance with the COPE guidelines.
Funding: No funding was provided for this manuscript.
References
Author notes
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
Comments