





Abstract
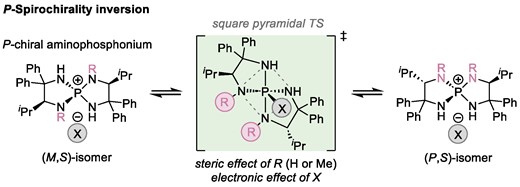
The key factors determining the feasibility of epimerization of P-spiro chiral tetraaminophosphoniums have been elucidated by DFT calculations. P-Spirochirality inversion proceeds through the formation of a pentacoordinate phosphorane by the nucleophilic attack of the counter ion and the subsequent pseudorotation via the square pyramidal transition state (TS). The TS energies are highly dependent on the nitrogen substituents of the aminophosphoniums and the counter ion structure. The energy barrier for the TS significantly increases by the introduction of methyl groups on two of the four nitrogen atoms of the aminophosphonium because of their steric repulsion. On the other hand, the energy barrier decreases when oxyanions, such as nitronate and phenoxide, are involved due to the effective interaction of the oxyanion p-orbitals with P–N σ* orbitals unlike the case with chloride ions. These profiles will account for our experimental observations.
The pseudorotation of pentacoordinate trigonal bipyramidal (TBP) phosphorus compounds (phosphoranes) has been extensively studied over the past decades. This is because the primary structure of phosphoranes is commonly found in the intermediates and transition states (TSs) of organic transformations, and their stereochemical stability is often relevant to understanding reaction outcomes.1 Through experimental and theoretical analyses of the pseudorotation mechanism, two representative processes, Berry pseudorotation (BPR) and turnstile rotation,2 have been proposed to account for the stereochemical scrambling of phosphoranes. However, while chirality inversion at the central element often occurs during the interconversion between stereoisomers of pentacoordinate phosphorus3 and silicon compounds4 via a square pyramidal (SP) TS, the racemization/epimerization of P-chiral nonracemic tetracoordinate phosphorus compounds5 via a pentacoordinate phosphorane intermediate, temporarily formed upon reaction with a nucleophile, remains underexplored (Fig. 1a).6 In particular, the effects of the nucleophile structure on the formation of pentacoordinate intermediates and the pseudorotation stages are virtually unknown. During our study on the development and applications of P-spiro chiral tetraaminophosphonium ions as organic molecular catalysts,7 we observed the epimerization of the aminophosphonium ion with four NH moieties on its P-spirocyclic core when paired with relatively nucleophilic anions. We also found that structurally similar aminophosphonium ions possessing methyl groups on two of the four nitrogen atoms exhibit contrasting robustness in the spirochirality. This observation led us to envisage that gaining insights into how the stereochemical lability of tetraaminophosphonium ions depends on their structures and pairing anions could enhance the understanding of the pseudorotation of P-chiral phosphorus compounds. Here, we describe our investigation of the epimerization processes of tetraaminophosphonium ions 1 and 2, which involve the formation of pentacoordinate phosphorane intermediates, in terms of the effects of nitrogen substituents (R) and the nature of the counter ions (X) (Fig. 1b).
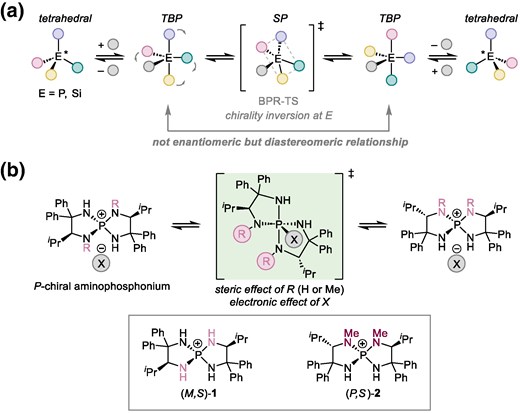
a) Racemization mechanism of tetracoordinate heavy main-group compounds. b) Investigation of the effects of nitrogen substituents (R) and anions (X) on BPR in the epimerization of tetraaminophosphonium 1 and 2.
In our previous study, P-spiro chiral tetraaminophosphonium chloride 1 was prepared from L-valine-derived diamine and phosphorus pentachloride, and a mixture of two diastereomers (M,S)- and (P,S)-1·Cl was obtained.8 The conjugate base of (M,S)-1·Cl, triaminoiminophosphorane, generated in situ via treatment with potassium tert-butoxide, acted as an effective catalyst for the anti and enantioselective Henry reaction. Although a high level of stereoselectivity was consistently achieved in reactions between various nitroalkanes and aldehydes under cryogenic conditions (−78 °C), the selectivity proved to be sensitive to the reaction temperature, which was inconsistent with the in-depth computational analysis reported by Simón and Paton.9 When the reaction was conducted by generating iminophosphorane at −30 °C in the presence of nitroalkane, a substantial decrease in enantioselectivity and the epimerization of P-spirochirality were observed as confirmed after the recovery of the aminophosphonium salt.10 Interestingly, while N-methylated (P,S)-2-derived iminophosphorane was found to be ineffective in terms of both reactivity and selectivity, no epimerization of the P-spirochirality was observed even after increasing the reaction temperature. These observations not only indicate a clear difference in the configurational stability between 1 and 2 but also suggest that the stereorandomization of (M,S)-1 is associated with the generation of aminophosphonium nitronate, possibly reflecting the counter ion-dependent stability of P-spirochirality. The consequence was the erosion of enantioselectivity caused by the intervention of diastereomeric iminophosphorane as a catalyst.
We then turned our attention to understanding the epimerization process, including the potential impact of N-substituents and the properties of the pairing anions. Our assumption was that epimerization would occur through the temporal formation of a pentacoordinate phosphorane by the nucleophilic attack of the anion on the stereogenic phosphorus center of the aminophosphonium ion. To this end, we conducted DFT calculations [B3LYP/6-31G(d) level] to investigate phosphorane formation and the subsequent pseudorotation step initially with aminophosphonium chloride, (M,S)-1·Cl, after organizing five possible TBP stereoisomers of the pentacoordinate phosphorane and their topological relationship. These stereoisomers were classified into two groups, depending on whether the chlorine atom adopts an apical or equatorial position (Fig. 2). In general, when two bidentate ligands share a central atom to form a respective five-membered ring, the stereoisomers with a monodentate ligand at the apical position (IV and V) are unstable because of the ring strain in the bidentate ligand that occupies the two equatorial positions. These stereoisomers are considered as TSs in the isomerization between stereoisomers with the monodentate ligand at the equatorial position, which are distinguished by the relative disposition of the two identical bidentate ligands (I, II, and III).4 Accordingly, we only considered three stereoisomers I, II, and III and calculated their Gibbs free energies, which revealed that III is the least stable isomer probably because both its isopropyl groups are oriented toward the equatorial chlorine atom. This result allowed us to focus on I and II as possible intermediates of the P-spirochirality interconversion.
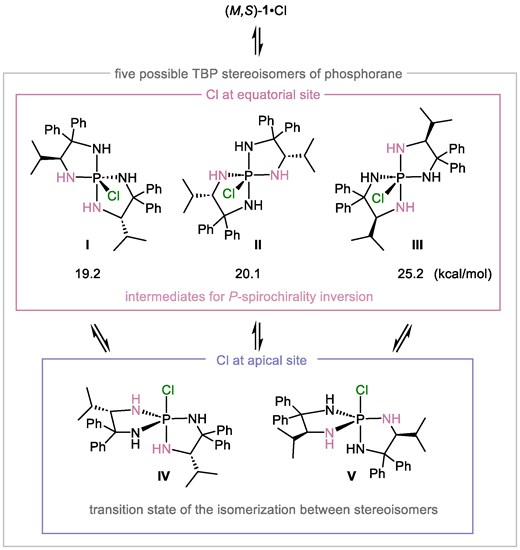
Five possible TBP stereoisomers of pentacoordinate phosphorane generated from (M,S)-1·Cl.
As illustrated in Fig. 3a, the energy barriers for the TSs to form the pentacoordinate stereoisomers I-B and II-B were 19.6 (I-A) and 25.1 kcal/mol (II-A), respectively, and I-B and II-B were in equilibrium via stereoisomer IV shown in Fig. 2. The SP TS energies of the P-spirochirality inversion were 27.4 kcal/mol (I-C) and 28.4 kcal/mol (II-C), and the TSs of subsequent chloride elimination (I-E and II-E) were lower in energy than those of the spirochirality inversion. Therefore, chirality inversion via TS I-C is the rate-determining step, and the energy barrier (27.4 kcal/mol) well accounts for the configurational stability of (M,S)-1·Cl during its preparation and storage.11 We also calculated the energy profile for the inversion of N-methylated (P,S)-2 to (M,S)-2 (Table 1, X = Cl, and Supplementary Fig. S4). This analysis revealed that P-spirochirality inversion occurs from stereoisomer II preferentially to circumvent steric repulsion between the methyl groups on nitrogen, as evident from comparing the structures of TSs I-C and II-C (Fig. 3c). At the same time, however, all the intermediates and TSs involved in the stereochemical flipping processes were located much higher in energy than 1·Cl. Especially in SP TSs, 2-II-C is more distorted (diagonal P–N–P angles: 154.1° and 163.9°) than 1-I-C (157.8° and 157.5°) due to the additional steric repulsion between the methyl and isopropyl groups (Fig. 3b and c). Consequently, the high energy barrier for TS 2-II-C (40.4 kcal/mol) hampered pseudorotation, rendering 2·Cl configurationally very stable.11,12
![a) P-spirochirality inversion process from (M,S)-1·Cl to (P,S)-1·Cl and the value of Gibbs free energy with (M,S)-1·Cl set to 0.0 (kcal/mol). [B3LYP/6-31G(d)] b) Structures of TBP intermediate 1-I-B and SP TS 1-I-C. c) Structures of 2-I-C and 2-II-C. d) Structure of 1-II-B·CH2NO2.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/chemlett/54/5/10.1093_chemle_upaf085/1/m_upaf085f3.jpeg?Expires=1750789219&Signature=P45Yu5KSLHkKcGUhThsdtizFvI1wY6F83ZHTQZkND1yjoJEezwivJ5Dt3kvUWTFwh0iQFbc61JnuoDoEGHGeF7x53z1u-uExBLSL~RZVem3VCqadpm2wwdeUlivggxD5VPAdLr~pCReIuKliPKl9a-BJ9OJDC3yLKCJ2JyrsEZSS2lk79yEB1IX5xrkqgn8mAXEIt~m8iOLP1Yjw62Ep0uhNHRGkeCh3pixISngU~Hv~r8WF1YUyjpr8zdv60tOI24xhQr4YfEuZnQ0hQbs9Uwtkdw26TZB7gyuO55Bxqt8FESbFlZ4ooTfbMiUov6JLQQhOVFoLDQzjHQrluqlURw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
a) P-spirochirality inversion process from (M,S)-1·Cl to (P,S)-1·Cl and the value of Gibbs free energy with (M,S)-1·Cl set to 0.0 (kcal/mol). [B3LYP/6-31G(d)] b) Structures of TBP intermediate 1-I-B and SP TS 1-I-C. c) Structures of 2-I-C and 2-II-C. d) Structure of 1-II-B·CH2NO2.
Gibbs free energies of stereoisomers and TSs involved in the process of P-spirochirality inversion [kcal/mol, B3LYP/6-31G(d) level].
| X | Phosphonium | isomer | (M,S) | A (TSadd) | B | C (TSinv)a | D | E (TSadd) | (P,S) |
|---|---|---|---|---|---|---|---|---|---|
| Cl | 1 | I | 0.0 | 19.6 | 19.2 | 27.4 | 20.7 | 22.0 | 1.2 |
| II | 25.1 | 20.1 | 28.4 | 22.8 | 26.0 | ||||
| 2 | I | 5.6 | 32.4 | 31.5 | 41.6 | 36.4 | 37.2 | 0.0 | |
| II | 31.6 | 30.2 | 40.4 | 28.8 | 37.5 | ||||
| CH2NO2 | 1 | I | 0.0 | 23.0 | 13.4 | 22.7 | 10.7 | 17.9 | 1.5 |
| II | 20.7 | 13.0 | 20.5 | 13.7 | 22.5 | ||||
| 2 | I | 10.8 | 32.2 | 26.8 | 42.7 | 26.4 | 28.0 | 0.0 | |
| II | 26.7 | 24.2 | 36.2 | 22.0 | 29.4 | ||||
| PhO | 1 | I | 0.0 | 16.4 | 1.9 | 8.8 | 0.4 | 19.1 | 1.3 |
| II | 18.4 | 3.4 | 10.3 | 2.1 | 15.5 | ||||
| 2 | I | 7.3 | 21.9 | 16.1 | 31.8 | 19.7 | 30.9 | 0.0 | |
| II | 31.7 | 21.4 | 28.5 | 10.9 | 25.2 |
| X | Phosphonium | isomer | (M,S) | A (TSadd) | B | C (TSinv)a | D | E (TSadd) | (P,S) |
|---|---|---|---|---|---|---|---|---|---|
| Cl | 1 | I | 0.0 | 19.6 | 19.2 | 27.4 | 20.7 | 22.0 | 1.2 |
| II | 25.1 | 20.1 | 28.4 | 22.8 | 26.0 | ||||
| 2 | I | 5.6 | 32.4 | 31.5 | 41.6 | 36.4 | 37.2 | 0.0 | |
| II | 31.6 | 30.2 | 40.4 | 28.8 | 37.5 | ||||
| CH2NO2 | 1 | I | 0.0 | 23.0 | 13.4 | 22.7 | 10.7 | 17.9 | 1.5 |
| II | 20.7 | 13.0 | 20.5 | 13.7 | 22.5 | ||||
| 2 | I | 10.8 | 32.2 | 26.8 | 42.7 | 26.4 | 28.0 | 0.0 | |
| II | 26.7 | 24.2 | 36.2 | 22.0 | 29.4 | ||||
| PhO | 1 | I | 0.0 | 16.4 | 1.9 | 8.8 | 0.4 | 19.1 | 1.3 |
| II | 18.4 | 3.4 | 10.3 | 2.1 | 15.5 | ||||
| 2 | I | 7.3 | 21.9 | 16.1 | 31.8 | 19.7 | 30.9 | 0.0 | |
| II | 31.7 | 21.4 | 28.5 | 10.9 | 25.2 |
aThe boldfaced values attribute to the lower-energy transition state among the possible two.
Gibbs free energies of stereoisomers and TSs involved in the process of P-spirochirality inversion [kcal/mol, B3LYP/6-31G(d) level].
| X | Phosphonium | isomer | (M,S) | A (TSadd) | B | C (TSinv)a | D | E (TSadd) | (P,S) |
|---|---|---|---|---|---|---|---|---|---|
| Cl | 1 | I | 0.0 | 19.6 | 19.2 | 27.4 | 20.7 | 22.0 | 1.2 |
| II | 25.1 | 20.1 | 28.4 | 22.8 | 26.0 | ||||
| 2 | I | 5.6 | 32.4 | 31.5 | 41.6 | 36.4 | 37.2 | 0.0 | |
| II | 31.6 | 30.2 | 40.4 | 28.8 | 37.5 | ||||
| CH2NO2 | 1 | I | 0.0 | 23.0 | 13.4 | 22.7 | 10.7 | 17.9 | 1.5 |
| II | 20.7 | 13.0 | 20.5 | 13.7 | 22.5 | ||||
| 2 | I | 10.8 | 32.2 | 26.8 | 42.7 | 26.4 | 28.0 | 0.0 | |
| II | 26.7 | 24.2 | 36.2 | 22.0 | 29.4 | ||||
| PhO | 1 | I | 0.0 | 16.4 | 1.9 | 8.8 | 0.4 | 19.1 | 1.3 |
| II | 18.4 | 3.4 | 10.3 | 2.1 | 15.5 | ||||
| 2 | I | 7.3 | 21.9 | 16.1 | 31.8 | 19.7 | 30.9 | 0.0 | |
| II | 31.7 | 21.4 | 28.5 | 10.9 | 25.2 |
| X | Phosphonium | isomer | (M,S) | A (TSadd) | B | C (TSinv)a | D | E (TSadd) | (P,S) |
|---|---|---|---|---|---|---|---|---|---|
| Cl | 1 | I | 0.0 | 19.6 | 19.2 | 27.4 | 20.7 | 22.0 | 1.2 |
| II | 25.1 | 20.1 | 28.4 | 22.8 | 26.0 | ||||
| 2 | I | 5.6 | 32.4 | 31.5 | 41.6 | 36.4 | 37.2 | 0.0 | |
| II | 31.6 | 30.2 | 40.4 | 28.8 | 37.5 | ||||
| CH2NO2 | 1 | I | 0.0 | 23.0 | 13.4 | 22.7 | 10.7 | 17.9 | 1.5 |
| II | 20.7 | 13.0 | 20.5 | 13.7 | 22.5 | ||||
| 2 | I | 10.8 | 32.2 | 26.8 | 42.7 | 26.4 | 28.0 | 0.0 | |
| II | 26.7 | 24.2 | 36.2 | 22.0 | 29.4 | ||||
| PhO | 1 | I | 0.0 | 16.4 | 1.9 | 8.8 | 0.4 | 19.1 | 1.3 |
| II | 18.4 | 3.4 | 10.3 | 2.1 | 15.5 | ||||
| 2 | I | 7.3 | 21.9 | 16.1 | 31.8 | 19.7 | 30.9 | 0.0 | |
| II | 31.7 | 21.4 | 28.5 | 10.9 | 25.2 |
aThe boldfaced values attribute to the lower-energy transition state among the possible two.
With insights into the stereorandomization of aminophosphonium chlorides 1·Cl and 2·Cl in hand, we focused on assessing the influence of anions (X) on epimerization. Similar calculations were performed to estimate the Gibbs free energies of stereoisomers and TSs involved in the P-spirochirality inversion processes. In the case of aminophosphonium nitronate (Table1, X = CH2NO2), generated from (M,S)-1-derived iminophosphorane and nitromethane, the most stable pentacoordinate phosphorane was formed by the attack of the nitronate oxygen to the phosphorus center via TS A (Fig. 3d). The energy barrier for the TS C of the subsequent pseudorotation was substantially lower (20.5 kcal/mol) than that of its chloride counterpart, and it was similar to the energy barrier for TS A of nitronate addition. This confirmed our assumption that P-spirochirality inversion occurred via the generation of aminophosphonium nitronate, which would cause the decrease in enantioselectivity when the Henry reaction was carried out at higher temperatures. We then moved on further analysis with aminophosphonium salts possessing other oxyanions, specifically, aminophosphonium phenoxide 1·OPh.13 As included in Table 1 (X = PhO), the energy barrier for TS C was even lower (8.8 kcal/mol), and the resulting pentacoordinate phosphorane (B and D) was very stable with its energy comparable to that of 1·Cl and 1·CH2NO2. Therefore, this step is no longer rate-determining and the nucleophilic attack of phenoxide via TS I-A becomes the rate-determining step. Noteworthy was that the energy barriers for the additions of the counter ions to (M,S)-1 were all similar for Cl, CH2NO2, and OPh (19.6, 20.7, and 16.4 kcal/mol, respectively), indicating that the nucleophilicity of the anion is not a key factor in facilitating the spirochirality inversion. Rather, the difference in their ability to exert stabilization effect on TS C is of critical importance. In contrast, the energies for the chirality inversion of 2 via TS C significantly increased to 36.2 kcal/mol for nitronate and 28.5 kcal/mol for phenoxide, which agrees well with the experimentally observed stereochemical robustness of 2.
To clarify the origin of the stability differences between aminophosphoniums 1 and 2 against P-spirochirality inversion depending on the presence of methyl groups on nitrogen and the nature of pairing anions, we performed distortion/interaction-activation strain (DIAS) analysis14 on the SP TS C for each combination of aminophosphonium and anion primarily for distinguishing steric and electronic effects (Table2). The difference in distortion energy between the parent ion pair and C (ΔEdis), an indicator of steric repulsion, was uniformly larger for 2 than for 1, irrespective of the anion (X). In addition, the difference in ΔEdis between 1 and 2 (ΔEdis2−ΔEdis1) increased with the size of the anion. On the other hand, while the absolute value of ΔEint, an indicator of electronic stabilization, was smaller for 2, the difference in ΔEint between 1 and 2 (ΔEint2−ΔEint1) was not as pronounced depending on the anion. Therefore, the difference in ΔEdis between 1 and 2 can be ascribed to steric effects due to the presence or absence of the methyl group rather than to the electronic properties of the anions. The marginal electronic effect was consistent with the outcome that the P–X bond lengths in TS C were almost identical for 1 and 2, regardless of the anion. However, the absolute value of ΔEint increased when the anion switched from chloride to nitronate or phenoxide. This change correlates with the electron-donating ability of these three anions, for which the P–X bond length is inversely proportional. In the TBP structure, the hyperconjugation of the lone pair of nitrogen atoms at the equatorial position with the equatorial P–O σ*-orbital contributes to structural stabilization.15 Hence, similar hyperconjugation might be responsible for stabilizing the SP TS.
DIAS analysis for SP TS C of P-spirochirality inversion.
| X | ΔEdis (kcal/mol) | ΔEint (kcal/mol) | P–X (Å) | |
|---|---|---|---|---|
| Cl | 1 | 54.6 | −28.9 | 2.179 |
| 2 | 59.4 | −22.7 | 2.216 | |
| CH2NO2 | 1 | 67.1 | −49.1 | 1.731 |
| 2 | 74.8 | −41.6 | 1.736 | |
| PhO | 1 | 62.3 | −56.4 | 1.689 |
| 2 | 81.7 | −53.2 | 1.701 |
| X | ΔEdis (kcal/mol) | ΔEint (kcal/mol) | P–X (Å) | |
|---|---|---|---|---|
| Cl | 1 | 54.6 | −28.9 | 2.179 |
| 2 | 59.4 | −22.7 | 2.216 | |
| CH2NO2 | 1 | 67.1 | −49.1 | 1.731 |
| 2 | 74.8 | −41.6 | 1.736 | |
| PhO | 1 | 62.3 | −56.4 | 1.689 |
| 2 | 81.7 | −53.2 | 1.701 |
DIAS analysis for SP TS C of P-spirochirality inversion.
| X | ΔEdis (kcal/mol) | ΔEint (kcal/mol) | P–X (Å) | |
|---|---|---|---|---|
| Cl | 1 | 54.6 | −28.9 | 2.179 |
| 2 | 59.4 | −22.7 | 2.216 | |
| CH2NO2 | 1 | 67.1 | −49.1 | 1.731 |
| 2 | 74.8 | −41.6 | 1.736 | |
| PhO | 1 | 62.3 | −56.4 | 1.689 |
| 2 | 81.7 | −53.2 | 1.701 |
| X | ΔEdis (kcal/mol) | ΔEint (kcal/mol) | P–X (Å) | |
|---|---|---|---|---|
| Cl | 1 | 54.6 | −28.9 | 2.179 |
| 2 | 59.4 | −22.7 | 2.216 | |
| CH2NO2 | 1 | 67.1 | −49.1 | 1.731 |
| 2 | 74.8 | −41.6 | 1.736 | |
| PhO | 1 | 62.3 | −56.4 | 1.689 |
| 2 | 81.7 | −53.2 | 1.701 |
To gain insights into whether hyperconjugation from the lone pair of oxygen atoms of the anion stabilizes the SP TS, we conducted natural bonding orbital analysis (Supplementary Figs. S5 and S6, and Supplementary Table S1). In all cases, hyperconjugations between two lone pairs on either chlorine or oxygen atom and P–N σ*-orbital existed, and the E2 values for 1·OPh were the largest. However, the additional stronger interaction was identified for 1·CH2NO2 and 1·OPh; that was the interaction between a bonding p-orbital (σ-orbital) on oxygen and P–N σ*-orbitals (Supplementary Fig. S6b). The strength of this interaction was larger in 1·OPh than that in 1·CH2NO2, reflecting the difference in electron-donating ability. In contrast, no such σ−σ* interaction was observed in 1·Cl. These results clearly indicate that the interaction of p-orbitals on anions with P–N σ*-orbitals is a critical factor for TS stabilization, where the electron-donating ability of a bonding p-orbital on oxygen plays the most significant role.
In summary, we elucidated the key elements determining the feasibility of chirality inversion of the tetracoordinate phosphorus center of P-spiro chiral tetraaminophosphoniums via BPR through the pentacoordinate SP TS after anion coordination. As the TS is stabilized by interactions between p-orbitals on the anion and P–N σ*-orbitals, its attenuation, along with the amplification of the distortion energy by the steric repulsion due to the nitrogen substituents, is critical for preventing P-spirochirality inversion. We anticipate that this study will provide a basis for the development of novel chiral catalysts by exploiting heteroatom-centered chirality.
Acknowledgments
Y.K. expresses gratitude to the Asahi Glass Foundation for generous financial support.
Supplementary data
Supplementary material is available at Chemistry Letters online.
Funding
This work was financially supported by JSPS KAKENHI grant number JP23K17914 (grant-in-Aid for Challenging Research Exploratory) and MEXT KAKENHI grant number JP22K21346 (International Leading Research) (T.O.).
References
Author notes
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}