





Abstract
This International Consensus Guideline was developed by experts in the field of small for gestational age (SGA) of 10 pediatric endocrine societies worldwide. A consensus meeting was held and 1300 articles formed the basis for discussions. All experts voted about the strengths of the recommendations. The guideline gives new and clinically relevant insights into the etiology of short stature after SGA birth, including novel knowledge about (epi)genetic causes. Further, it presents long-term consequences of SGA birth and also reviews new treatment options, including treatment with gonadotropin-releasing hormone agonist (GnRHa) in addition to growth hormone (GH) treatment, as well as the metabolic and cardiovascular health of young adults born SGA after cessation of childhood GH treatment in comparison with appropriate control groups.
To diagnose SGA, accurate anthropometry and use of national growth charts are recommended. Follow-up in early life is warranted and neurodevelopment evaluation in those at risk. Excessive postnatal weight gain should be avoided, as this is associated with an unfavorable cardiometabolic health profile in adulthood. Children born SGA with persistent short stature < −2.5 SDS at age 2 years or < −2 SDS at 3 to 4 years of age, should be referred for diagnostic workup. In case of dysmorphic features, major malformations, microcephaly, developmental delay, intellectual disability, and/or signs of skeletal dysplasia, genetic testing should be considered. Treatment with 0.033 to 0.067 mg GH/kg/day is recommended in case of persistent short stature at age of 3 to 4 years. Adding GnRHa treatment could be considered when short adult height is expected at pubertal onset. All young adults born SGA require counseling to adopt a healthy lifestyle.

Small for gestational age (SGA) is defined as a birth weight and/or length < −2 SDS
Follow-up in early life is warranted and neurodevelopment evaluation in children at risk
Excessive weight gain, particularly in early life, should be avoided as this is associated with an unfavorable health profile in adulthood
New (epi)genetic causes of short stature after SGA birth are increasingly found
Short children born SGA should be referred for diagnostic workup when height < −2.5 SDS at age 2 years or < −2 SDS at age 3 to 4 years
Start of GH treatment (dose of 0.033-0.067 mg/kg/day) is recommended in case of persistent short stature at age 3 to 4 years, as it is effective and safe, including in the long term
Routine health surveillance is not needed in all adults born SGA, but they require counseling to adopt a healthy lifestyle
Low birth weight is associated with both long-term and short-term consequences. Low birth weight is defined by the World Health Organization (WHO) as a birth weight of less than 2500 g (1). This definition includes, however, both preterm neonates, who are generally born with an appropriate size for their gestational age, and neonates with a low birth weight for gestational age. Low birth weight has been associated with an increased risk of type 2 diabetes mellitus and cardiometabolic diseases relatively early in adult life (2, 3). An extensive literature search and a meeting was organized to examine the current data relevant to diagnosis, etiology, and consequences of small for gestational age (SGA) birth and proposed clinical management. This International Consensus Guideline on SGA was developed with experts in the field of SGA, including representatives of the European Society for Paediatric Endocrinology (ESPE), Pediatric Endocrine Society (PES), Asia Pacific Pediatric Endocrine Society (APPES), Australasian Paediatric Endocrine Group (APEG), Sociedad Latino-Americana de Endocrinología Pediátrica (SLEP), Japanese Society for Pediatric Endocrinology (JSPE), Arab Society for Paediatric Endocrinology and Diabetes (ASPED), Chinese Society of Pediatric Endocrinology and Metabolism (CSPEM), Indian Society for Pediatric and Adolescent Endocrinology (ISPAE), and International Society for Pediatric and Adolescent Diabetes (ISPAD). This guideline gives special attention to new and clinically relevant insights into the etiology of short stature, including novel knowledge about (epi)genetic causes. Further, it presents the long-term consequences of SGA birth and new treatment options for short stature after SGA birth since the Consensus Meeting in 2006 (4). Moreover, it provides a review of the effects of treatment with gonadotropin-releasing hormone agonists (GnRHa) in addition to growth hormone (GH) treatment, and the metabolic and cardiovascular health of young adults born SGA after cessation of childhood GH treatment in comparison with appropriate controls groups.
Methods
The taskforce consisted of 26 members, who were invited through their societies based on their publication record and expertise in SGA. They included pediatric endocrinologists, clinical geneticists and neonatologists, and representatives nominated by 10 international pediatric endocrine societies. The consensus was supported by academic and society funding without pharmaceutical support or presence. A comprehensive literature search was conducted using the PubMed, Embase, and Cochrane databases, and the search terms “small for date infant” and “gestation.” Searches were performed in December 2019 and updated in March 2022. Additional relevant articles were identified by PubMed searches when supplementary information was necessary. A comprehensive review of 1300 articles formed the basis of discussion by 3 working groups: 1.) Diagnosis and etiology of SGA (M.B., K.K., Y.N., A.H-K., A.A.J., A.D.); 2.) Consequences of SGA (S.C., J.D., V.M., W.C., P.H., E.I., X.L.); and 3.) Clinical management of SGA (M.v.d.S., R.H., R.R., A.A., D.B., E.C., S.D.C., A.D., W.G., C.K.G, V.K., S.M., M.Y.). Preparations for the consensus included 2 preparatory meetings and regular teleconference discussions between the working group members. Propositions and recommendations were prepared and reviewed by participants prior to the consensus meeting. At the final consensus meeting, these were discussed in plenary sessions, enabling reformulation of the recommendations if required. Where published data were unavailable or insufficient, experts’ clinical experiences and opinions were considered. Finally, all experts voted on the recommendations using the following system: A. General agreement allows full agreement with the recommendation. B. General agreement is in favor of the recommendation. C. General agreement is weak for the recommendation. D. There is no general agreement with the recommendation. Depending on the proportion of votes received, the strength of the recommendation was recorded as follows: + 26% to 49% of the votes; ++ 50% to 69% of the votes; and +++ ≥ 70% of the votes.
Being born SGA is defined as having a birth weight and/or birth length below −2 SDS for gestational age.
Catch-up growth in height is defined as a growth rate (cm/year) of > 0 SDS, ie, more than the median for chronological age and sex.
Appropriate catch-up growth in height is defined as growing into the normal height range, taking into account the mid-parental height.
Short stature is defined as a height below −2.0 SDS for age and sex.
There was full agreement on these definitions among the taskforce members.
Diagnosis and Etiology of SGA
Intrauterine Growth
Normally, intrauterine linear and organ growth and weight gain occur in a balanced way to attend fetal demands while preserving the health of the mother. This involves a complex interaction of maternal, placental, and fetal factors. Maternal influences on fetal growth are determined by nutrient intake, health conditions, medication, habits, and genetic factors (eg, height, weight, and uterine capacity) (5). Biological and pathological conditions during any time of this process have the potential to interfere with growth potential and reduce size at birth. Intrauterine growth charts are used to interpret fetal growth. The interpretation of fetal growth is strongly dependent on the accuracy of gestational age determination. The most accurate method for the determination of gestational age is ultrasound assessment obtained in early pregnancy (6).
Definition of SGA and Intrauterine Growth Retardation
SGA describes neonates born with a birth weight and/or length below the normal range for gestational age. Although the terms intrauterine growth retardation (IUGR) and SGA are often used as synonyms, they are not interchangeable. IUGR refers to inappropriate gain in estimated fetal weight and abdominal circumference during a certain period of gestation based on 2 ultrasound measurements, irrespective of the size at birth (7). Most but not all infants with IUGR may be born SGA, depending on timing and severity of the intrauterine insult (8, 9). Conversely, many SGA infants will have not experienced IUGR. IUGR tends to cause a relatively large head and length relative to birth weight (9, 10).
The definition of SGA requires knowledge of gestational age, precise anthropometric measurements at birth, and appropriate reference data for birth weight and birth length. Country- or ethnic-specific normative data are important for identifying those at risk. The most commonly used reference data on birth size are derived from charts developed by Usher and McLean (11) or Niklasson (12), which have been used in large studies on SGA. Alternatively, the Intergrowth-21 birth weight and length standards can be used to identify SGA (13). We recommend the use of national growth charts, when available, or the careful selection of the most appropriate for the region and ethnic-specific population (14). Neonates can be subclassified into SGA for weight, SGA for length, or SGA for both weight and length (15). Neonates born SGA with a small head circumference should be identified, as this may point to specific etiologies. These subclassifications may help in understanding the mechanisms and implications of being born SGA.
In the previous consensus statement published in 2007, SGA was uniformly defined as a birth weight and/or length below −2.0 standard deviation scores (SDS) for gestational age (4). Although this definition is arbitrary, knowing that the long-term consequences of being born SGA are likely graded rather than binary, we recommend keeping the same definition of SGA as being born with birth weight and/or length below −2 SDS for gestational age according to national reference standards, since this definition identifies children at high risk of short adult stature.
Recommendation:1.For accurate classification of SGA, we recommend the use of national growth charts, when available, or the careful selection of the most appropriate ones for the region- and ethnic-specific population. (A +++)
Etiology of SGA: Maternal, Paternal, Fetal, and Environmental Factors
SGA comprises a heterogeneous group including children with different pathologies due to a complex interaction of maternal, placental, and fetal factors. They have a broad spectrum of clinical characteristics and underlying causes under the umbrella of SGA (9). Optimal nutritional status of women in the reproductive age group is one of the most important factors for prevention of SGA. The risk of SGA is significantly higher among mothers with chronic hypertension, preeclampsia, malnutrition, subclinical hypothyroidism (particularly in iodine-deficient areas), chronic infections, and malaria (16, 17). Although not all factors resulting in fetal growth restriction are modifiable, routine practices of birth spacing, treatment of maternal infections, and improvement of access to antenatal services are important to ensure optimal fetal growth.
Genetic causes of short stature after SGA birth will be discussed in more detail later in “(Epi)genetic causes of short stature after being born SGA.”
Consequences of Being Born SGA
Neonatal Period
Mortality risk of neonates born SGA is higher than of those born appropriate for gestational age (AGA), while neonates born both preterm and SGA have the highest risk (18). Approximately one-third of SGA infants experience hypoglycemia after birth (18). Causes for hypoglycemia include low glycogen stores and lower levels of free fatty acids and ketone bodies suggestive of reduced fat stores. They also have a higher likelihood of hypothermia compared to their AGA counterparts due to excessive heat loss through an increased body surface due to a relatively large head, increased transdermal insensible losses, and lower subcutaneous fat and reduced body fat stores. In addition, hypoglycemia can negatively affect the thermoregulatory response.
Infancy
Infants born SGA have a reduced total fat mass with a normal amount of visceral fat but a reduced amount of subcutaneous fat, causing an elevated ratio of visceral to subcutaneous fat (19, 20). However, the key risk factor for developing metabolic disease in later life is not the birth size but the postnatal growth rate (20-23). Catch-up in weight and length is highest in SGA-born infants between 3 and 6 months, with higher catch-up in weight than length (24). Accelerated postnatal weight gain increases the long-term risk of obesity, metabolic diseases, aging and coronary vascular disease of all infants, but particularly in those born SGA (21-23, 25). The increase in fat mass occurs earlier and is greater than the increase in muscle mass (26). In healthy catch-up growth, gain in weight, length, and lean body mass occur in parallel (27). Observations that weight acceleration increases the risk of metabolic disease in animals without prenatal growth restriction, and that the effects of low birth weight on the adult phenotype can be reversed by preventing accelerated postnatal weight gain, support the concept that accelerated postnatal weight gain per se is the key independent risk factor for later metabolic disease (28).
Childhood and Adolescence
Growth
As a group, children born SGA do not achieve their genetic height potential, falling on average 1 SD below mid-parental height/target height (28). At least 90% of children born SGA show spontaneous catch-up growth into the normal height range (> −2 SDS) during childhood. Catch-up growth is generally defined as a sustained growth rate (cm/year) > 0 SDS, that is, more than the median for chronological age and sex (4, 29). Sufficient catch-up is arbitrarily defined as growing into the normal height range. This process is more pronounced during the first 6 months and usually completed within the first 2 years of life, in which 85% of children show catch-up growth to a normal length (30). Catch-up might take longer in infants born preterm and SGA, up to the age of 4 years (31). Children born SGA with a height below −2 SDS at the age of 3 years, have a 7-fold increased risk of remaining short, whereas those born SGA with a birth weight below −2 SDS have a 5-fold increased risk (32, 33). The reasons for insufficient catch-up growth in children born SGA are poorly understood. Factors that positively influence childhood growth include female sex, multiple birth, taller parents, and more rapid gain in length during early life (34).
Neurodevelopment and cognition
During intrauterine life, fetal neurons proliferate, differentiate, and migrate, which is genetically determined but epigenetically directed and environmentally influenced (35). The majority of children born SGA have normal pre- and postnatal growth of the brain (with the head circumference being a proxy of brain growth) (36). Physiologically, the brain is spared as much as possible in cases of IUGR. However, certain areas, such as the cerebellum, hippocampus, and cerebral cortex, demonstrate more profound effects with an impact on memory and attention, as the developing brain prioritizes protein and energy during times of deficiency. During infancy, growth factors in the central nervous system stimulate the growth and elongation of synapses (visualized in a microscope as the so-called “neuronal forest”). Malnutrition downregulates the growth factors that are critical for normal development (37). Moreover, both malnutrition and prenatal stress exposure seem to alter synaptic plasticity in the hippocampus, which is one of several mechanisms for the impaired spatial learning and memory seen in IUGR-born offspring (38). Neonates born SGA have significantly different electroencephalograph activities compared with neonates born AGA, which persist into childhood and are associated with adverse neurodevelopmental outcomes (39, 40). Children born SGA, especially the very preterm neonates, are at increased risk of developmental delay, impaired cognition, attention deficit/hyperactivity disorder (ADHD), and learning difficulties (41, 42). Among Swedish adolescents, an increased risk of poor school performance was reported in all SGA children, with the risk increasing with severity of SGA and diminishing with adequate catch-up growth (43). Two studies found that children born SGA had lower cognitive scores and more behavioral problems than children born AGA (44, 45), while another study found only impaired cognitive function and delayed developmental outcomes in short children born SGA (46, 47). In contrast, it was reported that non-asphyxiated children born SGA had cognitive scores within the normal range, if adjustments for socioeconomic status were made, with only a trend toward lower scores (48). The average score of the full developmental quotient for children born SGA was lower than that of the reference population, but it should be stressed that the majority scored more than 85, and thus are within the normal range (49).
Pubertal maturation, gonadal function, and fertility
Most children born SGA have a normal pubertal maturation (50-54). Puberty starts slightly early but within the normal age range depending on ethnicity (50, 54, 55). Age at menarche is on average 5 to 6 months earlier and pubertal progression is on average slightly faster (50, 51, 53, 54). The slightly younger age at onset of puberty and accelerated tempo is often disadvantageous for adult stature (50, 51).
Premature adrenarche can occur in girls born SGA who experienced accelerated weight gain during early childhood (56). It has been hypothesized that an earlier pubertal maturation in girls born SGA ensues from a mismatch between reduced prenatal weight gain (with reduced adipogenesis, thus a reduced capacity for safe lipid storage) and accelerated postnatal weight gain (with augmented lipogenesis, and thus an augmented need for lipid storage) (57). This mismatch might explain why girls born SGA with accelerated postnatal weight gain have an increased risk of early onset of insulin resistance and dyslipidemia, exaggerated adrenarche and premature pubarche, a (slightly) younger age at menarche, and a higher incidence of polycystic ovary syndrome (57-59), in contrast to girls born SGA without accelerated postnatal weight gain.
There is some discordance about pubertal onset and biochemistry in girls born SGA. Pubic hair, axillary hair, or apocrine odor at the onset of puberty were similar in matched healthy girls either born SGA or AGA (52, 54). However, uterine and ovarian volumes may not be similar in adolescence and have been described as smaller in girls born SGA compared with girls born AGA (53, 60). Serum dehydroepiandrosterone sulfate (DHEA-S), androstenedione, inhibin B, follicle-stimulating hormone (FSH), luteinizing hormone (LH), and testosterone concentrations were similar in both groups (61), but girls born SGA had increased estradiol and stimulated estradiol and 17-hydroxyprogesterone concentrations (52). Consistent with a PCOS risk, serum SHBG concentrations were lower, and testosterone concentrations higher in girls born SGA who had experienced catch-up in weight, but LH, FSH and estradiol concentrations were similar in adolescent girls born SGA and those born AGA (53). Short pubertal SGA boys and age-matched AGA boys had similar inhibin B and anti-Mullerian hormone (AMH) concentrations (62).
A questionnaire-based study showed no significant difference in fertility between adults born SGA or AGA (63). Normal gonadal function was found in several SGA cohorts (62, 64, 65). In contrast, some studies conducted in small numbers of SGA subjects reported alterations in sex steroid synthesis and metabolism (66). No differences were found in serum inhibin B, testosterone, FSH, and LH between young SGA and AGA men (49), suggesting that being born SGA does not impair Sertoli and Leydig cell function. In young women born SGA, no differences were found in anti-Mullerian hormone levels compared with age-matched women born AGA (67). In a small study group of young women born SGA, elevated gonadotropin concentrations associated with reduced size of uterus and ovaries (60), and reduced ovulation rate (68) was found. However, overall, the available data do not support an impact of being born SGA on gonadal function and fertility.
Thyroid function
Preterm newborns born SGA have higher thyrotropin (thyroid-stimulating hormone; TSH) levels, albeit within the normal range, and an increased incidence of transient thyroid dysfunction (69). Term neonates born SGA and older children born SGA do not show hypothyroidism (70, 71).
Bone mineral density
Bone mineral density (BMD) is on average lower but within the normal range in children born SGA (72-74). Interestingly, among individuals born SGA, both those with persistent short stature and those with spontaneous catch-up growth to a normal stature have a lower BMD of the total body (BMDTB) compared with subjects born AGA, even after correction for height SDS, suggesting that BMDTB is not only reduced by short stature but also due to other determining factors that are disturbed in subjects born SGA (20, 75). Bone mineral apparent density of the lower spine (BMADLS) is similar in young adults born SGA or AGA (76).
Kidney function
Glomerular filtration rate, blood pressure and urinary albumin-to-creatinine ratio at the age of 21 years is similar between young adults born SGA and those born AGA (77).
Metabolic consequences
In children born SGA, there is a gradual development of insulin resistance from birth onwards, which is already detected by the age of 1 to 4 years in those with spontaneous catch-up in weight (78-80). The compensatory increase in insulin secretion maintains normoglycemia (78, 81, 82). The insulin resistance in childhood is sharply amplified by accelerated fat mass accumulation and obesity in those with rapid weight gain in the first 3 months of life, and in those with a high BMI (22, 78, 83, 84). There is, however, no evidence that type 2 diabetes mellitus, impaired glucose tolerance, clinically relevant dyslipidemia or hypertension occur more commonly among children born SGA than in those born AGA (81). The risk for metabolic disorders can, however, be amplified by the presence of risk factors such as overweight (BMI body mass index (BMI) ≥ 1 SDS), obesity (BMI ≥ 2 SDS), ethnicity, and family history. Routine evaluation of metabolic parameters is, therefore, not recommended for all children born SGA, but for those with one or more risk factors.
Adulthood
Cardiometabolic risk
Barker et al reported an inverse association between birth weight and risk for type 2 diabetes mellitus, hypertension, and cardiovascular disease in adult life, with the highest risk in term-born neonates with a birth weight below 2.5 kg (2, 3, 85-87). Epidemiological evidence suggests a U-shaped curve relating the size at birth with long-term morbidity, with both SGA and large for gestational age (LGA) associated with increased risk for cardiometabolic disease (88-91). A confounding factor is the use of different cutoffs for defining SGA in different studies (92). From the cardiometabolic perspective, it is reasonable to envisage that there is a continuum from an extreme intrauterine growth restriction to milder growth impairments associated with different degrees of long-term consequences. In this context, 4 longitudinal studies that include relatively large and homogenous cohorts of (young) adults born SGA indicate a higher prevalence of metabolic syndrome, insulin resistance (93-95), type 2 diabetes and parameters of cardiometabolic risk, and higher risk of metabolically unhealthy body composition (20, 22, 96, 97). These reports indicate that subjects born SGA have parameters suggestive of a higher cardiometabolic risk in adulthood when they experienced a rapid weight gain in early life (22, 97, 98). Adults born SGA, with a median age of 34 years, showed a markedly reduced exercise capacity compared to adults born AGA (99), but further research is warranted to find the cause and if there is an association with increased cardiovascular mortality. Finally, one large population-based study (118 634 women), using data of the Swedish Medical Birth Registry from 1973 to 2003, showed an increased risk of severe preeclampsia in women born SGA (odds ratio 1.62 [95% CI, 1.32-2.02]) (100). In conclusion, there is a possible association between being born SGA and cardiometabolic risk in later life, especially in those being born SGA with accelerated weight gain in early life and/or obesity in later life (101).
Cancer risk
Epidemiological studies showed an association between birth weight and risk of cancer, in particular hepatoblastoma (102-105). There is, however, only one case-control study reporting an association between hepatoblastoma and being born SGA (OR = 1.75; 95% CI, 1.25-2.45), which is less significant than the association with low birth weight (OR = 2.02; 95% CI, 1.29-3.15), very low birth weight (OR = 15.4; 95% CI, 10.7-22.3), and preterm birth (OR = 7.27; 95% CI, 5.00-10.6) (106). In conclusion, the evidence in favor of an association between SGA and cancer risk is weak and based on a single epidemiological study.
Clinical Management of Children Born SGA
Early Life and Childhood
During the first 2 years of life, the clinical management focuses on optimal nutrition to ensure optimal catch-up growth and prevention of hypoglycemia, excessive weight gain, neurodevelopmental delay, and exclusion of an underlying cause including genetic conditions. Some may also require transient diazoxide to treat hypoglycemia which has shown to be beneficial (18, 107). Recent reviews with longer-term follow-up of children born SGA concluded that breastfeeding promotes growth without causing adverse body composition or reduced insulin sensitivity and is, therefore, ideal for infants born SGA (47, 108). A balanced diet with age-appropriate macro- and micronutrients is recommended for optimal growth and development. Early growth monitoring provides useful information regarding the growth pattern of neonates born SGA and is a vital tool in clinical practice. Weight, length, head circumference, and weight for length or BMI should be monitored every 3 months during the first year of life, 6-monthly in the second year of life, and yearly thereafter until the child reaches their genetic height potential or a height in the normal range. The growth parameters should be plotted on an appropriate growth chart and assessed for adequacy. After the first 2 years of life, besides the key elements of nutrition, growth, and neurodevelopment, the clinical management also focuses on pubertal development, and metabolic parameters, when indicated. Evaluating pubertal onset and progression is important since this could influence treatment strategies (4). As bone age is inaccurate in predicting adult height in children born SGA (4), routine bone age assessment is not recommended. Those born very preterm (gestational age < 28 weeks) or severely SGA (birth weight and/or birth length < −3 SDS for gestational age), with short parents, or having a syndrome associated with growth retardation, should be monitored more closely and referred to a pediatric endocrinologist in the first year of life.
Excessive weight gain (a change in weight for length > 0.67 SDS, that is, passing weight SDS lines in upward direction), particularly during early life, should be avoided (27). A healthy lifestyle, including a balanced diet and regular physical activity must be encouraged. Metabolic parameters, such as fasting plasma glucose, oral glucose tolerance test, and lipid profile are only recommended in children with overweight (BMI ≥ 1 SDS) or obesity (BMI ≥ 2 SDS), those with a family history of type 2 diabetes mellitus, or when clinical signs suggest a metabolic disease. Those with a small head circumference have the highest risk of cognitive impairment (109). Early neurodevelopment evaluation and interventions are warranted in children at risk.
Additional challenges in low- and middle-income countries
In low- and middle-income countries, only 50% to 60% of infants born SGA show catch-up growth in either height, weight, or both, due to multiple factors, such as poor maternal and postnatal nutrition, poor hygiene, many infections, and factors associated with low socioeconomic status (110). Catch-up growth in countries with poor nutrition and relatively low protein dietary content may be delayed up to 4 to 5 years of age (111). Children of upper socioeconomic status show better catch-up growth (112). Appropriate local reference charts can be used to monitor growth of children born SGA. Using WHO charts in low-income countries has its problems, as many healthy children fall below the third percentile of WHO charts for both height and weight with relative sparing of weight-for-height. In SGA, this effect is more pronounced, and hence one should give more attention to the growth trajectory than to a single reading of a growth parameter for stunting and wasting (113).
Given the high prevalence of malnutrition and infections, recommendations may be different beyond the sensitive window of metabolic programming, specifically, the first 3 to 6 months of life. This may only be achieved by changing the feeding policy based on very close monitoring of the growth trajectory—allowing neither failure to thrive nor excessive catch-up in weight. Exclusive breastfeeding has been associated with less rapid catch-up and lower fasting insulin and glucose concentrations, at least in the short term (114), and is associated with a consistent protective effect against later obesity in numerous observational studies and meta-analyses (115-117). Therefore, current WHO policy recommends exclusive breastfeeding or using standard formulas rather than nutrient-enriched formulas up to the age of 6 months. For low-birth-weight infants in extremely impoverished environments, clearly the priority is to prevent malnutrition and growth faltering (118).
Recommendations:
2.It is recommended to improve nutrition, hygiene, antenatal care, and monitoring for subclinical hypothyroidism in pregnant women and also to prevent and treat malaria in countries where this is endemic, as this may prevent a substantial portion of SGA birth. (A +++)
3.Children born SGA should be carefully followed in the first years of life by a neonatologist or a pediatrician to evaluate their growth, weight gain, and neurodevelopment. (A +++)
4.Close monitoring of weight and length trajectories is needed to identify malnutrition, growth faltering, and excessive weight catch-up. (A +++)
5.It is recommended to avoid additional nutritional supplementation to healthy infants up to the age of 6 months unless the infant suffers from malnutrition. (A +++)
6.In children born SGA, excessive postnatal weight gain should be avoided, since it is associated with a less favorable cardiometabolic health profile in young adulthood. (A +++)
7.Weight, length, head circumference, and weight for length or BMI should be monitored every 3 months during the first year of life, 6-monthly in the second year of life, and yearly thereafter until the child reaches their genetic height potential or height is in the normal range. The parameters should be plotted on an appropriate growth chart and assessed for adequacy. (A +++)
8.When a child born SGA has persistent short stature < −2.5 SDS by the age of 2 years or < −2 SDS around the age of 3 to 4 years, without signs of catch-up growth during the previous 6 months, referral to a pediatric endocrinologist or a pediatrician with expertise in SGA is indicated. (A +++)
9.Those born very preterm (gestational age < 28 weeks) or severely SGA (birth weight and/or birth length < −3 SDS for gestational age) or with a suspicion of a syndrome associated with growth retardation should be monitored more closely, and referred to a pediatric endocrinologist early during infancy. (A +++)
10.Early screening for neurocognitive impairment is particularly indicated in those born preterm, those with a complicated perinatal period, or with a small head circumference, and special attention is required in case of developmental delay, impaired cognition, attention deficit/hyperactivity disorder (ADHD), or learning difficulties. (A +++)
11.Diagnostic tests to evaluate pubertal development are only indicated when pubertal development is abnormal. (A ++)
12.Assessment of fasting glucose and lipid concentrations is only recommended in children born SGA with one or more risk factors, such as overweight (body mass index [BMI] ≥ 1 SDS), obesity (BMI ≥ 2 SDS), and family history, or when clinical signs suggest a metabolic disease. (B ++)
13.Bone mineral density and kidney function should only be investigated when clinically indicated. (A +++)
Short Stature
Definition
Various definitions for short stature exist, but short stature is mostly defined as a height < −2 SDS for age, sex, and population. Low height velocity is mostly defined as a growth velocity < −0.7 SDS during 6 months. Accurate determination of short stature requires appropriate national growth charts. When these are not available, the most appropriate ones for the country should be used.
Recommendation:
14. For accurate determination of short stature, we recommend the use of national growth charts or the most appropriate growth chart for the country. (A +++)
(Epi)genetic causes of short stature after being born SGA
Children with short stature after SGA birth form a heterogeneous group with a wide spectrum of clinical symptoms under the umbrella of SGA. The increasing use of next-generation sequencing, whole exome sequencing (WES), chromosomal microarrays, RNA sequencing, and methylation arrays has resulted in the discovery of novel genetic and epigenetic causes of short stature in children born SGA. Some monogenic disorders are now found in children born SGA while previously considered to cause idiopathic short stature (ISS). Most monogenic primordial growth disorders have short stature after SGA birth as one of their clinical features. For clinical purposes, we first present the monogenic disorders with a normal head circumference and short stature (either proportionate or disproportionate) (Table 1), followed by monogenic primordial disorders with microcephaly (Table 2) and finally the currently known imprinting disorders and methylation disturbances (Table 3). This classification is arbitrary, because some genetic aberrations were first reported for children with a very characteristic phenotype (including microcephaly) while, over time, milder phenotypes were recognized. The genetic disorders in short SGA children can also be presented according to a pathophysiological classification (see review (9)).
Monogenic disorders in short children born SGA with normal head circumference
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with normal head circumference and proportionate short stature | |||||||
| GH deficiency | GH1, GHRHR, BTK | AR, AD X-linked | 1:5.000 | −0.9/−0.6 | Wide variation in height deficit | ↓GH peak during GH stimulation test, ↓IGF-I, ↓IGFBP-3, ↓ALS | GH treatment |
| Laron syndrome [262500] | GHR | AR or rarely AD | ≈500 cases | −0.6/−1.6 | Wide variation in height, midfacial hypoplasia | ↑GH, ↓IGF-I, ↓IGFBP-3, ↓ALS, variable GHBP | IGF-I treatment moderately effective |
| ALS deficiency [615961] | IGFALS | AR | ≈65 cases | −2.2 | Mild to moderate short stature | ↓IGF-I, ↓IGFBP-3, ↓ALS | No data |
| 3-M syndrome [273750, 612921, 614205] | CUL7, OBSL1, CCDC8 | AR | ≈200 cases | −3.1 | Facial features, normal mental development, long and slender tubular bones, reduced AP diameter of vertebral bodies, delayed bone age | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient |
| Silver-Russell variant [616489] | IGF2 | AD, paternal | 8 cases | −3.9/−4.6 | Dysmorphic features like SRS, fulfilling the Netchine-Harbison criteria for SRS, including relative macrocephaly | ↑/normal GH, normal IGF-I, ↑/normal IGFBP-3, ↓ IGF-II | GH treatment likely as effective as in other genetic variants of SRS |
| Floating Harbor syndrome [136140] | SCRAP | AD | ≈52 cases | −2.5 | Proportionate short stature, delayed bone age and speech, triangular face, deep-set eyes, long eyelashes, bulbous nose, wide columella, short philtrum, thin lips | GH deficiency described in some patients. ↑/normal IGF-I (mainly during rhGH treatment. Nl IGFBP-3 | Insufficient data |
| Noonan syndrome [163950] | PTPN11 and 12 other genes | AD or rarely AR | 1:1.000-2.500 | −1.0 | Short stature, facial dysmorphism, wide spectrum of congenital heart defects, pectus deformity, cryptorchidism, coagulation defect | Nl GH, IGFBP-3 ↓/normal IGF-I | GH treatment registered in US and EU |
| Monogenic disorders with normal head circumference and disproportionate short stature | |||||||
| SHOX-associated short stature [300582] | SHOX | AD | 2-17% of short stature | −0.4 −1.1 | Short forearm and lower leg, bowing of forearm and tibia, dislocation of ulna at elbow, Madelung deformity, muscular hypertrophy, radiologic signs at wrist and forearm | Nl GH, IGF-I, IGFBP-3 | GH has similar efficacy as in Turner syndrome; registered in many countries |
| Achondroplasia [100800] | Act FGFR3 | AD | 1:15.000-40.000 | −0.7/−1.0 | Rhizomelic limb shortening, frontal bossing, midface hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum, trident hand | Nl GH, IGF-I, IGFBP-3 | Effects of GH considered insufficient |
| Hypochondro-plasia [146000] | Act FGFR3 | 1:15.000-40.000 | — | Rhizomelic limb shortening, limitation of elbow extension, brachydactyly, relative macrocephaly, generalized laxity, specific radiologic features | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient | |
| Short stature with nonspecific skeletal abnormalities [616255] | NPR2 | AD | 1-2% of short SGA and ISS | −0.8/−2.3 | ↑ sitting height/height ratio, shortening of metacarpals, phenotypic or radiographic indicators of SHOX HI (but no Madelung deformity) | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Brachydactyly type A1 [1112500] | IHH | AD | 1.6% of short SGA and ISS | −/−1.4 | ↑ sitting height/height ratio, shortening of middle phalanx of 2nd and 5th fingers with cone-shaped epiphyses | Nl GH, IGF-I, IGFBP-3 | Preliminary data GH treatment positive |
| Short stature/early-onset osteoarthritis or osteochondritis [165800] | ACAN | AD | 1%-2% of short SGA and ISS | −0.7/−1.5 | Proportionate or disproportionate short stature, with or without advanced bone age, brachydactyly, early-onset osteoarthritis | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with normal head circumference and proportionate short stature | |||||||
| GH deficiency | GH1, GHRHR, BTK | AR, AD X-linked | 1:5.000 | −0.9/−0.6 | Wide variation in height deficit | ↓GH peak during GH stimulation test, ↓IGF-I, ↓IGFBP-3, ↓ALS | GH treatment |
| Laron syndrome [262500] | GHR | AR or rarely AD | ≈500 cases | −0.6/−1.6 | Wide variation in height, midfacial hypoplasia | ↑GH, ↓IGF-I, ↓IGFBP-3, ↓ALS, variable GHBP | IGF-I treatment moderately effective |
| ALS deficiency [615961] | IGFALS | AR | ≈65 cases | −2.2 | Mild to moderate short stature | ↓IGF-I, ↓IGFBP-3, ↓ALS | No data |
| 3-M syndrome [273750, 612921, 614205] | CUL7, OBSL1, CCDC8 | AR | ≈200 cases | −3.1 | Facial features, normal mental development, long and slender tubular bones, reduced AP diameter of vertebral bodies, delayed bone age | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient |
| Silver-Russell variant [616489] | IGF2 | AD, paternal | 8 cases | −3.9/−4.6 | Dysmorphic features like SRS, fulfilling the Netchine-Harbison criteria for SRS, including relative macrocephaly | ↑/normal GH, normal IGF-I, ↑/normal IGFBP-3, ↓ IGF-II | GH treatment likely as effective as in other genetic variants of SRS |
| Floating Harbor syndrome [136140] | SCRAP | AD | ≈52 cases | −2.5 | Proportionate short stature, delayed bone age and speech, triangular face, deep-set eyes, long eyelashes, bulbous nose, wide columella, short philtrum, thin lips | GH deficiency described in some patients. ↑/normal IGF-I (mainly during rhGH treatment. Nl IGFBP-3 | Insufficient data |
| Noonan syndrome [163950] | PTPN11 and 12 other genes | AD or rarely AR | 1:1.000-2.500 | −1.0 | Short stature, facial dysmorphism, wide spectrum of congenital heart defects, pectus deformity, cryptorchidism, coagulation defect | Nl GH, IGFBP-3 ↓/normal IGF-I | GH treatment registered in US and EU |
| Monogenic disorders with normal head circumference and disproportionate short stature | |||||||
| SHOX-associated short stature [300582] | SHOX | AD | 2-17% of short stature | −0.4 −1.1 | Short forearm and lower leg, bowing of forearm and tibia, dislocation of ulna at elbow, Madelung deformity, muscular hypertrophy, radiologic signs at wrist and forearm | Nl GH, IGF-I, IGFBP-3 | GH has similar efficacy as in Turner syndrome; registered in many countries |
| Achondroplasia [100800] | Act FGFR3 | AD | 1:15.000-40.000 | −0.7/−1.0 | Rhizomelic limb shortening, frontal bossing, midface hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum, trident hand | Nl GH, IGF-I, IGFBP-3 | Effects of GH considered insufficient |
| Hypochondro-plasia [146000] | Act FGFR3 | 1:15.000-40.000 | — | Rhizomelic limb shortening, limitation of elbow extension, brachydactyly, relative macrocephaly, generalized laxity, specific radiologic features | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient | |
| Short stature with nonspecific skeletal abnormalities [616255] | NPR2 | AD | 1-2% of short SGA and ISS | −0.8/−2.3 | ↑ sitting height/height ratio, shortening of metacarpals, phenotypic or radiographic indicators of SHOX HI (but no Madelung deformity) | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Brachydactyly type A1 [1112500] | IHH | AD | 1.6% of short SGA and ISS | −/−1.4 | ↑ sitting height/height ratio, shortening of middle phalanx of 2nd and 5th fingers with cone-shaped epiphyses | Nl GH, IGF-I, IGFBP-3 | Preliminary data GH treatment positive |
| Short stature/early-onset osteoarthritis or osteochondritis [165800] | ACAN | AD | 1%-2% of short SGA and ISS | −0.7/−1.5 | Proportionate or disproportionate short stature, with or without advanced bone age, brachydactyly, early-onset osteoarthritis | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
Abbreviations: act, activating; AD, autosomal dominant; ALS, acid labile subunit; AR, autosomal recessive; BL, birth length; BW, birth weight; GH, growth hormone; IGF, insulin-like growth factor; IGFBP, IGF binding protein; ISS, idiopathic short stature; SDS, standard deviation score; SGA, small for gestational age; SRS, Silver-Russell syndrome.
Monogenic disorders in short children born SGA with normal head circumference
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with normal head circumference and proportionate short stature | |||||||
| GH deficiency | GH1, GHRHR, BTK | AR, AD X-linked | 1:5.000 | −0.9/−0.6 | Wide variation in height deficit | ↓GH peak during GH stimulation test, ↓IGF-I, ↓IGFBP-3, ↓ALS | GH treatment |
| Laron syndrome [262500] | GHR | AR or rarely AD | ≈500 cases | −0.6/−1.6 | Wide variation in height, midfacial hypoplasia | ↑GH, ↓IGF-I, ↓IGFBP-3, ↓ALS, variable GHBP | IGF-I treatment moderately effective |
| ALS deficiency [615961] | IGFALS | AR | ≈65 cases | −2.2 | Mild to moderate short stature | ↓IGF-I, ↓IGFBP-3, ↓ALS | No data |
| 3-M syndrome [273750, 612921, 614205] | CUL7, OBSL1, CCDC8 | AR | ≈200 cases | −3.1 | Facial features, normal mental development, long and slender tubular bones, reduced AP diameter of vertebral bodies, delayed bone age | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient |
| Silver-Russell variant [616489] | IGF2 | AD, paternal | 8 cases | −3.9/−4.6 | Dysmorphic features like SRS, fulfilling the Netchine-Harbison criteria for SRS, including relative macrocephaly | ↑/normal GH, normal IGF-I, ↑/normal IGFBP-3, ↓ IGF-II | GH treatment likely as effective as in other genetic variants of SRS |
| Floating Harbor syndrome [136140] | SCRAP | AD | ≈52 cases | −2.5 | Proportionate short stature, delayed bone age and speech, triangular face, deep-set eyes, long eyelashes, bulbous nose, wide columella, short philtrum, thin lips | GH deficiency described in some patients. ↑/normal IGF-I (mainly during rhGH treatment. Nl IGFBP-3 | Insufficient data |
| Noonan syndrome [163950] | PTPN11 and 12 other genes | AD or rarely AR | 1:1.000-2.500 | −1.0 | Short stature, facial dysmorphism, wide spectrum of congenital heart defects, pectus deformity, cryptorchidism, coagulation defect | Nl GH, IGFBP-3 ↓/normal IGF-I | GH treatment registered in US and EU |
| Monogenic disorders with normal head circumference and disproportionate short stature | |||||||
| SHOX-associated short stature [300582] | SHOX | AD | 2-17% of short stature | −0.4 −1.1 | Short forearm and lower leg, bowing of forearm and tibia, dislocation of ulna at elbow, Madelung deformity, muscular hypertrophy, radiologic signs at wrist and forearm | Nl GH, IGF-I, IGFBP-3 | GH has similar efficacy as in Turner syndrome; registered in many countries |
| Achondroplasia [100800] | Act FGFR3 | AD | 1:15.000-40.000 | −0.7/−1.0 | Rhizomelic limb shortening, frontal bossing, midface hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum, trident hand | Nl GH, IGF-I, IGFBP-3 | Effects of GH considered insufficient |
| Hypochondro-plasia [146000] | Act FGFR3 | 1:15.000-40.000 | — | Rhizomelic limb shortening, limitation of elbow extension, brachydactyly, relative macrocephaly, generalized laxity, specific radiologic features | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient | |
| Short stature with nonspecific skeletal abnormalities [616255] | NPR2 | AD | 1-2% of short SGA and ISS | −0.8/−2.3 | ↑ sitting height/height ratio, shortening of metacarpals, phenotypic or radiographic indicators of SHOX HI (but no Madelung deformity) | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Brachydactyly type A1 [1112500] | IHH | AD | 1.6% of short SGA and ISS | −/−1.4 | ↑ sitting height/height ratio, shortening of middle phalanx of 2nd and 5th fingers with cone-shaped epiphyses | Nl GH, IGF-I, IGFBP-3 | Preliminary data GH treatment positive |
| Short stature/early-onset osteoarthritis or osteochondritis [165800] | ACAN | AD | 1%-2% of short SGA and ISS | −0.7/−1.5 | Proportionate or disproportionate short stature, with or without advanced bone age, brachydactyly, early-onset osteoarthritis | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with normal head circumference and proportionate short stature | |||||||
| GH deficiency | GH1, GHRHR, BTK | AR, AD X-linked | 1:5.000 | −0.9/−0.6 | Wide variation in height deficit | ↓GH peak during GH stimulation test, ↓IGF-I, ↓IGFBP-3, ↓ALS | GH treatment |
| Laron syndrome [262500] | GHR | AR or rarely AD | ≈500 cases | −0.6/−1.6 | Wide variation in height, midfacial hypoplasia | ↑GH, ↓IGF-I, ↓IGFBP-3, ↓ALS, variable GHBP | IGF-I treatment moderately effective |
| ALS deficiency [615961] | IGFALS | AR | ≈65 cases | −2.2 | Mild to moderate short stature | ↓IGF-I, ↓IGFBP-3, ↓ALS | No data |
| 3-M syndrome [273750, 612921, 614205] | CUL7, OBSL1, CCDC8 | AR | ≈200 cases | −3.1 | Facial features, normal mental development, long and slender tubular bones, reduced AP diameter of vertebral bodies, delayed bone age | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient |
| Silver-Russell variant [616489] | IGF2 | AD, paternal | 8 cases | −3.9/−4.6 | Dysmorphic features like SRS, fulfilling the Netchine-Harbison criteria for SRS, including relative macrocephaly | ↑/normal GH, normal IGF-I, ↑/normal IGFBP-3, ↓ IGF-II | GH treatment likely as effective as in other genetic variants of SRS |
| Floating Harbor syndrome [136140] | SCRAP | AD | ≈52 cases | −2.5 | Proportionate short stature, delayed bone age and speech, triangular face, deep-set eyes, long eyelashes, bulbous nose, wide columella, short philtrum, thin lips | GH deficiency described in some patients. ↑/normal IGF-I (mainly during rhGH treatment. Nl IGFBP-3 | Insufficient data |
| Noonan syndrome [163950] | PTPN11 and 12 other genes | AD or rarely AR | 1:1.000-2.500 | −1.0 | Short stature, facial dysmorphism, wide spectrum of congenital heart defects, pectus deformity, cryptorchidism, coagulation defect | Nl GH, IGFBP-3 ↓/normal IGF-I | GH treatment registered in US and EU |
| Monogenic disorders with normal head circumference and disproportionate short stature | |||||||
| SHOX-associated short stature [300582] | SHOX | AD | 2-17% of short stature | −0.4 −1.1 | Short forearm and lower leg, bowing of forearm and tibia, dislocation of ulna at elbow, Madelung deformity, muscular hypertrophy, radiologic signs at wrist and forearm | Nl GH, IGF-I, IGFBP-3 | GH has similar efficacy as in Turner syndrome; registered in many countries |
| Achondroplasia [100800] | Act FGFR3 | AD | 1:15.000-40.000 | −0.7/−1.0 | Rhizomelic limb shortening, frontal bossing, midface hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum, trident hand | Nl GH, IGF-I, IGFBP-3 | Effects of GH considered insufficient |
| Hypochondro-plasia [146000] | Act FGFR3 | 1:15.000-40.000 | — | Rhizomelic limb shortening, limitation of elbow extension, brachydactyly, relative macrocephaly, generalized laxity, specific radiologic features | Nl GH, IGF-I, IGFBP-3 | Effect of GH considered insufficient | |
| Short stature with nonspecific skeletal abnormalities [616255] | NPR2 | AD | 1-2% of short SGA and ISS | −0.8/−2.3 | ↑ sitting height/height ratio, shortening of metacarpals, phenotypic or radiographic indicators of SHOX HI (but no Madelung deformity) | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
| Brachydactyly type A1 [1112500] | IHH | AD | 1.6% of short SGA and ISS | −/−1.4 | ↑ sitting height/height ratio, shortening of middle phalanx of 2nd and 5th fingers with cone-shaped epiphyses | Nl GH, IGF-I, IGFBP-3 | Preliminary data GH treatment positive |
| Short stature/early-onset osteoarthritis or osteochondritis [165800] | ACAN | AD | 1%-2% of short SGA and ISS | −0.7/−1.5 | Proportionate or disproportionate short stature, with or without advanced bone age, brachydactyly, early-onset osteoarthritis | Nl GH, IGF-I, IGFBP-3 | Insufficient data |
Abbreviations: act, activating; AD, autosomal dominant; ALS, acid labile subunit; AR, autosomal recessive; BL, birth length; BW, birth weight; GH, growth hormone; IGF, insulin-like growth factor; IGFBP, IGF binding protein; ISS, idiopathic short stature; SDS, standard deviation score; SGA, small for gestational age; SRS, Silver-Russell syndrome.
Monogenetic disorders in short children born SGA with microcephaly
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with microcephaly | |||||||
| IGF-I deficiency [608747] | IGF1 | AR, AD | 4 homozyg 7 heterozyg | hom −3.7 het −1.9 | Microcephaly, deafness, reduced mental development | ↑GH, variable IGF-I, ↑IGFBP-3, | IGF-I treatment moderately effective |
| Resistance to IGF-I [1270450] | IGF1R | AD | 1-2% of short SGA | −2.1/−2.7 | Microcephaly, reduced mental development | ↑/normal GH, ↑/normal IGF-I, ↑/normal IGFBP-3 | GH treatment moderately effective |
| PAPP-A2 deficiency | PAPPA2 [homozyg] | AR | 5 cases | −1.6/−1,3 | Microcephaly, skeletal abnormalities | ↑GH, ↑IGF-I, ↑IGFBP-3, ↑IGFBP-5, ↑ALS | IGF-I treatment possibly effective |
| Primordial dwarfism with microcephaly | |||||||
| Cornelia de Lange syndrome (1-5) [122470] | NIPBL, SMC1A,SMC3, RAD21, HDAC8 | AD | 1/40.000 | −3.4/- | Low anterior hairline, connected eyebrows, ante-verted nares, maxillary prognatism, long philtrum, thin lips, “carp” mouth | Not available | No data on GH, likely ineffective |
| Meier-Gorlin syndrome (1-5) [224690] | ORC1, ORC4, ORC6, CDT1, CDC6 | AR | ≈67 cases | −3.8/- | Bilateral microtia, aplasia or hypoplasia of the patellae, normal intelligence | Not available | No data on GH, likely ineffective |
| MOPD I [210710] | RNU4ATAC | AR | <1/1.000.000 | Extremely low | Neurologic abnormalities, including intellectual disability, brain malformations, ocular or auditory sensory deficits | Not available | No data on GH, likely ineffective |
| MOPD II [210720] | PCNT | AR | Extremely rare | −3.9 | No or mild mental impairment, truncal obesity, DM, moyamoya disease, small loose teeth, radiologic abnormalities | Not available | No data on GH, likely ineffective |
| Seckel syndrome (1-8) [210600] | ATR, RBBP8, CENPJ, CEP152, CEP63, NIN, DNA2, ATRIP | AR | <1/1.000.000 | Extremely low | Microcephaly, intellectual disability, characteristic “bird-headed” face (receding forehead and micrognathia) | Not available | No data on GH, likely ineffective |
| Smit-Lemli-Opitz [270400] | DHCR7 | AR | 1/50.000 (mainly in Caucasians) | Extremely low | Microcephaly, moderate to severe mental retardation, dysmorphic features and organ malformations (heart, palate, syndactyly 2nd and 3rd toes, underdeveloped genitalia boys) | Not available | No data on GH, likely ineffective |
| DNA repair defects with microcephaly | |||||||
| Bloom syndrome [210900] | RECQL3 | AR | 1/48.000 (Ashk Jews, but also others) | −4.7/−4.8 | Microcephaly, sun-sensitivity, telangiectatic, hypo- and hyperpigmented skin lesions, predisposition to cancer, maturity-onset DM | Not available | GH not recommended |
| Fanconi anemia [many] | FANCA and 14 other genes | AR or X-linked | 1/160.000 | −1.8/−2.1 | Microcephaly, genomic instability, hypo- and hyperpigmentation, skin lesions abnormalities in major organ systems, bone marrow failure, predisposition to cancer | Not available | GH contra-indicated |
| Nijmegen breakage Syndrome [#251260] | NBN | AR | Extremely rare | −1.8/−2.2 | Microcephaly, mild to moderate intellectual disability, immunodeficiency, predisposition to cancer | Not available | GH contra-indicated |
| LIG 4 syndrome [606593] | LIG4 | AR | Extremely rare | −3.0/−3.8 | Microcephaly, sun-sensitive, combined immunodeficiency | Not available | GH contra-indicated |
| XRCC4 syndrome | XRCC4 | AR | Extremely rare | −1.6/−2.5 | Microcephaly, progressively short, hyper gonadotropic hypogonadism, multinodular goiter, diabetes mellitus | Not available | GH contra-indicated |
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with microcephaly | |||||||
| IGF-I deficiency [608747] | IGF1 | AR, AD | 4 homozyg 7 heterozyg | hom −3.7 het −1.9 | Microcephaly, deafness, reduced mental development | ↑GH, variable IGF-I, ↑IGFBP-3, | IGF-I treatment moderately effective |
| Resistance to IGF-I [1270450] | IGF1R | AD | 1-2% of short SGA | −2.1/−2.7 | Microcephaly, reduced mental development | ↑/normal GH, ↑/normal IGF-I, ↑/normal IGFBP-3 | GH treatment moderately effective |
| PAPP-A2 deficiency | PAPPA2 [homozyg] | AR | 5 cases | −1.6/−1,3 | Microcephaly, skeletal abnormalities | ↑GH, ↑IGF-I, ↑IGFBP-3, ↑IGFBP-5, ↑ALS | IGF-I treatment possibly effective |
| Primordial dwarfism with microcephaly | |||||||
| Cornelia de Lange syndrome (1-5) [122470] | NIPBL, SMC1A,SMC3, RAD21, HDAC8 | AD | 1/40.000 | −3.4/- | Low anterior hairline, connected eyebrows, ante-verted nares, maxillary prognatism, long philtrum, thin lips, “carp” mouth | Not available | No data on GH, likely ineffective |
| Meier-Gorlin syndrome (1-5) [224690] | ORC1, ORC4, ORC6, CDT1, CDC6 | AR | ≈67 cases | −3.8/- | Bilateral microtia, aplasia or hypoplasia of the patellae, normal intelligence | Not available | No data on GH, likely ineffective |
| MOPD I [210710] | RNU4ATAC | AR | <1/1.000.000 | Extremely low | Neurologic abnormalities, including intellectual disability, brain malformations, ocular or auditory sensory deficits | Not available | No data on GH, likely ineffective |
| MOPD II [210720] | PCNT | AR | Extremely rare | −3.9 | No or mild mental impairment, truncal obesity, DM, moyamoya disease, small loose teeth, radiologic abnormalities | Not available | No data on GH, likely ineffective |
| Seckel syndrome (1-8) [210600] | ATR, RBBP8, CENPJ, CEP152, CEP63, NIN, DNA2, ATRIP | AR | <1/1.000.000 | Extremely low | Microcephaly, intellectual disability, characteristic “bird-headed” face (receding forehead and micrognathia) | Not available | No data on GH, likely ineffective |
| Smit-Lemli-Opitz [270400] | DHCR7 | AR | 1/50.000 (mainly in Caucasians) | Extremely low | Microcephaly, moderate to severe mental retardation, dysmorphic features and organ malformations (heart, palate, syndactyly 2nd and 3rd toes, underdeveloped genitalia boys) | Not available | No data on GH, likely ineffective |
| DNA repair defects with microcephaly | |||||||
| Bloom syndrome [210900] | RECQL3 | AR | 1/48.000 (Ashk Jews, but also others) | −4.7/−4.8 | Microcephaly, sun-sensitivity, telangiectatic, hypo- and hyperpigmented skin lesions, predisposition to cancer, maturity-onset DM | Not available | GH not recommended |
| Fanconi anemia [many] | FANCA and 14 other genes | AR or X-linked | 1/160.000 | −1.8/−2.1 | Microcephaly, genomic instability, hypo- and hyperpigmentation, skin lesions abnormalities in major organ systems, bone marrow failure, predisposition to cancer | Not available | GH contra-indicated |
| Nijmegen breakage Syndrome [#251260] | NBN | AR | Extremely rare | −1.8/−2.2 | Microcephaly, mild to moderate intellectual disability, immunodeficiency, predisposition to cancer | Not available | GH contra-indicated |
| LIG 4 syndrome [606593] | LIG4 | AR | Extremely rare | −3.0/−3.8 | Microcephaly, sun-sensitive, combined immunodeficiency | Not available | GH contra-indicated |
| XRCC4 syndrome | XRCC4 | AR | Extremely rare | −1.6/−2.5 | Microcephaly, progressively short, hyper gonadotropic hypogonadism, multinodular goiter, diabetes mellitus | Not available | GH contra-indicated |
Abbreviations: AD, autosomal dominant; ALS, acid labile subunit; AR, autosomal recessive; BL, birth length; BW, birth weight; GH, growth hormone; IGF, insulin-like growth factor; IGFBP, IGF binding protein; ISS, idiopathic short stature; SDS, standard deviation score; SGA, small for gestational age; SRS, Silver-Russell syndrome.
Monogenetic disorders in short children born SGA with microcephaly
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with microcephaly | |||||||
| IGF-I deficiency [608747] | IGF1 | AR, AD | 4 homozyg 7 heterozyg | hom −3.7 het −1.9 | Microcephaly, deafness, reduced mental development | ↑GH, variable IGF-I, ↑IGFBP-3, | IGF-I treatment moderately effective |
| Resistance to IGF-I [1270450] | IGF1R | AD | 1-2% of short SGA | −2.1/−2.7 | Microcephaly, reduced mental development | ↑/normal GH, ↑/normal IGF-I, ↑/normal IGFBP-3 | GH treatment moderately effective |
| PAPP-A2 deficiency | PAPPA2 [homozyg] | AR | 5 cases | −1.6/−1,3 | Microcephaly, skeletal abnormalities | ↑GH, ↑IGF-I, ↑IGFBP-3, ↑IGFBP-5, ↑ALS | IGF-I treatment possibly effective |
| Primordial dwarfism with microcephaly | |||||||
| Cornelia de Lange syndrome (1-5) [122470] | NIPBL, SMC1A,SMC3, RAD21, HDAC8 | AD | 1/40.000 | −3.4/- | Low anterior hairline, connected eyebrows, ante-verted nares, maxillary prognatism, long philtrum, thin lips, “carp” mouth | Not available | No data on GH, likely ineffective |
| Meier-Gorlin syndrome (1-5) [224690] | ORC1, ORC4, ORC6, CDT1, CDC6 | AR | ≈67 cases | −3.8/- | Bilateral microtia, aplasia or hypoplasia of the patellae, normal intelligence | Not available | No data on GH, likely ineffective |
| MOPD I [210710] | RNU4ATAC | AR | <1/1.000.000 | Extremely low | Neurologic abnormalities, including intellectual disability, brain malformations, ocular or auditory sensory deficits | Not available | No data on GH, likely ineffective |
| MOPD II [210720] | PCNT | AR | Extremely rare | −3.9 | No or mild mental impairment, truncal obesity, DM, moyamoya disease, small loose teeth, radiologic abnormalities | Not available | No data on GH, likely ineffective |
| Seckel syndrome (1-8) [210600] | ATR, RBBP8, CENPJ, CEP152, CEP63, NIN, DNA2, ATRIP | AR | <1/1.000.000 | Extremely low | Microcephaly, intellectual disability, characteristic “bird-headed” face (receding forehead and micrognathia) | Not available | No data on GH, likely ineffective |
| Smit-Lemli-Opitz [270400] | DHCR7 | AR | 1/50.000 (mainly in Caucasians) | Extremely low | Microcephaly, moderate to severe mental retardation, dysmorphic features and organ malformations (heart, palate, syndactyly 2nd and 3rd toes, underdeveloped genitalia boys) | Not available | No data on GH, likely ineffective |
| DNA repair defects with microcephaly | |||||||
| Bloom syndrome [210900] | RECQL3 | AR | 1/48.000 (Ashk Jews, but also others) | −4.7/−4.8 | Microcephaly, sun-sensitivity, telangiectatic, hypo- and hyperpigmented skin lesions, predisposition to cancer, maturity-onset DM | Not available | GH not recommended |
| Fanconi anemia [many] | FANCA and 14 other genes | AR or X-linked | 1/160.000 | −1.8/−2.1 | Microcephaly, genomic instability, hypo- and hyperpigmentation, skin lesions abnormalities in major organ systems, bone marrow failure, predisposition to cancer | Not available | GH contra-indicated |
| Nijmegen breakage Syndrome [#251260] | NBN | AR | Extremely rare | −1.8/−2.2 | Microcephaly, mild to moderate intellectual disability, immunodeficiency, predisposition to cancer | Not available | GH contra-indicated |
| LIG 4 syndrome [606593] | LIG4 | AR | Extremely rare | −3.0/−3.8 | Microcephaly, sun-sensitive, combined immunodeficiency | Not available | GH contra-indicated |
| XRCC4 syndrome | XRCC4 | AR | Extremely rare | −1.6/−2.5 | Microcephaly, progressively short, hyper gonadotropic hypogonadism, multinodular goiter, diabetes mellitus | Not available | GH contra-indicated |
| Syndrome [OMIM] | Genetic defect | Inheritance | Incidence | Mean BW/BL SDS | Clinical features | Laboratory data | Treatment |
|---|---|---|---|---|---|---|---|
| Monogenic disorders with microcephaly | |||||||
| IGF-I deficiency [608747] | IGF1 | AR, AD | 4 homozyg 7 heterozyg | hom −3.7 het −1.9 | Microcephaly, deafness, reduced mental development | ↑GH, variable IGF-I, ↑IGFBP-3, | IGF-I treatment moderately effective |
| Resistance to IGF-I [1270450] | IGF1R | AD | 1-2% of short SGA | −2.1/−2.7 | Microcephaly, reduced mental development | ↑/normal GH, ↑/normal IGF-I, ↑/normal IGFBP-3 | GH treatment moderately effective |
| PAPP-A2 deficiency | PAPPA2 [homozyg] | AR | 5 cases | −1.6/−1,3 | Microcephaly, skeletal abnormalities | ↑GH, ↑IGF-I, ↑IGFBP-3, ↑IGFBP-5, ↑ALS | IGF-I treatment possibly effective |
| Primordial dwarfism with microcephaly | |||||||
| Cornelia de Lange syndrome (1-5) [122470] | NIPBL, SMC1A,SMC3, RAD21, HDAC8 | AD | 1/40.000 | −3.4/- | Low anterior hairline, connected eyebrows, ante-verted nares, maxillary prognatism, long philtrum, thin lips, “carp” mouth | Not available | No data on GH, likely ineffective |
| Meier-Gorlin syndrome (1-5) [224690] | ORC1, ORC4, ORC6, CDT1, CDC6 | AR | ≈67 cases | −3.8/- | Bilateral microtia, aplasia or hypoplasia of the patellae, normal intelligence | Not available | No data on GH, likely ineffective |
| MOPD I [210710] | RNU4ATAC | AR | <1/1.000.000 | Extremely low | Neurologic abnormalities, including intellectual disability, brain malformations, ocular or auditory sensory deficits | Not available | No data on GH, likely ineffective |
| MOPD II [210720] | PCNT | AR | Extremely rare | −3.9 | No or mild mental impairment, truncal obesity, DM, moyamoya disease, small loose teeth, radiologic abnormalities | Not available | No data on GH, likely ineffective |
| Seckel syndrome (1-8) [210600] | ATR, RBBP8, CENPJ, CEP152, CEP63, NIN, DNA2, ATRIP | AR | <1/1.000.000 | Extremely low | Microcephaly, intellectual disability, characteristic “bird-headed” face (receding forehead and micrognathia) | Not available | No data on GH, likely ineffective |
| Smit-Lemli-Opitz [270400] | DHCR7 | AR | 1/50.000 (mainly in Caucasians) | Extremely low | Microcephaly, moderate to severe mental retardation, dysmorphic features and organ malformations (heart, palate, syndactyly 2nd and 3rd toes, underdeveloped genitalia boys) | Not available | No data on GH, likely ineffective |
| DNA repair defects with microcephaly | |||||||
| Bloom syndrome [210900] | RECQL3 | AR | 1/48.000 (Ashk Jews, but also others) | −4.7/−4.8 | Microcephaly, sun-sensitivity, telangiectatic, hypo- and hyperpigmented skin lesions, predisposition to cancer, maturity-onset DM | Not available | GH not recommended |
| Fanconi anemia [many] | FANCA and 14 other genes | AR or X-linked | 1/160.000 | −1.8/−2.1 | Microcephaly, genomic instability, hypo- and hyperpigmentation, skin lesions abnormalities in major organ systems, bone marrow failure, predisposition to cancer | Not available | GH contra-indicated |
| Nijmegen breakage Syndrome [#251260] | NBN | AR | Extremely rare | −1.8/−2.2 | Microcephaly, mild to moderate intellectual disability, immunodeficiency, predisposition to cancer | Not available | GH contra-indicated |
| LIG 4 syndrome [606593] | LIG4 | AR | Extremely rare | −3.0/−3.8 | Microcephaly, sun-sensitive, combined immunodeficiency | Not available | GH contra-indicated |
| XRCC4 syndrome | XRCC4 | AR | Extremely rare | −1.6/−2.5 | Microcephaly, progressively short, hyper gonadotropic hypogonadism, multinodular goiter, diabetes mellitus | Not available | GH contra-indicated |
Abbreviations: AD, autosomal dominant; ALS, acid labile subunit; AR, autosomal recessive; BL, birth length; BW, birth weight; GH, growth hormone; IGF, insulin-like growth factor; IGFBP, IGF binding protein; ISS, idiopathic short stature; SDS, standard deviation score; SGA, small for gestational age; SRS, Silver-Russell syndrome.
Imprinting disorders and methylation disturbances in short children born SGA
| Syndrome [OMIM] | Epigenetic defect | Incidence | Mean BW/BL SDS | Clinical features besides SGA | Treatment |
|---|---|---|---|---|---|
| Silver-Russell syndrome [180860] | 11p15 LOM (30%-60%), upd(7)mat (5-10%), upd(20)mat, upd(16)mat act CDKN1C, HMAG2, PLAG1 and CNVs Exception # Paternal IGF2 mutation (see Table 1) | 1:30,000-100,000 | 11p15:−3.2/−4.5 UPD7:−2.3/−2.5 Clinical: −2.7/−1.8 | Relative macrocephaly, protruding forehead, body asymmetry, feeding problems and/or low BMI | GH effective (in label of registered GH treatment for short SGA) |
| Temple syndrome [616222] | 14q32 abnormalities: upd(14)mat, paternal microdeletions, hypomethylation of DLK1/GTL2 IG-DMR | < 100 cases | −1.9/−1.6 | Postnatal growth failure, hypotonia, delayed development of motor skills, feeding problems in infancy, early puberty, broad forehead, short nose with wide nasal tip, small hands and feet | Insufficient data |
| IMAGe syndrome [614732] | Maternally inherited activating mutations in CDKN1C | ≈ 15 cases | −2.0 to −4.0 | Relative macrocephaly at birth, no or mild intellectual disability, frontal bossing, low-set ears, flat nasal bridge, short nose, congenital adrenal hypoplasia, metaphyseal and/or epiphyseal dysplasia, male genital anomalies, early-onset type 1 DM | Insufficient data |
| Prader-Willi syndrome [176270] | Paternal 15q11.2q13 deletion (60%), upd(15)mat (40%), or imprinting center mutation (1-3%). Loss of SNRPN and NDN expression | 1:16.000 | −1.2/−1.1 | Diminished fetal activity, obesity, muscular hypotonia, intellectual disability, short stature, hypogonadotropic hypogonadism, small hands and feet | GH registered for PWS |
| Pseudohypopara-thyroidism type 1a/ca [103580] | Heterozygous GNAS1 mutation inherited from the mother | 1:150.000 | −0.6/−1.1 35% born SGA | Resistance to PTH and other hormones (TSH, LH, FSH and GHRH), Albright hereditary osteodystrophy (short stature, obesity, round face, subcutaneous ossifications, brachydactyly, mild intellectual disability) | Insufficient data |
| Pseudopseudo-hypoparathyroi-dism [612463]) | Heterozygous GNAS1 mutation inherited from the father | 1:150.000 | −2.7/−3.0 95% born SGA | Albright hereditary osteodystrophy without multiple hormone resistance and no hypocalcemia | Insufficient data |
| Syndrome [OMIM] | Epigenetic defect | Incidence | Mean BW/BL SDS | Clinical features besides SGA | Treatment |
|---|---|---|---|---|---|
| Silver-Russell syndrome [180860] | 11p15 LOM (30%-60%), upd(7)mat (5-10%), upd(20)mat, upd(16)mat act CDKN1C, HMAG2, PLAG1 and CNVs Exception # Paternal IGF2 mutation (see Table 1) | 1:30,000-100,000 | 11p15:−3.2/−4.5 UPD7:−2.3/−2.5 Clinical: −2.7/−1.8 | Relative macrocephaly, protruding forehead, body asymmetry, feeding problems and/or low BMI | GH effective (in label of registered GH treatment for short SGA) |
| Temple syndrome [616222] | 14q32 abnormalities: upd(14)mat, paternal microdeletions, hypomethylation of DLK1/GTL2 IG-DMR | < 100 cases | −1.9/−1.6 | Postnatal growth failure, hypotonia, delayed development of motor skills, feeding problems in infancy, early puberty, broad forehead, short nose with wide nasal tip, small hands and feet | Insufficient data |
| IMAGe syndrome [614732] | Maternally inherited activating mutations in CDKN1C | ≈ 15 cases | −2.0 to −4.0 | Relative macrocephaly at birth, no or mild intellectual disability, frontal bossing, low-set ears, flat nasal bridge, short nose, congenital adrenal hypoplasia, metaphyseal and/or epiphyseal dysplasia, male genital anomalies, early-onset type 1 DM | Insufficient data |
| Prader-Willi syndrome [176270] | Paternal 15q11.2q13 deletion (60%), upd(15)mat (40%), or imprinting center mutation (1-3%). Loss of SNRPN and NDN expression | 1:16.000 | −1.2/−1.1 | Diminished fetal activity, obesity, muscular hypotonia, intellectual disability, short stature, hypogonadotropic hypogonadism, small hands and feet | GH registered for PWS |
| Pseudohypopara-thyroidism type 1a/ca [103580] | Heterozygous GNAS1 mutation inherited from the mother | 1:150.000 | −0.6/−1.1 35% born SGA | Resistance to PTH and other hormones (TSH, LH, FSH and GHRH), Albright hereditary osteodystrophy (short stature, obesity, round face, subcutaneous ossifications, brachydactyly, mild intellectual disability) | Insufficient data |
| Pseudopseudo-hypoparathyroi-dism [612463]) | Heterozygous GNAS1 mutation inherited from the father | 1:150.000 | −2.7/−3.0 95% born SGA | Albright hereditary osteodystrophy without multiple hormone resistance and no hypocalcemia | Insufficient data |
Abbreviations: act, activating; BL, birth length; BMI, body mass index; BW, birth weight; DM, diabetes mellitus; FSH, follicle-stimulating hormone; LH, luteinizing hormone; LOM, loss of methylation; SGA, small for gestational age; upd()mat, maternal uniparental disomy.
Pseudohypoparathyroidism type 1b [OMIM 603233] is associated with normal or increased birth weight and overgrowth in childhood.
Imprinting disorders and methylation disturbances in short children born SGA
| Syndrome [OMIM] | Epigenetic defect | Incidence | Mean BW/BL SDS | Clinical features besides SGA | Treatment |
|---|---|---|---|---|---|
| Silver-Russell syndrome [180860] | 11p15 LOM (30%-60%), upd(7)mat (5-10%), upd(20)mat, upd(16)mat act CDKN1C, HMAG2, PLAG1 and CNVs Exception # Paternal IGF2 mutation (see Table 1) | 1:30,000-100,000 | 11p15:−3.2/−4.5 UPD7:−2.3/−2.5 Clinical: −2.7/−1.8 | Relative macrocephaly, protruding forehead, body asymmetry, feeding problems and/or low BMI | GH effective (in label of registered GH treatment for short SGA) |
| Temple syndrome [616222] | 14q32 abnormalities: upd(14)mat, paternal microdeletions, hypomethylation of DLK1/GTL2 IG-DMR | < 100 cases | −1.9/−1.6 | Postnatal growth failure, hypotonia, delayed development of motor skills, feeding problems in infancy, early puberty, broad forehead, short nose with wide nasal tip, small hands and feet | Insufficient data |
| IMAGe syndrome [614732] | Maternally inherited activating mutations in CDKN1C | ≈ 15 cases | −2.0 to −4.0 | Relative macrocephaly at birth, no or mild intellectual disability, frontal bossing, low-set ears, flat nasal bridge, short nose, congenital adrenal hypoplasia, metaphyseal and/or epiphyseal dysplasia, male genital anomalies, early-onset type 1 DM | Insufficient data |
| Prader-Willi syndrome [176270] | Paternal 15q11.2q13 deletion (60%), upd(15)mat (40%), or imprinting center mutation (1-3%). Loss of SNRPN and NDN expression | 1:16.000 | −1.2/−1.1 | Diminished fetal activity, obesity, muscular hypotonia, intellectual disability, short stature, hypogonadotropic hypogonadism, small hands and feet | GH registered for PWS |
| Pseudohypopara-thyroidism type 1a/ca [103580] | Heterozygous GNAS1 mutation inherited from the mother | 1:150.000 | −0.6/−1.1 35% born SGA | Resistance to PTH and other hormones (TSH, LH, FSH and GHRH), Albright hereditary osteodystrophy (short stature, obesity, round face, subcutaneous ossifications, brachydactyly, mild intellectual disability) | Insufficient data |
| Pseudopseudo-hypoparathyroi-dism [612463]) | Heterozygous GNAS1 mutation inherited from the father | 1:150.000 | −2.7/−3.0 95% born SGA | Albright hereditary osteodystrophy without multiple hormone resistance and no hypocalcemia | Insufficient data |
| Syndrome [OMIM] | Epigenetic defect | Incidence | Mean BW/BL SDS | Clinical features besides SGA | Treatment |
|---|---|---|---|---|---|
| Silver-Russell syndrome [180860] | 11p15 LOM (30%-60%), upd(7)mat (5-10%), upd(20)mat, upd(16)mat act CDKN1C, HMAG2, PLAG1 and CNVs Exception # Paternal IGF2 mutation (see Table 1) | 1:30,000-100,000 | 11p15:−3.2/−4.5 UPD7:−2.3/−2.5 Clinical: −2.7/−1.8 | Relative macrocephaly, protruding forehead, body asymmetry, feeding problems and/or low BMI | GH effective (in label of registered GH treatment for short SGA) |
| Temple syndrome [616222] | 14q32 abnormalities: upd(14)mat, paternal microdeletions, hypomethylation of DLK1/GTL2 IG-DMR | < 100 cases | −1.9/−1.6 | Postnatal growth failure, hypotonia, delayed development of motor skills, feeding problems in infancy, early puberty, broad forehead, short nose with wide nasal tip, small hands and feet | Insufficient data |
| IMAGe syndrome [614732] | Maternally inherited activating mutations in CDKN1C | ≈ 15 cases | −2.0 to −4.0 | Relative macrocephaly at birth, no or mild intellectual disability, frontal bossing, low-set ears, flat nasal bridge, short nose, congenital adrenal hypoplasia, metaphyseal and/or epiphyseal dysplasia, male genital anomalies, early-onset type 1 DM | Insufficient data |
| Prader-Willi syndrome [176270] | Paternal 15q11.2q13 deletion (60%), upd(15)mat (40%), or imprinting center mutation (1-3%). Loss of SNRPN and NDN expression | 1:16.000 | −1.2/−1.1 | Diminished fetal activity, obesity, muscular hypotonia, intellectual disability, short stature, hypogonadotropic hypogonadism, small hands and feet | GH registered for PWS |
| Pseudohypopara-thyroidism type 1a/ca [103580] | Heterozygous GNAS1 mutation inherited from the mother | 1:150.000 | −0.6/−1.1 35% born SGA | Resistance to PTH and other hormones (TSH, LH, FSH and GHRH), Albright hereditary osteodystrophy (short stature, obesity, round face, subcutaneous ossifications, brachydactyly, mild intellectual disability) | Insufficient data |
| Pseudopseudo-hypoparathyroi-dism [612463]) | Heterozygous GNAS1 mutation inherited from the father | 1:150.000 | −2.7/−3.0 95% born SGA | Albright hereditary osteodystrophy without multiple hormone resistance and no hypocalcemia | Insufficient data |
Abbreviations: act, activating; BL, birth length; BMI, body mass index; BW, birth weight; DM, diabetes mellitus; FSH, follicle-stimulating hormone; LH, luteinizing hormone; LOM, loss of methylation; SGA, small for gestational age; upd()mat, maternal uniparental disomy.
Pseudohypoparathyroidism type 1b [OMIM 603233] is associated with normal or increased birth weight and overgrowth in childhood.
Monogenic disorders with normal head circumference and proportionate short stature (Table 1)
Children with GH deficiency (GHD) due to a GH-1 gene mutation or GH insensitivity as a result of an inactivating mutation of GHR, or PIK3R1, or an activating STAT3 mutation, have a lower mean birth weight and birth length, but most of them do not fulfill the criteria for SGA. However, in case of a very low serum insulin-like growth factor 1 (IGF-I) and low GH peak concentrations during a provocative test or in case of lack of growth response during GH treatment in a short SGA child, one may consider testing for these genes, because short SGA does not exclude an abnormality in the GH-IGF pathway.
Heterozygous IGFALS mutations are found in short SGA children (119-121). Serum IGF-I and IGFBP-3 concentrations are low. In cases of homozygous IGFALS mutations (OMIM 615961), the absence of the acid labile subunit (ALS) results in low IGF-I and very low IGFBP-3 concentrations with birth weights varying from −3.7 to −0.1 SDS (120).
The 3-M syndrome is characterized by pre- and postnatal growth failure and is caused by mutations in CUL7 (OMIM 273750), OBSL1 (OMIM 610991) or CCDC8 (OMIM 614205) (122,123). It is associated with reduced IGF2 expression and increased H19 expression, as also found in Silver-Russell syndrome (SRS). Characteristically, birth size is severely affected although individuals with milder intrauterine growth restriction have been reported (124).
Recently, a paternal IGF2 gene mutation was reported, which leads to features resembling SRS. GH treatment is likely to be as effective as in other genetic variants of SRS (125,126).
Floating Harbor syndrome occurs due to heterozygous mutations in the SRCAP gene (OMIM 136140). Only 26% of such children are born SGA. Recognition can be difficult, especially in young children (before the age of 4 years) and/or with mild phenotype (127).
Noonan syndrome may also be the cause of short stature after being born SGA. Activation of the RAS/Mitogen activated protein kinase (MAPK) signaling pathway results in a number of overlapping syndromes, the so-called “RASopathies”, including Noonan (OMIM 163950), Leopard (OMIM 151100), Costello (OMIM 218040), cardio-facio-cutaneous (OMIM 115150), and neurofibromatosis-Noonan syndromes (OMIM 601321) (128). All have a varying degree of postnatal growth failure and, sometimes, there are no obvious clinical features.
Monogenic disorders with normal head circumference and disproportionate short stature (Table 1)
In most genetic disorders associated with disproportionate short stature, the genes are already abnormally expressed during fetal life, which often results in a lower birth length SDS than birth weight SDS. The more severe forms of skeletal dysplasia can be easily diagnosed in young children, but clinical features can be so mild that many children are initially labeled as short SGA of unknown origin. Body disproportion and skeletal abnormalities can become more abnormal when the child becomes older. When an SGA child has disproportionate short stature (sitting height/height ratio > 2 SDS and/or arm-span width > height), skeletal survey analysis and/or genetic testing can be an efficient diagnostic approach. A description of all skeletal dysplasias and their underlying genetic aberrations is beyond the scope of this guideline, but the most frequent ones in relation to SGA and short stature are presented in Table 1.
Turner syndrome (TS), either due to 45X or mosaicism, may explain disproportionate short stature after SGA. Birthweight is lower than normal, but only 30% are born SGA. It is nevertheless important to exclude Turner syndrome in all girls born SGA with unexplained growth failure or pubertal delay or any constellation of the Turner syndrome clinical findings. The disturbed prenatal and postnatal growth is caused by short-stature-homeobox (SHOX) haploinsufficiency (129). A full description of Turner syndrome is beyond the scope of this guideline.
Mutations or deletions in the SHOX gene can be present in short SGA children. These are located at the upper end of the short arm of the X and Y chromosome, and they are transmitted in a pseudoautosomal fashion (130). This mutation can lead to SGA, with minor or no skeletal findings, and no or mild body disproportion (OMIM 300582), particularly SHOX enhancer deletions. Birth length and weight show wide variations (−4.3 to 1.5, and −3.3 to 3.2 SDS, respectively) (131).
Heterozygous activating mutations in FGFR3 lead to a wide range of disorders, including achondroplasia (OMIM 100800), hypochondroplasia (OMIM 146000), and even proportionate short stature. Neonates with achondroplasia have a birth length approximately −1 SDS, but SGA is possible (132).
Children with hypochondroplasia (OMIM 146000) have rhizomelic limb shortening, limitation of elbow extension, brachydactyly, and relative macrocephaly. It is a relatively frequent genetic mutation in children with SGA and ISS (133).
Heterozygous carriers of NPR2 mutations (OMIM 616255) show a similar phenotype to SHOX haploinsufficiency with short forearms and short lower legs (mesomelia) but without Madelung deformity (134). NPR2 mutations explain ∼2% of cases with short stature due to SGA or ISS (135).
Heterozygous mutations of the Indian hedgehog gene (IHH) are associated with brachydactyly type A1 (OMIM 1112500) but may also cause short stature classified as ISS or SGA with mild disproportion (136). Most children are born SGA for length, and 50% have shortening of the middle phalanx of the second and fifth fingers with cone-shaped epiphyses.
A heterozygous mutation of ACAN (encoding aggrecan) leads to abnormal cartilage matrix formation, with mild skeletal dysplasia, spondyloepiphyseal dysplasia (OMIM 608361), or short stature without radiographic skeletal dysplasia (OMIM 165800) (137). Approximately 30% to 40% of children are born SGA and 14% of short SGA children with a bone age advancement of 6 or more months had a heterozygous ACAN mutation (138). However, bone age can be normal. Birth length SDS is always lower than birth weight SDS, and patients often have early-onset osteoarthritis and/or osteochondritis dissecans.
Monogenic disorders with microcephaly and short stature (Table 2)
Children with biallelic (homozygous or compound heterozygous) IGF1 mutations (OMIM-608747) are born with a very low birth weight and length and microcephaly, although patients homozygous for less disruptive IGF1 mutations or harboring heterozygous IGF1 mutations have a milder phenotype. Recombinant IGF-I treatment is moderately effective (139, 140). Children with complete loss-of-function mutations also have sensorineural deafness (OMIM-608747) (141).
Children with a heterozygous IGF1R mutation (MIM 270450) have a wide range of birth weight (−3.5 to −1.5 SDS), birth length (−5.0 to 0.3 SDS) and head circumference (−3.0 to 0 SDS) (142,143). The prevalence of heterozygous IGF1R mutations or deletions is estimated at 1% to 2% in short SGA children (143). Homozygous mutations are extremely rare and possibly not compatible with life, although one case was reported with severe pre- and postnatal growth failure (< −4.5 SDS) and congenital malformation) (144). The response to recombinant GH treatment is usually moderate and associated with high IGF-I concentrations (145, 146).
Terminal 15q deletion with allelic loss of IGF1R leads to pre- and postnatal growth retardation and cardiac symptoms, intellectual disability, diaphragmatic hernia, hearing problems, aortic root dilatation, neonatal lymphedema, and aplasia cutis (147).
Homozygous pregnancy-associated-plasma-protein-A2 (PAPPA2) mutations were recently found in some short children born SGA (148). They have progressive growth failure, moderate microcephaly, thin long bones, mildly decreased bone density, and elevated concentrations of serum IGF-I, IGFBP-3, IGFBP-5, ALS, and IGF-2. Lack of PAPP-A2 protein decreases the liberation of IGF-I from the ternary complex and likely results in lower IGF bioavailability.
Primordial dwarfism is a group of rare genetic disorders characterized by severe IUGR and SGA, extreme short stature, and distinct microcephaly, which occur as a result of disorganized molecular and genomic changes during embryonic development. Most children can be easily recognized, like those with Cornelia de Lange syndrome (OMIM 122470), Meier-Gorlin syndrome (OMIM 224690), microcephalic osteodysplastic primordial dwarfism (MOPD) types I (OMIM 210710) and II (OMIM 210720), and Seckel syndrome (OMIM 210600) (149).
Smith-Lemli-Opitz syndrome (OMIM 270400) is an autosomal recessive disorder characterized by SGA, short stature, microcephaly, dysmorphic features, mild to severe mental retardation and multiple malformations. Patients have decreased serum cholesterol concentrations due a deficient cholesterol synthesis because of mutations of the 3beta-hydroxysterol-delta7 reductase gene (DHCR7) (138).
Disorders of DNA repair or genomic instability are frequently associated with SGA, short stature, and microcephaly. Bloom syndrome (OMIM 210900) is a DNA repair disorder caused by a mutation in the gene encoding DNA helicase RecQ protein-like-3 (RECQL3) and features include skin hypersensitivity to sunlight, particularly recognizable on the face. Fanconi syndrome (OMIM 227650) has genomic instability, and clinical features include an irregular pattern of skin pigmentation which increases over time, abnormalities in major organ systems, early-onset bone marrow failure, and a high predisposition to cancer. Other syndromes in this category are Nijmegen breakage syndrome (OMIM#251260), LIG4 (OMIM 606593) and XRCC4 mutations (OMIM 616541), Cockayne syndrome (OMIM 216400), and Rothmund-Thomson syndrome (OMIM 268400) (139).
Imprinting disorders and methylation disturbances (Table 3)
Imprinting disorders are characterized by molecular disturbances affecting genomically imprinted chromosomal regions and genes. Genomic imprinting describes the expression of specific genes in a parent-of-origin specific manner, that is, they are expressed only from the maternal or from the paternal gene copy (150). Silver-Russell syndrome (SRS) is well-known, but there are several other imprinting disorders associated with short stature after being born SGA (151, 152). It was recently reported that copy number variations (CNVs), methylation disturbances in imprinted and non-imprinted genes, and sequence variants all contribute to prenatal growth failure (153).
Silver-Russell syndrome
Children with SRS (OMIM 180860) are born SGA, have short stature and relative macrocephaly, a triangular shaped face with frontal bossing, clinodactyly, and asymmetry of face and/or extremities (126). Severe feeding difficulties are often present, especially during infancy. Mean adult height is around −4 SDS. SRS is primarily a clinical diagnosis, which can be established with the Netchine-Harbison clinical scoring system (126).
In approximately 60% of children with SRS, an underlying cause can be identified. Fifty percent are caused by a loss of methylation (LOM) of the telomeric domain in the 11p15.5 region, resulting in downregulation of paternal IGF2 expression (126). Approximately 5% to 10% of children with SRS have a maternal uniparental disomy of chromosome 7 (upd(7)mat), with potential causative genes including GRB10 (7p12.1) and MEST (7q32) (126). In 35%, the genetic cause is yet unknown, namely, clinical SRS. Patients with 11p15 loss of methylation have a more “classic” SRS phenotype with a higher prevalence of asymmetry than those with upd(7)mat, but the latter carries a higher risk of behavioral problems (126). More widespread use of methylation studies will probably uncover more epigenetic disorders in short SGA children, as exemplified by a study in patients with suspected SRS or unexplained short stature/IUGR, among whom 37% showed abnormal methylation in 11 imprinted loci (154). More information can be found in the SRS Consensus Statement of 2016 (126).
Other imprinting and methylation disorders
Besides upd(7)mat, there are several IDs and methylation disorders associated with SGA and short stature (155). Maternal uniparental disomy of chromosome 20, upd(20)mat, causes SGA, short stature, and prominent feeding difficulties with failure to thrive, but the patients differ from those with SRS because there is no asymmetry and no prominent forehead correlative macrocephaly (156). Temple syndrome (OMIM 616222) due to maternal uniparental disomy 14 (upd14mat), paternal deletion, distal 14q duplication, or loss of methylation at the intergenic differentially methylated region, is characterized by hypotonia, early puberty, short stature, and often born SGA (157), and the clinical phenotype can overlap with SRS.
Stepwise diagnostic approach, including genetic and epigenetic testing, in short children born SGA
Early referral of children born SGA with dysmorphic features, malformations, microcephaly, developmental delay, intellectual disability, and/or signs of skeletal dysplasia to a pediatric endocrinologist or pediatrician with SGA expertise is very important. Referral is also indicated when height is < −2.5 SDS before the age of 2 years or height is < −2 SDS around the age of 3 to 4 years (4). The clinical assessment consists of a complete medical history, prenatal and birth history, family history, a detailed physical examination and analysis of the individual growth curve aimed at collecting diagnostic clues that point in the direction of a primary or secondary growth disorder (158). Because short children born SGA might have a form of skeletal dysplasia, measuring sitting height (SH) to height ratio, relationships between arm-span and height and head circumference and height, may be helpful in identifying skeletal dysplasia and other genetic syndromes (158, 159). Both parents’ heights should be measured and mid-parental height along with the target height documented. The parameters should be plotted on an appropriate growth chart and assessed for adequacy. Growth of children with known syndromes should be plotted on syndrome-specific growth charts.
Screening laboratory blood tests are performed to exclude non-endocrine causes, such as anemia, chronic renal failure, celiac disease, electrolyte disturbances, and bone metabolism. Systemic diseases, such as renal tubular acidosis and inflammatory bowel disease, should be treated before assessing the need for GH therapy (9). Endocrine causes, such as GHD and hypothyroidism should be considered. The diagnostic approach to the evaluation of the hypothalamic-pituitary-growth axis is controversial in children born SGA, as wide variations of IGF1 and IGFBP-3 concentrations and GH responses to stimulation tests are seen in children born SGA (160, 161). GH stimulation tests are not required in all short children born SGA but remain part of the comprehensive evaluation of those suspected of GHD. In Japan, however, a GH stimulation test is required when GH therapy is considered because short children born SGA with GHD are treated with a lower GH dose than used in those without GHD. Skeletal imaging and genetic testing might reveal anatomical abnormalities associated with skeletal dysplasia, particularly when the sitting height/height ratio is >2 SDS.
Figure 1 presents a stepwise diagnostic approach for short children born SGA. The main objective is to identify a specific condition that guides genetic investigation and a gene or a set of genes that need to be evaluated, as well as the most appropriate molecular technique for this investigation (162-164). When there are signs of a secondary growth disorder, conditions such as hypothyroidism, celiac disease, GH-IGF-axis disturbance, or chronic disease should be evaluated. A skeletal survey is useful to obtain a specific diagnosis of skeletal dysplasia in short children with body disproportion and/or skeletal deformities. When there are dysmorphic features, major malformations, microcephaly, developmental delay, intellectual disability, and/or signs of skeletal dysplasia, one may consider evaluation by a clinical geneticist (162-164). Repeatedly high serum IGF-I concentrations (>1 SDS) in a child with microcephaly requires genetic testing of the IGF1R gene (147, 165). In all girls with unexplained growth failure, Turner syndrome should be excluded (129). When there are signs of a primary growth disorder and Turner syndrome is excluded, further genetic testing should be performed.
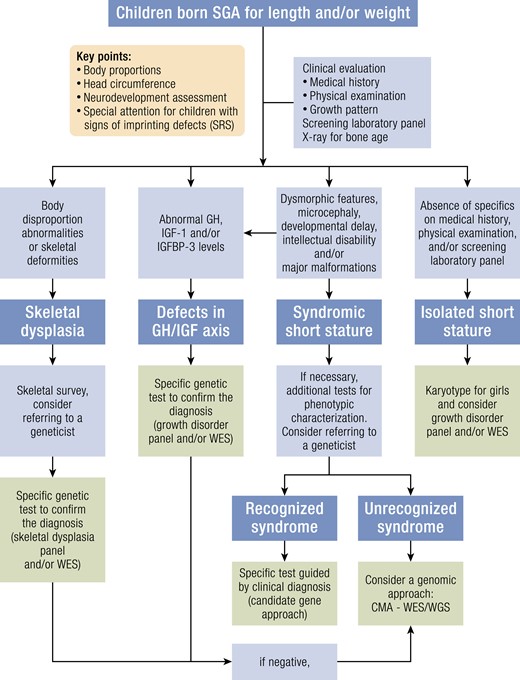
Genetic testing. Abbreviations: CMA, chromosomal microarray analysis; GH, growth hormone; IGF-I, Insulin-like growth factor-I; IGFBP-3, Insulin-like growth factor-binding protein-3; WES, whole exome sequencing; SRS, Silver-Russell syndrome; WGS, whole genome sequencing.
An important point before starting genetic investigation is the recognition of patients who may have imprinting defects. For investigating imprinting disorders such as SRS, specific molecular tests for methylation defects (epimutations) or uniparental disomy are recommended. A further step is multilocus methylation analysis. In case of a positive Netchine-Harbison score (126), genetic testing for SRS is also indicated, as mutations in the HMAG2 or IGF2 gene are increasingly found (125, 166).
Figure 1 presents also a flowchart of (epi)genetic testing in children born SGA. If there is a strong suspicion of a genetic condition, DNA sequencing and/or an MLPA (multiplex ligation-dependent probe amplification) test of one specific gene can be performed (“candidate gene approach”), but one may also decide to perform a growth-specific gene panel or a specific exome-based gene panel targeted to growth-related genes (167-169). The multigene sequencing analysis is preferred in heterogeneous genetic disorders, such as SGA with short stature, which are caused by multiple genes. In patients with a nonspecific phenotype, which does not allow for a candidate gene approach, but with a high probability of a monogenic condition, a genomic approach is a cost-effective strategy to obtain a diagnosis (164, 168, 169).
The clinical application of genetic investigations is rapidly expanding due to the development of new molecular techniques and increasing knowledge of the genetic basis of different conditions. As a result, the diagnostic yield is also rapidly increasing over time. Currently, chromosomal microarray (CMA), such as comparative genomic hybridization (CGH) array or single nucleotide polymorphism (SNP) array, can identify a pathogenic copy number variation (CNVs, deletion or duplication) in 15% of short children born SGA with unrecognized syndromic conditions (170-172). Whole exome sequencing (WES) is effective to obtain a definitive diagnosis in up to 45% of children with a high degree of suspicion for a monogenic condition, based on the presence of skeletal dysplasia or syndromic phenotype, history of consanguinity, clear autosomal dominant inheritance pattern, and/or severe short stature (168, 172, 173). The trio-WES (patient and both parents or including siblings) can increase the diagnostic yield. In children born SGA with short stature as the only manifestation, the diagnostic yield was approximately 15% with a multigene approach (WES or growth-specific gene panel) (173, 174). These children represent the mild forms of skeletal dysplasias (heterozygous defects in ACAN, SHOX, NPR2, IHH) or syndromic conditions (Noonan syndrome or neurofibromatosis type 1). As a last step, whole genome sequencing (WGS) or a methylation array could be considered in special cases, although these approaches are primarily restricted for research purposes. However, as genetic techniques are becoming more commonly available, a cause will be identified in an increasing number of short SGA children, and it might turn out that many cases of SGA are caused by a combination of multiple (epi)genetic variants (175). When ordering genetic testing, one should consider the likelihood of finding a causal mutation in any specific patient (ie, the pre-test probability) vs the likelihood of getting a false positive especially when employing large-scale sequencing methods. Additionally, in the case of a hypothesis-free approach (WGS or WES) in children with a complex phenotype, the result must guide a review of the patient's clinical findings to reassure its causal relationship.
Recommendations:
15. Endocrine workup to determine the cause of short stature includes thyroid hormone and serum IGF-I concentrations. In case of low IGF-I concentrations, it is useful to add IGFBP-3. A GH stimulation test is only indicated in those with suspected GH deficiency. (A ++)
16. When there are dysmorphic features, major malformations, microcephaly, developmental delay, intellectual disability, and/or signs of skeletal dysplasia, evaluation by a clinical geneticist should be considered. (A +++)
17. Genetic testing should be considered in children born SGA with persistent short stature with or without skeletal disproportion for whom no other cause is known. (A ++)
Growth Hormone Treatment
Indications for GH treatment
After GHD, short stature after SGA birth is the second most common indication for GH treatment. Treatment initiation at a young age is one of the most important factors predicting a favorable growth response. Therefore, early identification and referral of children with persistent short stature after SGA birth is very important. In the United States and Latin America, GH treatment can be initiated from an age of 2 years, while children may be treated after the age of 3 years and 4 years in Japan and Europe, respectively (176-178). The indication of GH treatment based on height SDS varies between countries with cutoffs between −2.0 and −2.5 SDS (178), whereas no height SDS criterion is set by the Food and Drug Administration (FDA) in the United States.
Growth velocity < 0 SDS is required by the European Agency for the Evaluation of Medical Products (EMA) and Japanese agency (PMDA), but not in the United States and Latin America (176-178). EMA requires, in addition, a distance to mid-parental height (MPH) of −1 SDS. Table 4 shows the criteria for GH treatment for short children born SGA in various areas of the world. These are mostly defined as: documented small birth weight and/or birth length (< −2 SDS for gestational age), persistent short stature (< −2.0 SDS), at an age after which catch-up growth is unlikely to occur (3-4 years), growing at an average or subnormal rate for age, provided that other causes for short stature have been ruled out, and that there are no clinical features suggestive of a dysmorphic syndrome (with the exception of SRS) (29). GH treatment is not recommended in several disorders with a high predisposition to develop cancer, such as chromosomal breakage syndromes and DNA repair disorders (179). Diagnosing these syndromes can be challenging but identifying the genetic etiology before start of GH treatment is important for health prognosis, genetic counseling, and treatment options.
Indications for growth hormone treatment in short children born SGA
| United States | Europe | Japan | Latin America | |
|---|---|---|---|---|
| Earliest age at start (years) | 2 | 4 | 3 | 2 |
| Height SDS | <−2.0 | <−2.5 | <−2.5 | <−2.0 |
| Growth velocity | No indication | <0 SDS | <0 SDS | No indication |
| Reference to mid-parental height | No indication | < −1.0 SDS from MPH | No indication | No indication |
| United States | Europe | Japan | Latin America | |
|---|---|---|---|---|
| Earliest age at start (years) | 2 | 4 | 3 | 2 |
| Height SDS | <−2.0 | <−2.5 | <−2.5 | <−2.0 |
| Growth velocity | No indication | <0 SDS | <0 SDS | No indication |
| Reference to mid-parental height | No indication | < −1.0 SDS from MPH | No indication | No indication |
Indications for growth hormone treatment in short children born SGA
| United States | Europe | Japan | Latin America | |
|---|---|---|---|---|
| Earliest age at start (years) | 2 | 4 | 3 | 2 |
| Height SDS | <−2.0 | <−2.5 | <−2.5 | <−2.0 |
| Growth velocity | No indication | <0 SDS | <0 SDS | No indication |
| Reference to mid-parental height | No indication | < −1.0 SDS from MPH | No indication | No indication |
| United States | Europe | Japan | Latin America | |
|---|---|---|---|---|
| Earliest age at start (years) | 2 | 4 | 3 | 2 |
| Height SDS | <−2.0 | <−2.5 | <−2.5 | <−2.0 |
| Growth velocity | No indication | <0 SDS | <0 SDS | No indication |
| Reference to mid-parental height | No indication | < −1.0 SDS from MPH | No indication | No indication |
In low- and middle-income countries, GH treatment in children born SGA is a great challenge and remains an unfilled gap. The high cost of GH constitutes a major obstacle for treatment access. In these countries, only a minority of society can afford such a treatment.
Recommendations:
18. GH treatment is recommended in children born SGA with persistent short stature at an age after which catch-up growth is unlikely to occur (which is in most children at the age of 3 to 4 years) provided other common causes for short stature have been ruled out. (A +++)
19. GH treatment is not recommended in several genetic disorders with a high predisposition to develop cancer, such as chromosomal breakage syndromes and DNA repair disorders. (A +++)
Effects during GH treatment
Growth and adult height
GH treatment with a dose of 0.033 mg/kg/day (1 mg/m2/day) induces catch-up growth and improves adult height of short children born SGA (180-186). A systematic review published in 2009 identified 4 high-quality trials with adult height outcome in 391 short children born SGA treated with GH and concluded that the mean height gain was on average 1.25 SDS (184). When GH treatment is started before puberty, adult height results are similar in children treated with a GH dose of 0.033 mg/kg/day or 0.067 mg/kg/day (182). The GH-induced catch-up growth is accompanied by normal body proportions and proportional head growth (10, 181). The GH-induced growth response is, however, highly variable (182). Several studies identified clinical predictors for growth response to GH treatment (30, 185, 187-190). A lower height SDS at start of GH, more height gain during the first year on GH, longer duration of treatment, a higher length SDS at birth and a higher height of the mother associate with a higher adult height SDS and explained 70% of the variability of adult height (189). The growth response to GH treatment is similar between those born preterm or term (191, 192). The large variability in growth response to GH is also likely to be associated with multiple gene variants resulting in SGA birth and short stature (193). Thus, a more complete understanding of the specific genetic variations that affect growth and the response to growth hormone will improve the management of affected individuals.
Pubertal maturation
GH treatment neither influences the onset and duration of puberty nor the pubertal height gain, regardless of GH dose (54, 194).
Glucose metabolism
Short children born SGA have reduced insulin sensitivity before the start of GH treatment (195). GH treatment results in a further decrease in insulin sensitivity with a compensatory increase in insulin secretion due to the insulin-antagonistic effects of GH (196-202). However, long-term GH treatment in large study groups showed that glycated hemoglobin (HbA1c) concentrations remained within the normal range, and that none of the GH-treated SGA-born subjects developed type 2 diabetes mellitus (196, 203, 204). Insulin sensitivity increases after GH cessation (186, 187).
Body composition
Children born SGA show significant changes in body composition during GH treatment, with a decrease in fat mass and an increase in lean body mass due to the lipolytic and anabolic effects of GH, respectively (201). This shift in body composition is considered healthier and is maintained throughout the course of GH treatment.
Blood pressure and lipid concentrations
Blood pressure SDS decreases in GH-treated children born SGA and becomes significantly lower than in untreated short children born SGA (200, 203, 205-207), which might be explained by a decrease in matrix metalloproteinases (208). Total cholesterol and low-density lipoprotein cholesterol (LDLc) decrease during GH treatment (203, 205, 207, 209), indicating a beneficial effect of GH treatment on lipid metabolism.
Thyroid function
GH treatment leads to lower serum free thyroxine (T4) levels in 14% of children born SGA in the presence of normal TSH levels, which is due an increased conversion of free T4 to biologically active free triiodothyronine (T3) (210). GH treatment does not result in thyroid dysfunction (210, 211).
Bone mineral density
Short children born SGA have on average a lower BMD, albeit in the normal range, also after adjustment for short stature (72-74). During GH treatment, BMD of the total body and lumbar spine increases (75), reflecting the significant role of the GH-IGF-I axis in the accrual of bone mass.
Cognition and health-related quality of life
The presence of GH receptors in the brain implies that the brain is a target for GH, and that GH treatment could affect brain function. GH-treated children born SGA show modest improvement in cognitive function, IQ scores, and psychosocial functioning (9, 212, 213). A small study during 2 years of GH treatment showed no beneficial effect on IQ score (214). Long-term GH treatment improved health-related quality of life (HRQoL) in children born SGA (215-218). GH-treated adolescents born SGA scored higher on physical abilities, contact with adults and on body image, compared to untreated short adolescents born SGA (216).
Clinical management during GH treatment (Table 5)
We recommend treating short children born SGA with a GH dose of 0.033 mg/kg/d and only increasing the GH dose if the growth response is unsatisfactory and causes of a poor growth response, such as nonadherence, hypothyroidism, unknown disease, or syndrome are ruled out. In case of a poor growth response, the GH dose can be increased within the dose range until there is a satisfying growth response and serum IGF-I concentrations remain ≤ 2 SDS. We do not recommend starting with a GH dose of 0.067 mg/kg/d as this resulted in high serum IGF-I concentrations (around or above +2 SDS) and a similar adult height as treatment with a dose of 0.033 mg/kg/d (180,182,203). GH dosing based on IGF-I titration results in a diminished growth response than a treatment based on a fixed GH dose (219). Be aware that the IGF-I SDS is dependent on pubertal development in addition to age.
During GH treatment, it is essential to monitor height, sitting height, weight, and pubertal Tanner stage regularly (Table 5). The first-year increase in height under GH treatment should be more than 0.5 SDS (220). A French study showed that cessation of GH treatment after a positive growth response for 3 years leads to catch-down growth and is, therefore, not recommended (221). If there is an inadequate growth response, reevaluation is indicated and the decision to discontinue GH treatment should be considered. When adolescents have attained near adult height, defined as a growth rate < 1 cm/6 months in combination with closed epiphyses, GH treatment should be discontinued.
Monitoring during growth hormone treatment in short children born SGA
| At start of GH treatment | Every 3 to 4 months during GH treatment | Yearly during GH treatment | At cessation of GH treatment | |
|---|---|---|---|---|
| Height | X | X | X | X |
| Weight | X | X | X | X |
| Sitting height | X | — | — | — |
| Blood pressure | X | X | X | X |
| Tanner stage | X | X | X | — |
| IGF-1 | X | — | X | — |
| IGFBP-3 | X | — | — | — |
| Free T4 | X | — | X | X |
| TSH | X | — | X | X |
| Fasting lipid levels | X | — | — | X |
| Fasting glucose, insulin, HbA1c | X | — | — | — |
| Bone age with X-ray hand/wrist | X | — | —a | X |
| At start of GH treatment | Every 3 to 4 months during GH treatment | Yearly during GH treatment | At cessation of GH treatment | |
|---|---|---|---|---|
| Height | X | X | X | X |
| Weight | X | X | X | X |
| Sitting height | X | — | — | — |
| Blood pressure | X | X | X | X |
| Tanner stage | X | X | X | — |
| IGF-1 | X | — | X | — |
| IGFBP-3 | X | — | — | — |
| Free T4 | X | — | X | X |
| TSH | X | — | X | X |
| Fasting lipid levels | X | — | — | X |
| Fasting glucose, insulin, HbA1c | X | — | — | — |
| Bone age with X-ray hand/wrist | X | — | —a | X |
Bone age: Bone maturation is a poor predictor of pubertal timing and height attainment in SGA children. It is, however, worth performing a bone age evaluation around the age of 8 years and regularly from the onset of puberty onwards, as an advanced bone age could be a warning sign for the presence of an underlying genetic variation, such as an ACAN mutation.
Monitoring during growth hormone treatment in short children born SGA
| At start of GH treatment | Every 3 to 4 months during GH treatment | Yearly during GH treatment | At cessation of GH treatment | |
|---|---|---|---|---|
| Height | X | X | X | X |
| Weight | X | X | X | X |
| Sitting height | X | — | — | — |
| Blood pressure | X | X | X | X |
| Tanner stage | X | X | X | — |
| IGF-1 | X | — | X | — |
| IGFBP-3 | X | — | — | — |
| Free T4 | X | — | X | X |
| TSH | X | — | X | X |
| Fasting lipid levels | X | — | — | X |
| Fasting glucose, insulin, HbA1c | X | — | — | — |
| Bone age with X-ray hand/wrist | X | — | —a | X |
| At start of GH treatment | Every 3 to 4 months during GH treatment | Yearly during GH treatment | At cessation of GH treatment | |
|---|---|---|---|---|
| Height | X | X | X | X |
| Weight | X | X | X | X |
| Sitting height | X | — | — | — |
| Blood pressure | X | X | X | X |
| Tanner stage | X | X | X | — |
| IGF-1 | X | — | X | — |
| IGFBP-3 | X | — | — | — |
| Free T4 | X | — | X | X |
| TSH | X | — | X | X |
| Fasting lipid levels | X | — | — | X |
| Fasting glucose, insulin, HbA1c | X | — | — | — |
| Bone age with X-ray hand/wrist | X | — | —a | X |
Bone age: Bone maturation is a poor predictor of pubertal timing and height attainment in SGA children. It is, however, worth performing a bone age evaluation around the age of 8 years and regularly from the onset of puberty onwards, as an advanced bone age could be a warning sign for the presence of an underlying genetic variation, such as an ACAN mutation.
Bone maturation is a poor predictor of pubertal timing and height attainment in SGA children (180). It is, however, worth performing a bone age evaluation around the age of 8 years and regularly from the onset of puberty onwards, as an advanced bone age could be a warning sign for the presence of an underlying genetic variation, such as an ACAN mutation (138).
It is recommended to determine the serum IGF-I concentrations at 3 to 6 months after the start of treatment to evaluate whether the GH dose requires adjustment. Thereafter, it is advised to determine the serum IGF-I concentrations at least annually. Be aware that high total IGF-I levels during GH treatment do not always parallel high IGF bioactivity in children born SGA (222). When the IGF-I concentrations remain high even with a relatively low GH dose, one should consider performing IGF1R mutational analysis and to examine the presence of a possible underlying dysmorphic syndrome, such as Bloom syndrome or Fanconi syndrome (179). As GH treatment could potentially increase the intrinsic tumor risk in such conditions, this should be thoroughly discussed with parents and treatment options based on shared decision making (179). It is recommended to perform thyroid function tests annually, since GH treatment can lead to decreased serum free T4 levels (206). This is mostly due to the increased conversion to free T3 and accompanied by normal TSH levels. In case of a low free T4 and elevated TSH levels, hypothyroidism might be present, which would require treatment with levothyroxine.
GH treatment has limited effects on cardiometabolic parameters during treatment and no reported long-term adverse cardiometabolic effects. We, therefore, do not recommend routine evaluation of metabolic parameters for all children born SGA treated with GH, but for those with risk factors, such as overweight, obesity, ethnicity, and family history.
Recommendations:
20. The recommended GH dose should cover the range 0.033 to 0.067 mg/kg/day, with a starting dose of 0.033 mg/kg/day. The GH dose can be increased within the dose range when the growth response is unsatisfactory (delta height gain <0.5 SDS) during the first year of treatment, and a higher starting dose within the dose range could be considered in those with the most marked growth retardation (A ++)
21. GH dosing based on IGF-I titration alone is not recommended. (A +++)
22. It is recommended to determine serum IGF-I at 3 to 6 months after the start of GH treatment to evaluate adherence to the treatment and whether the GH dose requires adjustment for safety reasons. Thereafter, it is advised to determine the serum IGF-I concentrations at least annually. (A +++)
23. Bone maturation is a poor predictor of pubertal timing and height attainment in SGA children. Therefore, during GH treatment, yearly bone age assessment is not recommended in prepubertal children. (A ++) It is, however, worth performing a bone age assessment around the age of 8 years and regularly from the onset of puberty onwards, as an advanced bone age could be a warning sign for the presence of an underlying genetic variation, such as an ACAN mutation.
24. During GH treatment, annual assessment of serum concentrations of thyroid hormone (free T4, TSH) is recommended. Routine evaluation of metabolic parameters such as fasting serum lipid and glucose/insulin/HbA1c concentrations is only recommended for children born SGA treated with GH with risk factors, such as overweight, obesity, and family history (A +++)
25. GH treatment is discontinued in case of (near-)adult height attainment defined as growth rate less than 1 cm in 6 months in combination with closed epiphyses or when there are repeated and clear signs of nonadherence or no response to GH treatment. (A +++)
Addition of 2 years of gonadotropin-releasing hormone agonist treatment to GH treatment
Effect on adult height
When GH-treated children born SGA are still short at the onset of puberty with an expected short adult height (< −2.5 SDS), postponement of puberty with a gonadotropin-releasing hormone analogue (GnRHa) for 2 years results in an average improvement in adult height of 6.6 cm, in boys and girls alike, compared with those who did not receive GnRHa treatment (223). Thus, addition of 2 years of GnRHa treatment is worth considering for SGA children who are still short at the onset of puberty. Further longitudinal studies should validate the benefits of GnRHa treatment in children born SGA.
Safety of combined GH/GnRHa treatment
Studies in children with central precocious puberty, who were treated with GnRHa, reported a decrease in insulin sensitivity, a gain in weight and fat mass (224-227), and a decrease in bone turnover and BMD (228-231). Most studies, however, evaluated these parameters only in girls with central precocious puberty during GnRHa treatment. As both GH and GnRHa could potentially have an adverse effect on cardiometabolic health, the combination of both treatments did raise concerns. Longitudinal studies have now shown that GH treatment in combination with 2 years of GnRHa treatment has no long-term negative effects on body composition, insulin sensitivity, blood pressure, and lipid concentrations, up to 5 years after cessation of GH compared with subjects who did not receive GnRHa treatment (74, 232-234). Cognitive functioning and quality of health was similar in young adults born SGA following combined GH/GnRHa treatment compared with those treated with GH only (218, 235-237). Problematic behavior and self-perception were similar at adult height in the subjects additionally treated with GnRHa treatment (237).
Recommendation:
26. In short children born SGA, addition of GnRHa treatment to GH treatment could be considered if the expected adult height is below −2.5 SDS at the onset of puberty; further longitudinal studies should validate the benefits of GnRHa treatment. (A ++)
Effects after cessation of long-term GH treatment
Postmarketing GH registries and the SAGhE study
The GH safety workshop position paper of 2016 concluded that GH continues to have a good safety record when used for approved indications and at recommended doses (238). Over the years, many papers have shown that GH treatment is well-tolerated, and that serious adverse effects are uncommon (186). The Safety and Appropriateness of Growth hormone treatments in Europe (SAGhE) project, initiated in 8 European Union countries, evaluated the long-term mortality in patients treated with GH during childhood (239). Preliminary French data suggested an increased risk of cardiovascular mortality in subjects born SGA (240). However, the preliminary data from Belgium, The Netherlands, and Sweden, participating in the same project, did not confirm the French findings (241). The SAGhE project showed no raised overall cancer risk in subjects born SGA but increased mortality for diseases of circulatory system (242, 243). Recently, a nationwide population-based Swedish cohort study showed an increased risk of cardiovascular events in all adults treated with GH in childhood. The hazard ratio for subjects born SGA was 1.97 (95% CI, 1.28-3.04), but the absolute risk was low and comparable with the hazard ratio of patients with GH deficiency or ISS (244). One of the main limitations of all these observational studies is that data of participants were compared with national reference data or healthy age- and sex-matched regional controls and not with an appropriate control group of untreated short subjects born SGA.
Body composition and cardiometabolic health after cessation of long-term GH treatment in childhood
There are few papers about the long-term cardiometabolic health of young adults born SGA after cessation of GH treatment due to adult height attainment in comparison with appropriate control groups (75, 77, 204, 245). Since GH treatment has cardiometabolic effects and raises serum concentrations of IGF-I concentrations, which is mitogenic and antiapoptotic, there have been concerns about the long-term side effects of GH treatment even after cessation of treatment. Until now, there is one large Dutch cohort of 199 previously GH-treated young adults born SGA, who were followed for 5 to 12 years after cessation of childhood GH treatment (75, 77, 204, 245, 246). At 5 and 12 years after cessation, at the age of 21 and 30 years, their data were also compared with 285 age-matched untreated controls, consisting of SGA-born subjects with either persistent short stature or normal stature after spontaneous catch-up during childhood and AGA-born young adults (Fig. 2). Cessation of GH treatment was followed by a significant and persistent increase in fat mass, whereas lean body mass decreased only during the first 6 months after GH cessation and remained stable thereafter. Fat mass at 5 and 12 years after GH treatment cessation was, however, similar compared to untreated controls. At 5 and 12 years after GH cessation, lean body mass was lower in all subjects born SGA compared to subjects born AGA, suggesting that lean body mass is reprogrammed during fetal and early life rather than by postnatal growth or GH treatment (204, 246). Frequently sampled intravenous glucose tolerance tests showed that the GH-induced lower insulin sensitivity was reversible after GH cessation and at 5 and 12 years thereafter, and insulin sensitivity and beta-cell function were similar compared to untreated controls (203, 204, 246). GH cessation had no adverse effect on systolic and diastolic blood pressure (203, 245, 246). Carotid intima media thickness, a surrogate marker of cardiovascular risk, did not change during 5 years after GH cessation and both blood pressure and carotid intima media thickness in previously GH-treated subjects born SGA were similar to untreated controls (245). The beneficial effect of GH treatment on serum lipid concentrations was sustained during 5 years after GH cessation (245). In contrast, the beneficial effects of GH treatment on total-body BMD and BMD of the lower spine were lost after GH cessation, showing a gradual deterioration but remaining above −1 SDS (75). At 5 years after GH cessation, previously GH-treated subjects born SGA had similar lipid concentrations, BMD, and a prevalence of metabolic syndrome compared to untreated controls (75, 203, 204, 245). Glomerular filtration rate (GFR) decreased after GH cessation but remained within the normal range (77) and GFR and urinary albumin-to-creatinine ratio were similar in previously GH-treated subjects born SGA and untreated controls (77).
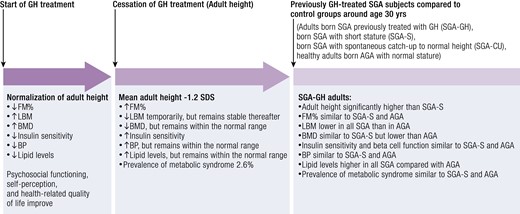
Effects of GH treatment during and after cessation, compared with untreated controls. Abbreviations: AGA, appropriate for gestational age; BMD, bone mineral density; BP, blood pressure; FM, fat mass; FM%, fat mass percentage; GH, growth hormone; LBM, lean body mass; SDS, standard deviation score; SGA, small for gestational age.
In summary, the above-mentioned data provide important insights into the cardiometabolic health after cessation of GH treatment of subjects born SGA, mitigating the previous concerns raised about the safety of this treatment. Based on these results, GH can be safely administered in clinical practice. Longer-term follow-up of previously GH-treated older adults born SGA is necessary to gain more insight into the beneficial impact of GH treatment.
Suggestions for follow-up of previously GH-treated adults born SGA
As the majority of previously GH-treated SGA-born adults are not routinely followed-up after cessation of GH treatment, there is limited literature regarding this topic. We would advise to counsel previously GH-treated adults born SGA to adopt a healthy diet and lifestyle with particular emphasis on regular exercise to avoid weight gain and maintain or preferably improve lean body mass. They should seek medical consultation in case of chronic fatigue, considerable weight gain, or hypertension, particularly in case of a family history of diabetes mellitus or cardiovascular disease (247). Registries at the national and international level to report on long-term health issues of adults born SGA, whether treated with GH or not, are of utmost importance to clarify their future health status. Moreover, it is important to inform neonatologists, general practitioners, pediatricians, and parents of infants born SGA about the importance of healthy catch-up, that is, a balanced gain in weight and length, particularly during the first months of life. As those with spontaneous catch-up growth to a normal stature might have a higher risk of metabolic and cardiovascular diseases in later life than those who remain short, similar counseling concerning lifestyle, exercise, and medical consultation should be offered to them.
Recommendations:
27. Upon discontinuation of GH treatment, adults born SGA should be counseled to adopt a healthy diet and lifestyle with regular exercise to avoid excessive weight gain and to maintain or preferably improve lean body mass. (A +++)
28. Long-term medical and metabolic follow-up of previously GH-treated young adults born SGA is recommended in those with risk factors for metabolic and/or cardiovascular disease. (A ++)
29. Registries at the national and international level to report on long-term health issues of adults born SGA, whether treated with GH or not, are of utmost importance to clarify the future health of subjects born SGA. (A +++)
General Conclusions
We recommend defining SGA as being born with a birth weight and/or birth length below −2 SDS for gestational age.
Children born SGA should be carefully followed in the first years of life by a neonatologist or a pediatrician to evaluate their growth, weight gain, and neurodevelopment. Excessive postnatal weight gain should be avoided since it is associated with a less favorable cardiometabolic health profile in adulthood. It is recommended to avoid additional nutritional supplementation to healthy infants up to the age of 6 months unless the infant suffers from malnutrition. We do not recommend routine investigations of all children born SGA. However, in case of persistent short stature < −2.5 SDS at the age of 2 years or a height < −2 SDS around the age of 3 to 4 years, without signs of catch-up growth, referral to a pediatric endocrinologist or a pediatrician with SGA expertise is indicated. Those born very preterm or severely SGA, with a complicated perinatal period, small head circumference, or with a suspicion of a syndrome associated with SGA, should be monitored more closely and referred to a pediatric endocrinologist early during infancy. Early screening for neurocognitive impairment is warranted for children at risk.
The diagnostic workup of short stature after SGA birth includes standard investigations. In addition, genetic testing should be considered since an increasing number of underlying (epi)genetic causes can be found.
GH treatment (0.033 to max 0.067 mg/kg/day) is recommended for children born SGA with persistent short stature as it is effective and safe, including in the long-term after GH cessation. Addition of 2 years of GnRHa treatment to GH treatment could be considered if the expected adult height is below −2.5 SDS at the onset of puberty. We recommend determining serum IGF-I concentrations at 3 to 6 months after the start of GH treatment to evaluate adherence to the treatment and for safety reasons, as well as assessing serum free T4 and TSH concentrations annually, but routine evaluation of other metabolic parameters is only recommended for children with risk factors, such as overweight, obesity, and family history.
Upon discontinuation of GH treatment, young adults born SGA should be counseled to adopt a healthy diet and lifestyle with regular exercise to avoid excessive weight gain and to maintain or preferably improve lean body mass. Long-term medical and metabolic follow-up of previously GH-treated young adults born SGA is only recommended in those with risk factors for metabolic and/or cardiovascular disease. Registries are needed to report on the long-term health of older adults born SGA, whether treated with GH during childhood or not.
Acknowledgments
The authors wish to thank W.M. Bramer, biomedical information specialist, from the Medical Library of Erasmus University Medical Center, The Netherlands, for developing and updating the search strategies.
Author Contributions
Three working groups formed the basis of this International Consensus Statement. These groups focused on diagnosis and etiology of SGA (working group 1: M.B., K.K., Y.N., A.H-K., A.A.J., A.D.), consequences of SGA birth (working group 2: S.C., J.D., V.M., W.C., P.H., E.I., X.L.), clinical management of children born SGA (working group 3: M.v.d.S., R.H., R.R., A.A., D.B., E.C., S.D.C., A.D., W.G., C.K.G., V.K., S.M., M.Y.). All authors contributed to the manuscript and participated in the consensus meeting to discuss and vote on recommendations. A.H.-K. and M.v.d.S. drafted the final manuscript. All authors read and approved the final manuscript.
Disclosures
A.H-K. was a recipient of investigator-initiated independent research grants from Novo Nordisk and Pfizer, served on advisory boards of Novo Nordisk and has received lecture fees from Merck-Serono, Novo Nordisk, and Pfizer; M.B. served as an occasional advisory board member and speaker for Novo Nordisk, Pfizer, and Merck; S.C. served on advisory boards of Sandoz and Novo Nordisk, and his Unit has received research grant support from the Italian Ministry of Health, EU and EC, Eli Lilly, MSD, Novo Nordisk, and Sanofi; J.D. received honoraria by Merck, Nestlé, Novo Nordisk, and Pfizer; R.H. served in advisory boards of Novo Nordisk, Pfizer/OPKO, and LUMOS Pharma, received lecture fees from Novo Nordisk, Pfizer, and Ascendis and research grants from Novo Nordisk, JCR, and Sandoz; S.C. served on advisory boards of BioMarin and Sandoz, and received research grants from Ascendis and Novo Nordisk; A.A.J. received consulting fees from Novo Nordisk and an independent research grant from BioMarin.
References
Abbreviations
- AGA
appropriate for gestational age
- BMD
bone mineral density
- BMI
body mass index
- FSH
follicle-stimulating hormone
- GH
growth hormone
- GHD
growth hormone deficiency
- GnRHa
gonadotropin-releasing hormone agonist
- IGF-I
insulin-like growth factor 1
- IGFBP
IGF binding protein
- ISS
idiopathic short stature
- IUGR
intrauterine growth restriction
- LH
luteinizing hormone
- SAGhE
Safety and Appropriateness of Growth hormone treatments in Europe
- SDS
standard deviation score
- SGA
small for gestational age
- SRS
Silver-Russell syndrome
- T3
triiodothyronine
- T4
thyroxine
- TSH
thyrotropin (thyroid-stimulating hormone)
- upd
uniparental disomy
- WES
whole exome sequencing
- WGS
whole genome sequencing
- WHO
World Health Organization
Author notes
Anita C. S. Hokken-Koelega and Manouk van der Steen joint first author.

{kind=link}
{kind=link}
{kind=link}