





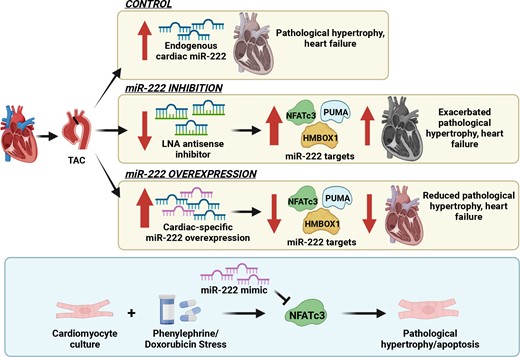
Abstract
Physiological cardiac hypertrophy occurs in response to exercise and can protect against pathological stress. In contrast, pathological hypertrophy occurs in disease and often precedes heart failure. The cardiac pathways activated in physiological and pathological hypertrophy are largely distinct. Our prior work demonstrated that miR-222 increases in exercised hearts and is required for exercise-induced cardiac hypertrophy and cardiomyogenesis. Here, we sought to define the role of miR-222 in pathological hypertrophy.
We found that miR-222 also increased in pathological hypertrophy induced by pressure overload. To assess its functional significance in this setting, we generated a miR-222 gain-of-function model through cardiac-specific constitutive transgenic miR-222 expression (TgC-miR-222) and used locked nucleic acid anti-miR specific for miR-222 to inhibit its effects. Both gain- and loss-of-function models manifested normal cardiac structure and function at baseline. However, after transverse aortic constriction (TAC), miR-222 inhibition accelerated the development of pathological hypertrophy, cardiac dysfunction, and heart failure. Conversely, miR-222-overexpressing mice had less pathological hypertrophy after TAC, as well as better cardiac function and survival. We identified p53-up-regulated modulator of apoptosis, a pro-apoptotic Bcl-2 family member, and the transcription factors, Hmbox1 and nuclear factor of activated T-cells 3, as direct miR-222 targets contributing to its roles in this context.
While miR-222 is necessary for physiological cardiac growth, it inhibits cardiac growth in response to pressure overload and reduces adverse remodelling and cardiac dysfunction. These findings support the model that physiological and pathological hypertrophy are fundamentally different. Further, they suggest that miR-222 may hold promise as a therapeutic target in pathological cardiac hypertrophy and heart failure.
Time for primary review: 53 days See the editorial comment for this article ‘The dual effects of miR-222 in cardiac hypertrophy: bridging pathological and physiological paradigms’, by D. Kesidou et al., https://doi.org/10.1093/cvr/cvae033.
1. Introduction
Cardiac hypertrophy is a common precursor to heart failure1 but also occurs in response to exercise, where it does not generally lead to adverse consequences and is associated with protection against pathological stress.2,3 Through unbiased gene expression profiling of transcriptional components4 and long non-coding RNAs,5 we have found that the few transcripts that significantly changed in both pathological and physiological forms of hypertrophy generally change in opposite directions, suggesting that the two forms of growth are mechanistically distinct. Moreover, cardiac growth with physiological or pathological features can be experimentally induced by mimicking many of the transcriptional changes induced by exercise or pathological stress, respectively. Of particular note, the long non-coding RNA lncExACT1 was up-regulated in the heart in human and experimental heart failure and down-regulated in exercise training, and its inhibition or overexpression was sufficient to toggle the heart between physiological or pathological hypertrophy, respectively.5
Using unbiased transcriptional profiling, we previously identified miR-222 as one of 16 miRNAs concordantly regulated in both swim training and voluntary wheel running.6 Prior reports demonstrated increased circulating miR-222 in healthy athletes after both acute and chronic exercise,7 and we extended this observation to heart failure patients after acute exercise.6 We further found that the inhibition of miR-222 in mice with a locked nucleic acid (LNA)8,9–modified anti-miR completely blocked cardiac growth and cardiomyogenesis in response to exercise.6,10 Although transgenic overexpression of miR-222 in the heart was not sufficient to induce cardiac hypertrophy at baseline, miR-222 overexpressing mice subjected to ischaemic injury exhibited less fibrosis and ventricular dilatation with better cardiac function.6
In the present study, we report that miR-222 expression is also increased in a transverse aortic constriction (TAC) model of pressure-overload-induced pathological hypertrophy and heart failure. However, in contrast to its demonstrated role driving physiological cardiac growth, miR-222 actually inhibits pathological growth and the associated adverse remodelling and cardiac dysfunction by targeting a largely distinct set of genes. These data provide new support for an emerging model in which fundamental differences, driven in part by non-coding RNA mechanisms, distinguish pathological and physiological cardiac growth.
2. Methods
2.1 Mouse model of TAC
All rodents were maintained and studied using protocols approved by the Animal Care and Use Committee of Massachusetts General Hospital (protocols 2015N000029 and 2015N000070) and in accordance with NIH guidelines. Mice aged 8–16 weeks were subjected to TAC as described.11 During surgery, the mice were anaesthetized with ketamine (80–100 mg/kg) and xylazine (12 mg/kg), delivered intraperitoneally. If required, supplemental ketamine was administered in increments of 1/3 the original dose. Analgesia consisted of 0.05–0.1 mg/kg buprenorphine delivered subcutaneously 30 min before surgery and once every 8–12 h for 72 h after surgery. Adequacy of anaesthesia was monitored by respiratory rate, toe pinch, jaw laxity, and muscle relaxation. Animals were euthanized by ketamine/xylazine anaesthetic overdose: 240–300 mg/kg ketamine + 15–30 mg/kg xylazine, intraperitoneally. Detailed methods can be found in the Supplementary Material online.
2.2 Generation of Tg-miR-222 mice
A tetracycline-off binary α-myosin heavy chain (αMHC) transgene system was used as previously described.6,12 For the responder mouse line, a 388 bp fragment containing mmu-miR-222 was amplified from mouse genomic DNA, confirmed by sequencing and sub-cloned into a vector generously provided by Dr Jeffrey Robbins. A Not I fragment was micro-injected into FVB oocytes and transferred to pseudo-pregnant mice. After confirmation of stable Mendelian transmission, positive mice were bred to the appropriate driver line12 and cardiac-specific, constitutive miR-222 expression was confirmed in line 8 (TgC-miR-222), which was used for all the experiments presented.
2.3 Injections of LNA-anti-miR
LNA-anti-miR injections were performed as described.13 Detailed methods can be found in the Supplementary Material online.
2.4 Immunofluorescent staining and confocal microscopy
Frozen heart sections were stained with Masson’s trichrome stain (MTS, for fibrosis), wheat germ agglutinin (WGA, for cell size), or TUNEL (for apoptosis). Slides were imaged on a Leica DM 5000 B microscope using standard procedures.6 For quantification of MTS, images were quantified using BZ-X Analyzer software. For quantification of WGA, images were quantified using CellProfiler software. Troponin I- and TUNEL-double positive cells were quantified using ImageJ. All imaging was performed and analysed by investigators blinded to the treatment group. At least 30 random images were obtained from each group.
2.5 Echocardiography
Echocardiography was performed on mice anaesthetized with 2.5% isoflurane delivered using a precision vaporizer. Adequacy of anaesthesia was monitored by respiratory rate, toe pinch, jaw laxity, and muscle relaxation. A Vevo 3100 with MX250s probe (15–30 mHz) (FUJIFILM VisualSonics, Canada) was used as described.14,15 Echocardiography data were analysed by investigators blinded to treatment and genotype. The average of at least three measurements was used for every data point from each mouse. Detailed methods can be found in the Supplementary Material online.
2.6 Microarray
Heart samples from miR-222 mice and wild-type (WT) mice (n = 3–4 per group) 1 week after TAC were collected and microarray was performed by Dana-Farber/Harvard Cancer Center DNA Resource core following standard procedures.
2.7 Quantitative real-time polymerase chain reaction
RNA was isolated from both tissue and cell samples using TRIzol (Invitrogen) following the manufactures’ manual. Quantitative real-time polymerase chain reaction (qRT-PCR) for microRNA was performed on cDNA generated from 100 ng of total RNA using the TaqMan microRNA assay protocol (Invitrogen). qRT-PCR for all other transcripts was performed on cDNA generated from 200 ng of total RNA using the protocol of a high-capacity cDNA reverse transcription kit (Invitrogen). Amplification and detection of specific products were performed on a Bio-Rad CFX384 qPCR System. U6 was used as an internal control for microRNA template normalization and 18S or U6 was used for other template normalization. The sequences of the primers used are listed in the Supplementary material online, Table S1. The relative gene expression was calculated by comparing cycle times (Ct) for each target PCR as described.16
2.8 Luciferase assays
A reporter plasmid was constructed by inserting a fragment of the 3ʹ-UTR of nuclear factor of activated T-cells 3 (NFATc3) mRNA containing the putative miR-222 binding site into the luciferase reporter plasmid psiCHECK-2 (Promega). As a mutated control, a construct containing a mutated fragment of the 3ʹ-UTR of NFATc3 mRNA without the putative miR-222 binding site was made by using the QuikChange site-directed mutagenesis kit (Agilent). The primers used for the constructs and mutation are shown in the Supplementary material online, Table S2. COS7 cells were co-transfected with the reporter plasmid or the mutated construct (100 ng) and Ambion scrambled pre-miR miRNA or miR-222 precursor (1 µM) using lipofectamine 3000 according to the manufacturer’s transfection procedure (Invitrogen). Forty-eight hours after transfection, cells were lysed. With isolated cell lysates, relative luciferase expression was measured on a multi-mode multi-format reader SpectraMax M5 by using a dual luciferase reporter system (Promega).
2.9 Western blotting
Proteins were isolated from cultured neonatal rat ventricular myocytes (NRVMs) and hearts and protein levels were determined by performing a western blot analysis. Briefly, equal amounts of protein determined by using the BCA protein assay kit (Pierce) were subjected to SDS-PAGE. The standard western blot analysis was conducted using NFATc3 (1:500, R&D Systems AF5834), and HMBOX1 (1:500, Abcam ab101140). GAPDH (1:1000, Cell Signaling #2118) or Vinculin (1:1000, Sigma V9264) was used as a loading control.
2.10 Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) unless otherwise indicated. An unpaired, two-tailed Student’s t-test was used when indicated, with P < 0.05 considered statistically significant. When assessing multiple groups, one-way analysis of variance (ANOVA) was utilized with Tukey’s post hoc test. Statistical analysis was performed by using GraphPad Prism software (GraphPad Software Inc.).
3. Results
3.1 Cardiac miR-222 expression is increased in pathological cardiac hypertrophy and heart failure
We first examined cardiac miR-222 expression in pathological cardiac hypertrophy and heart failure induced by TAC. Heart weight (HW) to tibial length (TL) ratios were increased 7 and 14 days after TAC (Figure 1A) without an increase in lung weight (LW) to TL ratios (Figure 1B), indicating the absence of pulmonary congestion or heart failure. Cardiac gene expression measured by qRT-PCR was consistent with pathological hypertrophy, with increased natriuretic peptide A (ANP), natriuretic peptide B (BNP), and β-MHC (Myh7) (Figure 1C). Cardiac miR-222 expression was increased 7 and 14 days after TAC (Figure 1D). Interestingly, the primary miR-222 transcript, pri-miR-222, was increased as early as 3 days after TAC, with a non-significant trend to increase at subsequent time points (Figure 1E), suggesting that an early increase in miR-222 transcription leads to subsequent increased expression. By ∼42 days after TAC, mice had developed substantial cardiac dysfunction as indicated by a substantial decline in ejection fraction (EF) (P < 0.05, see Supplementary material online, Figure S1). At this time point, the mice showed an even greater increase in HW/TL (Figure 1F), reflecting ventricular dilation (see Supplementary material online, Figure S1). LW/TL was also substantially increased (Figure 1G), indicating pulmonary congestion consistent with overt heart failure. In failing hearts, miR-222 expression was also increased approximately two-fold compared with sham controls (Figure 1H). Thus, miR-222 increased both in pathological hypertrophy without heart failure, as early as 7 days after TAC, and in overt heart failure, 6 weeks after TAC.
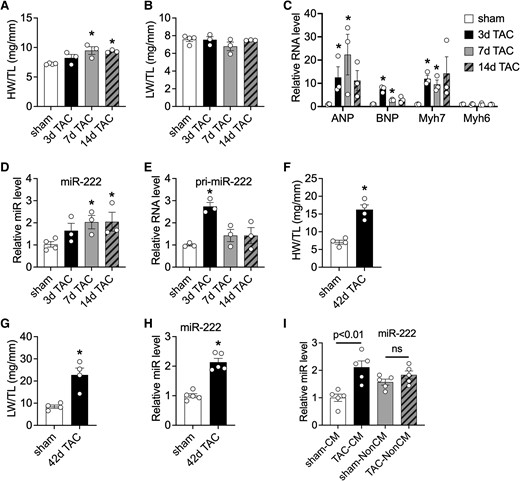
Cardiac miR-222 increases in pathological cardiac hypertrophy and heart failure induced by pressure overload. (A and B) HW/TL (A) and LW/TL (B) ratios of sham or TAC mice at 3, 7, and 14 days after TAC (n = 3–4 in each group). (C–E) A qRT-PCR analysis of hypertrophy markers (C), miR-222 (D), and pri-miR-222 (E) in hearts from sham or TAC mice at different time points. Data are shown as fold induction of gene or miR expression normalized to GAPDH (C), U6 (D), and GAPDH (E), respectively (n = 3–4 hearts in each group). (F and G) HW/TL (F) and LW/TL (G) ratios of mice 42 days after sham and TAC surgery (n = 3–4 hearts in each group). (H) A qRT-PCR analysis of miR-222 in hearts from mice 42 days after sham or TAC. Data are shown as fold induction of gene or miR expression normalized to U6 (n = 3–4 hearts in each group). (I) A qRT-PCR analysis of miR-222 in adult cardiomyocytes and non-cardiomyocytes isolated from mice 2 weeks after sham and TAC surgery. Data shown as fold induction of gene or miRNA expression normalized to U6 (n = 5 in each group). The error bars represent SEM. *P < 0.05 compared with respective controls using Student’s t-test or one-way ANOVA with Tukey’s post hoc test.
To identify the source of increased miR-222, we isolated cardiomyocytes and non-cardiomyocytes 2 weeks after TAC or sham surgery (see Supplementary material online, Figure S2) when the animals exhibited cardiac hypertrophy. The expression of lineage markers confirmed that isolated populations represented predominantly the appropriate cell populations, and this was further confirmed by probing for miR-133a and miR-208b, two miRNAs predominantly expressed in cardiomyocytes.17 As a positive control, we also confirmed that miR-208b was increased in cardiomyocytes after TAC, which was consistent with prior work.18 As previously reported,6 miR-222 expression was higher in non-cardiomyocytes than in cardiomyocytes after sham surgery. However, after TAC, we observed a substantial (approximately two-fold) increase in miR-222 expression in cardiomyocytes without a significant change in non-cardiomyocytes (Figure 1I). The primary miR-222 transcript, pri-miR-222, was also more abundant in non-cardiomyocytes than in cardiomyocytes in sham animals, but unlike the mature transcript, its expression pattern was not significantly altered by TAC (see Supplementary material online, Figure S2). Interestingly, miR-221, a miRNA in the same cluster as miR-222 that shares the same seed sequence, was expressed at higher levels in cardiomyocytes than in non-cardiomyocytes and, like miR-222, its expression was increased specifically in cardiomyocytes after TAC (see Supplementary material online, Figure S2).
3.2 miR-222 inhibition accelerates the development of pathological cardiac hypertrophy and heart failure
To determine the functional role of increased miR-222 in pathological hypertrophy and heart failure, we treated mice with LNA-anti-miR-222, which we had previously shown effectively and specifically inhibits miR-222,6 or a scrambled control LNA-anti-miR. qRT-PCR confirmed effective inhibition of miR-222 by LNA-anti-miR-222 both 1 and 3 weeks after injection (see Supplementary material online, Figure S3A and B). While miR-222 inhibition had no effect on cardiac function or structure in sham-operated mice (Figure 2A–C), LNA-anti-miR-222-treated animals showed dramatically accelerated functional decline after TAC (Figure 2A). This was accompanied by reduced wall thickness (Figure 2B) and increased left ventricular dilation (Figure 2C and Supplementary material online, Table S4). Of note, in mice with TAC, compared with LNA-control, the inhibition of miR-222 did not affect the pressure gradient across TAC (see Supplementary material online, Figure S3C) or diastolic function (see Supplementary material online, Table S4). Similarly, gravimetric measurements in sham-operated animals were not affected by LNA-anti-miR-222 treatment compared with control LNA, demonstrating no difference in HW/TL or LW/TL ratios 1 and 3 weeks after treatment. However, after TAC, although HW/TL increased in all groups, mice treated with LNA-anti-miR-222 increased cardiac mass more than control LNA–treated animals did at 1 but not 3 weeks after TAC (Figure 2D). To determine whether the increased heart mass reflected cardiomyocyte hypertrophy or simply the ventricular dilatation documented above by echo, we measured cardiomyocyte size. Cardiomyocytes from LNA-anti-miR-222-treated mice were substantially larger 1 week after TAC than those from control LNA–treated animals (Figure 2E). LW was also increased 3 weeks after TAC in mice treated with LNA-anti-miR-222 compared with that in control LNA–treated animals (Figure 2F), suggesting increased pulmonary congestion and heart failure, consistent with the accelerated cardiac dysfunction evident on echo (Figure 2A). Programmed cell death (apoptosis) plays an important role in the progression from pathological cardiac hypertrophy to heart failure. TUNEL staining showed that the inhibition of miR-222 led to an approximately two-fold increase in apoptosis in comparison with control anti-miR-treated animals 3 weeks after TAC (Figure 2G), demonstrating that miR-222 inhibits apoptosis in pathological hypertrophy and heart failure. In contrast, the inhibition of miR-222 did not alter fibrosis 3 weeks after TAC (Figure 2H). Taken together, these data demonstrate that the increased miR-222 seen after TAC helps to slow the development of pathological hypertrophy and protect against apoptosis and the progression to heart failure.
![The inhibition of miR-222 accelerates cardiac dysfunction induced by pressure overload. (A–C) Echocardiography analyses and representative images [A: EF, B: left ventricular posterior wall diastole (LVPWd), C: left ventricular internal dimension diastole (LVIDd)] of scrambled control LNA-anti-miR (ctl-anti) or specific LNA-anti-miR-222 (anti-222)-treated mice after sham or TAC surgery, *P < 0.05 vs. ctl-anti sham, #P < 0.05 vs. ctl-anti TAC. These include EF (A) LVPWd (B) LVIDd (C). Representative images are from 3 weeks after TAC. (D) HW/TL ratios of scrambled control (ctl-anti) and LNA-anti-miR-222 (anti-222) mice 1 and 3 weeks after sham or TAC (*P < 0.05 vs. ctl-anti-sham, #P < 0.05 vs. ctl-anti-TAC). (E) Quantification of the cardiomyocyte area from heart sections stained with WGA (n = 4, ∼500 cells per animal) demonstrates that miR-222 inhibition exaggerates pathological cardiomyocyte hypertrophy 1 week after TAC. (F) LW/TL ratios of scrambled control (ctl-anti) and LNA-anti-miR-222 (anti-222) mice 1 and 3 weeks after sham or TAC (*P < 0.05 vs. ctl-anti-sham, #P < 0.05 vs. ctl-anti-TAC). (G) An increase in the number of TUNEL-troponin I double-positive cells normalized to total 4',6-diamidino-2-phenylindole (DAPI)-labelled cells demonstrates increased apoptosis in miR-222-expressing hearts 3 weeks after TAC (n = 4 hearts in each group). (H) Representative images and quantification of the fibrotic area in Masson trichrome–stained heart sections (n = 3–7 hearts in each group) demonstrating no effect of miR-222 inhibition on fibrosis 1 week after TAC. n = 3 per group in ctl-anti-sham and anti-222 sham; n = 9 per group in ctl-anti-TAC and anti-222 TAC. The error bars represent SEM. *P < 0.05 compared with respective controls using Student’s t-test or one-way ANOVA with Tukey’s post hoc test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/120/3/10.1093_cvr_cvad184/2/m_cvad184f2.jpeg?Expires=1749982017&Signature=U1Q7P998FsSvnSfrLuPK~DBToJU5YNnj-iYA0Wo6VTvYvxh0Qi~BRoatEeyilx02S-UNZ~3y5FDF7mghfNn2t839aNSCEtYtCuhjJkLCjJZfeeGkcVSxIj1dM6ObUo~uuLTEnJNiv04YQhhDlec~qYyibP~N8TyGOk9aIsXKNBAVK8mbXE2MgtLRgY68asPNWVRKXs1v2Bp~NekhJstYW3L8kGmHX4KYekuuPuqqFC-KY4s0fxMtSnzW81VGYWEE1Gj5WtyQHu--nZuNrlWudpJnMferDkkZe6O3eMicN3dv4ER-WA~h2I4y-Y2HCx1l1UDDfMXyD-NfzEsoEbJ2Zw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The inhibition of miR-222 accelerates cardiac dysfunction induced by pressure overload. (A–C) Echocardiography analyses and representative images [A: EF, B: left ventricular posterior wall diastole (LVPWd), C: left ventricular internal dimension diastole (LVIDd)] of scrambled control LNA-anti-miR (ctl-anti) or specific LNA-anti-miR-222 (anti-222)-treated mice after sham or TAC surgery, *P < 0.05 vs. ctl-anti sham, #P < 0.05 vs. ctl-anti TAC. These include EF (A) LVPWd (B) LVIDd (C). Representative images are from 3 weeks after TAC. (D) HW/TL ratios of scrambled control (ctl-anti) and LNA-anti-miR-222 (anti-222) mice 1 and 3 weeks after sham or TAC (*P < 0.05 vs. ctl-anti-sham, #P < 0.05 vs. ctl-anti-TAC). (E) Quantification of the cardiomyocyte area from heart sections stained with WGA (n = 4, ∼500 cells per animal) demonstrates that miR-222 inhibition exaggerates pathological cardiomyocyte hypertrophy 1 week after TAC. (F) LW/TL ratios of scrambled control (ctl-anti) and LNA-anti-miR-222 (anti-222) mice 1 and 3 weeks after sham or TAC (*P < 0.05 vs. ctl-anti-sham, #P < 0.05 vs. ctl-anti-TAC). (G) An increase in the number of TUNEL-troponin I double-positive cells normalized to total 4',6-diamidino-2-phenylindole (DAPI)-labelled cells demonstrates increased apoptosis in miR-222-expressing hearts 3 weeks after TAC (n = 4 hearts in each group). (H) Representative images and quantification of the fibrotic area in Masson trichrome–stained heart sections (n = 3–7 hearts in each group) demonstrating no effect of miR-222 inhibition on fibrosis 1 week after TAC. n = 3 per group in ctl-anti-sham and anti-222 sham; n = 9 per group in ctl-anti-TAC and anti-222 TAC. The error bars represent SEM. *P < 0.05 compared with respective controls using Student’s t-test or one-way ANOVA with Tukey’s post hoc test.
3.3 Cardiac-specific expression of miR-222 ameliorates pathological cardiac hypertrophy and heart failure after TAC
Since miR-222 appears necessary to prevent pathological hypertrophy and heart failure after TAC, we next asked whether increasing miR-222 is sufficient to protect against these conditions. We had previously generated transgenic mice with a conditional cardiac-specific expression of miR-222 using a tetracycline-off binary α-MHC transgene system.6 For the current studies, we used the same system, in the absence of tetracycline treatment, to produce mice with constitutive miR-222 expression throughout their lifespan. We confirmed miR-222 expression ∼10-fold above endogenous levels (TgC-miR-222, see Supplementary material online, Figure S4A). Transgene expression was predominantly increased in the heart, although QPCR also showed a modest increase in the skeletal muscle (see Supplementary material online, Figure S4B). As previously described, even an ∼10-fold transgenic miR-222 overexpression did not change the baseline cardiac structure or function (see Supplementary material online, Table S3), and TgC-miR-222 showed no difference in HW/TL or LW/TL ratios and no discernable difference in heart size compared with littermate controls (see Supplementary material online, Figure S4C–E).
We next subjected 4-month-old TgC-miR-222 mice and littermate controls to TAC. Compared with WT littermate subjected to TAC, the overexpression of miR-222 did not affect the pressure gradient across TAC (see Supplementary material online, Figure S4F). Echocardiographic analyses demonstrated that as early as 2 weeks after TAC, TgC-miR-222 mice had better cardiac function (P < 0.01, Figure 3A), greater wall thickness (P < 0.01, Figure 3B), less chamber dilation (P < 0.01 for both, Figure 3C and D), and slightly but not significantly improved diastolic function than control WT littermates (see Supplementary material online, Table S5). Reflecting the reduced dilation, heart weight was also reduced in TgC-miR-222 mice (HW/TL, P < 0.05, Figure 3E). Interestingly, miR-222 expression completely blocked the cardiomyocyte hypertrophy seen 2 weeks after TAC in WT control animals (Figure 3F). Moreover, the increased LW seen in WT mice after TAC indicative of heart failure was dramatically reduced in TgC-miR-222 mice (Figure 3G). TgC-miR-222 mice also had substantially better survival rates (78 vs. 22% P < 0.05) 120 days after TAC compared with WT age- and gender-matched controls (Figure 3H).
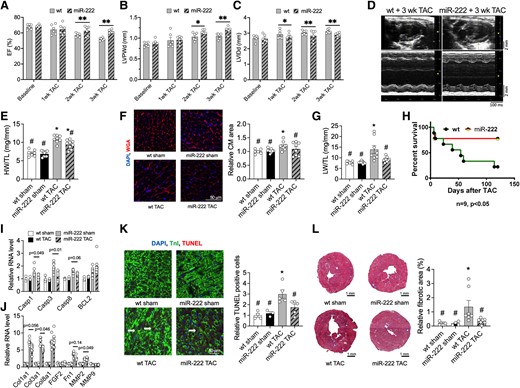
Cardiac-specific expression of miR-222 prevents cardiac dysfunction induced by pressure overload and improves survival. (A–D) Echocardiography analyses and representative images of 4-month-old WT and miR-222 transgenic mice at baseline, 1, 2, and 3 weeks after TAC (n = 6 hearts in each group, *P < 0.05 and **P < 0.01 vs. corresponding WT control group by Student’s t-test). These include EF (A) LVPWd (B) LVIDd (C) representative images (D). These data demonstrate that miR-222 protects against pathological cardiac hypertrophy. (E) HW/TL ratios of 4-month-old WT and miR-222 transgenic mice 2 weeks after sham or TAC surgery (n = 7–9 hearts in each group, *P < 0.05 vs. WT sham, #P < 0.05 vs. WT TAC using one-way ANOVA with Tukey’s post hoc test). (F) Quantification of the cardiomyocyte area from heart sections stained with WGA (n = 4, ∼500 cells per animal, *P < 0.05 vs. WT sham, #P < 0.05 vs. WT TAC) demonstrate that miR-222 blocks TAC-induced cardiomyocyte hypertrophy. (G) LW/TL ratios of 4-month-old WT and miR-222 transgenic mice 2 weeks after sham or TAC surgery (n = 7–9 hearts in each group, *P < 0.05 vs. WT sham, #P < 0.05 vs. WT TAC). (H) Survival analysis shows that 4-month-old miR-222 transgenic mice had improved survival rates compared with WT controls 4 months after TAC (n = 9 in each group, *P < 0.05 vs. WT). (I and J) A qRT-PCR analysis of apoptosis markers (I) and fibrosis markers (J) in hearts from 4-month-old WT and miR-222 transgenic mice 2 weeks after sham or TAC surgery (n = 4–5 hearts in each group). These data demonstrate that miR-222 inhibits fibrosis and pro-apoptosis gene expression. (K) A decrease in the number of TUNEL-troponin I double-positive cells normalized to total DAPI-labelled cells demonstrates reduced apoptosis in cardiomyocytes in miR-222-expressing hearts 2 weeks after TAC (n = 4 hearts in each group). (L) MTS for the fibrosis area from heart sections and quantification (n = 4 hearts in each group) shows that miR-222 expression significantly inhibits fibrosis formation 2 weeks after TAC. *P < 0.05, vs. WT sham; #P < 0.05 vs. WT TAC using one-way ANOVA with Tukey’s post hoc test.
To explore the potential underlying mechanisms, we performed qRT-PCR for the markers of fibrosis and apoptosis, such as Cal3a1, MMP2, Casp1, and Casp3. The TAC-induced increase in these markers was largely, although not completely, blunted in TgC-miR-222 mice (Figure 3I and J). Correspondingly, TUNEL staining showed that apoptosis was substantially reduced in TgC-miR-222 compared with WT mice 2 weeks after TAC (Figure 3K). Furthermore, miR-222 expression completely blocked TAC-induced cardiac fibrosis 2 weeks after TAC (Figure 3L). Taken together, these data demonstrate that cardiac miR-222 overexpression is sufficient to inhibit pathological hypertrophy and protect against TAC-induced apoptosis, fibrosis, and heart failure.
3.4 miR-222 targets HMBOX1, NFATc3, and p53-up-regulated modulator of apoptosis
To investigate the downstream mechanisms responsible for the effects of miR-222 after TAC, we performed microarray-based transcript profiling on 4-month-old TgC-miR-222 transgenics in comparison with WT controls 1 week after TAC. At this point after TAC, cardiac function begins to separate between the transgenic and the WT animals, suggesting that an analysis of these tissues might yield greater insight into primary mechanisms. Principal component analysis showed that the transcriptional profiles clearly distinguish among these states (Sham, TAC, and TAC with miR-222 overexpression; data not provided). We compared the expression profiling results with the profiling we had previously performed in miR-222-expressing rat cardiomyocytes (Gene Expression Omnibus accession number: GSE59641)6 as well as with miR-222 targets predicted by two bioinformatic programmes (Targetscan, Pictar) and known miR-222 targets (data not provided). These analyses identified three relevant potential miR-222 targets: the transcription factors Homeobox containing 1 (Hmbox1), which we previously identified as a direct target of miR-222 that inhibits cardiomyocyte growth,6 NFATc3, not previously recognized as a target of miR-222, and p53-up-regulated modulator of apoptosis (PUMA), a pro-apoptotic19,20 BH3-only member of the Bcl-2 protein family and known target of miR-22221,22 that was not implicated in the prior exercise studies. Of note, genetic deletion of PUMA protects the heart against apoptosis and consequent dysfunction in response to TAC.23 Prior work by the Molkentin laboratory has demonstrated that genetic disruption of NFATc3 reduces pathological cardiac hypertrophy after TAC.24,25 Thus, coordinated miR-222 targeting of Hmbox1, NFATc3, and PUMA would be expected to mitigate pathological cardiac hypertrophy and reduce cardiomyocyte apoptosis and heart failure.
Since Hmbox1 and PUMA are established targets of miR-222,6,21 we next focused on determining whether NFATc3 is also a direct target of miR-222. We cloned the WT NFATc3 3ʹ-UTR and a 3ʹ-UTR in which the putative miR-222 binding sites had been mutated, downstream of luciferase in the reporter plasmid psiCHECK-2 (Promega). These constructs were then co-transfected into COS7 cells along with miR-222 precursor miRNA or scrambled pre-miR. miR-222 expression had no effect on luciferase activity of a control reporter without a miR-222 binding site (data not provided). miR-222 expression induced a reduction in luciferase activity for the WT 3ʹUTR construct but had no effect when the miR-222 binding site was mutated (Figure 4A). These data indicate that the NFATc3 mRNA is a direct target of miR-222. To examine the functional role of NFATc3 in miR-222’s effects in cardiomyocytes, primary cardiomyocytes were treated with the miR-222 mimic without or with NFATc3 overexpression by adenovirus and exposed to either doxorubicin (to induce apoptosis) or phenylephrine (to induce pathological hypertrophy). As indicated by increased lactate dehydrogenase release, doxorubicin increased cardiomyocyte cell death and decreased cell viablity, indicated by reduced ATP content. These pathological effects were reduced by miR-222 overexpression, and the effect of miR-222 was attenuated by NFATc3 overexpression (Figure 4B and C). Similarly, phenylephrine induced an increase in cardiomyocyte size with a gene expression pattern consistent with pathological hypertrophy (increased ANP, BNP, β/αMHC ratio, and reduced PGC1α) (Figure 4D and E). These changes were attenuated by miR-222 overexpression but not when NFATc3 was also overexpressed (Figure 4D and E). These data indicate that NFATc3 works as a direct downstream target of miR-222 regulating cardiomyocyte survival and hypertrophy.
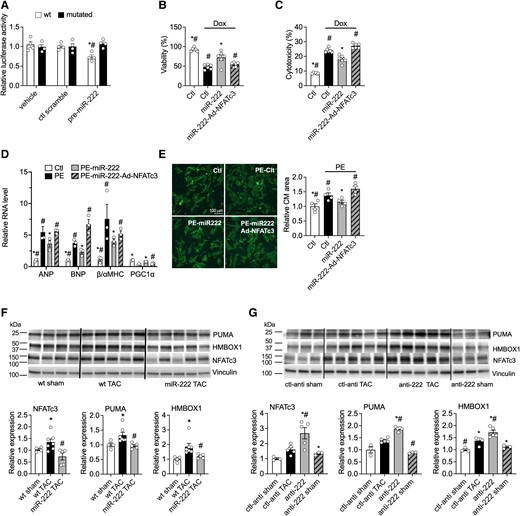
miR-222 directly targets HMBOX1, NFATc3, and PUMA in vitro and in vivo. (A) Luciferase assays in COS7 cells co-transfected with a control precursor (ctl scramble) or miR-222 precursor (pre-222) and reporter plasmids containing a fragment of the mouse NFATc3 3′UTR sequence including the WT or mutated miR-222 binding site. These data demonstrate that NFATc3 is a direct target of miR-222. (B and C) Cell viability and cytotoxicity in neonatal rat ventricular cardiomyocytes (NRVMs) treated with scrambled control (Ctl), miR-222 mimic (miR-222), or NFATc3 adenovirus (Ad-NFATc3) in the absence or presence of doxorubicin (Dox). (n = 4 in each group, *P < 0.05 vs. Ctl-Dox, #P < 0.05 vs. miR-222-Dox using one-way ANOVA with Tukey’s post hoc test). (D) mRNA expression of ANP, BNP, β/αMHC, and PGC1α in NRVMs treated with Ctl, miR-222, and Ad-NFATc3 in the absence or presence of phenylephrine (PE). (E) Representative images and quantification of NRVMs treated with Ctl, miR-222, and Ad-NFATc3 in the absence or presence of PE. (n = 4 in each group, *P < 0.05 vs. Ctl-PE, #P < 0.05 vs. miR-222-PE using one-way ANOVA with Tukey’s post hoc test). (F) Protein levels of the putative miR-222 targets in heart samples taken 1 week after sham or TAC surgery from WT or miR-222 transgenic mice. (G) Protein levels of the putative miR-222 targets in heart samples taken 1 week after sham or TAC surgery from mice treated with specific LNA-anti-miR-222 inhibitor (anti-222) or a scrambled control anti-miR (ctl-anti). Cumulative data are quantified below immunoblots and represented as fold change in protein expression, normalized to vinculin. These data demonstrate that miR-222 reduces the protein levels of all three targets in vivo. *P < 0.05 compared with WT sham or ctl-anti-sham and #P < 0.05 compared with WT TAC or ctl-anti TAC using Student’s t-test or one-way ANOVA.
To determine whether miR-222 regulates these targets in vivo, we performed immunoblotting on cardiac samples from miR-222 gain- and loss-of-function murine models after TAC. Transgenic miR-222 expression inhibited the TAC-induced increase in NFATc3, PUMA, and Hmbox1 protein level 1 week after TAC (Figure 4F). Conversely, LNA inhibition of miR-222 exaggerated the TAC-induced increase in NFATc3, PUMA, and HMBOX1 1 week after TAC (Figure 4G). Interestingly, the expression of p27, a known direct target of miR-222 in a variety of cells and tissues,6,16 which we had previously shown to be modulated by miR-222 after ischaemic injury,6 was not altered by miR-222 expression or inhibition in the context of TAC (see Supplementary material online, Figure S5). These data suggest that Hmbox1, PUMA, and NFATc3 are direct targets of miR-222 and likely contribute to miR-222’s effects in pathological cardiac hypertrophy and heart failure induced by pressure overload.
4. Discussion
Pathological cardiac hypertrophy commonly leads to heart failure,1 a major cause of morbidity and mortality throughout the world. In contrast, physiological cardiac growth, as seen in response to exercise, appears to protect the heart against injury and heart failure.2,3 Although transcriptional pathways altered in pathological and physiological heart growth are mostly distinct,4,5,26,27 miR-222 is unusual in that it is up-regulated in both physiological and pathological growth states. We have shown that miR-222 is necessary for physiological hypertrophy6 and cardiomyogenesis10 and sufficient to confer resistance to cardiac injury,16 but there is conflicting evidence concerning the role of miR-222 in the heart’s structural and functional response to ongoing pathological stress.17,28,29 This question has important implications for the mechanistic distinction between physiological and pathological growth states and for potential therapeutic modulation of these states.
Here, we used the TAC model of pressure overload to induce pathological cardiac hypertrophy and heart failure. We found that in both hypertrophy and heart failure, cardiac miR-222 expression was increased to levels comparable to those previously reported in exercise. Of note, the primary miR-222 transcript, pri-miR-222, increased early after TAC, followed by an increase in the mature miRNA, suggesting that early transcriptional up-regulation is a major contributor to increased expression of the mature miR-222. The expression of miR-222 was higher in non-cardiomyocytes than in cardiomyocytes at baseline, as noted previously,6 but the TAC-induced increase in cardiac miR-222 was seen only in cardiomyocytes.
While the concordant increase in cardiac miR-222 seen after both exercise and TAC would seem an exception to the theme that these interventions activate distinct pathways, functional studies demonstrated that miR-222’s role is dramatically different in these contexts. Whereas miR-222 is required for physiological cardiac growth and cardiomyogenesis in response to exercise,6,10 the gain- and loss-of-function studies reported here demonstrate that miR-222 actually inhibits pathological cardiac growth and progression to cardiac failure. miR-222 was necessary and sufficient to slow the pathological growth of cardiomyocytes, as well as reduce apoptosis, in response to pressure overload. These findings are reminiscent of those seen with the serine-threonine kinase, Akt1,27,30–32 which is similarly required for exercise-induced cardiac growth but inhibits pathological growth after TAC. Thus, although miR-222 expression increases in both physiological and pathological cardiac hypertrophy, the functional consequences of this expression for heart growth differ. Overall, these results are consistent with the model that physiological and pathological cardiac growth involve distinct underlying mechanisms.
Notably, our data suggest that different mechanisms underlie miR-222’s effects under pathological and physiological conditions. We previously identified p27, HIPK1, and HMBOX1 as cardiac miR-222 targets in exercise-induced hypertrophy, but the profiling of putative miR-222 target transcripts after TAC yielded a different set of targets, reinforcing the notion that although miR-222 is increased in both physiological and pathological hypertrophy, it functions in distinct ways. We narrowed these miR-222 targets down to three direct targets of miR-222 that appeared particularly relevant to its effects in the context of pressure-overload-induced pathological hypertrophy and heart failure. These include HMBOX1, which we previously implicated as an inhibitor of physiological cardiomyocyte growth,6 PUMA, which regulates apoptosis,23 and a novel miR-222 target, NFATc3. We confirmed that all three targets were up-regulated in mouse hearts in response to TAC, and miR-222 overexpression blocked this increase, while its inhibition exaggerated it. In contrast, p27, a known miR-222 target6,16 which we had previously shown to be modulated by miR-222 after ischaemic injury and to be sufficient to mimic miR-222’s cardiomyogenic effects,6 was not altered by miR-222 expression or inhibition in the context of TAC. NFATc3 has been shown to drive pathological hypertrophy and heart failure25,33 but had not previously been identified as a direct target of miR-222. Our data demonstrated that NFATc3 is a direct target of miR-222 and mediates its effects on both hypertrophy and cell survival in cardiomyocytes. Thus, these studies extend our understanding of the mechanisms regulating pathological hypertrophy in general and NFATc3 in particular. While there is overlap among the miR-222 targets relevant to pressure overload and exercise6 (e.g. HMBOX1), there are also important differences. Unravelling the mechanisms by which different subsets of miR-222 targets are modulated in these different settings is an important topic for future investigation.
It is unclear why a substantial overexpression of miR-222 has no discernable effect until the introduction of an additional stimulus (TAC in the current study and exercise or ischaemic injury in our prior work6). It is possible that these stimuli induce additional signals required to work in concert with miR-222. Alternatively, miR-222 (or its relevant targets) may be held in an inactive state (e.g. through sequestration by binding partners such as lncRNAs), which is released by the appropriate pathophysiological signals. Our recent work showed that cardiac lncExACT1 was increased in heart failure where it conferred its effects partly through binding to miR-222.5 Whatever the underlying mechanisms contributing to the lack of baseline effects with miR-222 expression, it suggests that miR-222 overexpression presents a potential therapeutic strategy that would lay dormant with little baseline effect but become functionally active when needed to counteract pathological stress.
These findings resonate with the aspects of a previous study of miR-222 and miR-221 in an angiotensin II infusion model and extend this study in important ways. Verjans et al.17 found that a concurrent inhibition of miR-221 and miR-222 exacerbated fibrosis and cardiac dysfunction in a mouse model of angiotensin II-infusion. Here, we demonstrate that the inhibition of miR-222 alone is sufficient to aggravate cardiac dysfunction in pressure overload–induced hypertrophy and heart failure. More importantly, we also demonstrate that enhanced miR-222 expression significantly mitigates cardiac remodelling in pressure overload. The inhibition of miR-222 alone did not significantly increase fibrosis after TAC in our study, but we did detect a decrease in fibrosis after TAC with miR-222 overexpression. Verjans et al. did not see an impact of miR-222/miR-221 inhibition on cardiac hypertrophy, while we did see an important modulation of hypertrophy with both the expression and the inhibition of miR-222. Possible explanations for this difference include the different models employed (infusion of a single pharmacological agent vs. pressure overload induced by aortic constriction) and the specificity of our interventions, which only altered miR-222 rather than also changing miR-221.
Verjans et al.17 also found that miR-222 levels were lower in cardiac samples from patients with severe fibrosis and cardiomyopathy or aortic stenosis. While it is possible that this reflects a different between species, it seems plausible that the difference in disease stage and chronicity play an important role. The DCM subjects had an average age in the 50s and the aortic stenosis patients in the 70s.17 We hypothesize that miR-222 increases early in pathological states as a compensatory mechanism but that this is not sustained and likely decreases at later stages, contributing to the decline seen in such patients.
It is worth noting that others have reported cardiac dysfunction, rather than protection, with comparable cardiac miR-222 overexpression.28 However, this observation was based on one transgenic line. Thus, deleterious positional effects produced by transgene insertion cannot be excluded. In contrast, we generated multiple independent miR-222 transgenic lines with results consistent with those reported here. The only instance in which we observed cardiac dysfunction in miR-222 transgenic mice was in very high-expressing lines with a >200-fold increase in miR-222 expression. Of note, a high-level expression of irrelevant genes (including GFP) has similarly been reported to induce heart failure in such transgenic systems.34 Thus, the available data suggest that miR-222 does not cause cardiac dysfunction when overexpressed at reasonable levels in the heart. Nevertheless, as with any medical intervention, future studies of miR-222 as a therapeutic target should carefully consider dose, timing, and delivery in evaluating its potential benefits.
Of note, our cardiomyocyte-specific miR-222 overexpression model demonstrated the beneficial effect of miR-222 on cardiomyocytes in protecting against pressure overload–induced heart failure. We also demonstrated that the inhibition of miR-222 exacerbated heart failure. However, it must be noted that the inhibition of miR-222 in the current study by LNA-anti-miR was systemic. It is possible that the adverse effects observed with miR-222 inhibition after TAC may result not only from the inhibition of miR-222 within cardiomyocytes but also from the inhibition of miR-222 in other cell types in the heart, or in other tissues. Further studies will be needed to establish the effects of cardiomyocyte-specific miR-222 inhibition in pathological hypertrophy and heart failure. In addition, our results demonstrated that the overexpression of miR-222 improved EF, although it is not clear whether this reflects a change in cardiomyocyte contractility or calcium transients, the two fundamental physiological functions of cardiomyocytes.
In summary, these studies provide the first demonstration that miR-222 is up-regulated in cardiomyocytes in pathological cardiac hypertrophy and heart failure, and functions to inhibit pathological cardiomyocyte growth, apoptosis, and heart failure. Direct targets that appear to contribute to these effects include HMBOX1, NFATc3, and PUMA. Taken together, these data provide further support for the model that pathological and physiological hypertrophy employ distinct downstream mechanisms. Moreover, they implicate miR-222 expression as a potential therapeutic modality that causes little or no effects at baseline but can protect against adverse remodelling in response to pressure overload or ischaemic injury.
We report that miR-222 was necessary and sufficient to limit cardiac growth, cardiomyocyte cell death, adverse ventricular remodelling, and cardiac dysfunction in response to pressure overload. This suggests possible therapeutic value, particularly as miR-222 is conserved between mice and humans and regulated by exercise in both.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
X.L., H.L., and A.R. conceived and designed the study. H.L., F.D., C.P., C.L., H.Z., X.P.W., A.Y., and P.M. performed, analysed, and interpreted the experiments. C.X. performed the TAC surgeries. X.L., H.L., M.H.H., and A.R. wrote the manuscript.
Acknowledgements
We thank Dr Ling Li for technical support. The graphical abstract was created with BioRender.com.
Funding
This work was supported by the American Heart Association (20CDA35310184 to H.L., 23MERIT1038415 to A.R.); National Institutes of Health (R21AG077040 to H.L., R01AG061034 and R35HL155318 to A.R., K08HL161405 to H.Z., T32GM007226 to C.P., 5T32HL007374-37 to F.D.). C.L. was supported by the German Center for Cardiovascular Research (DZHK), the Medical Faculty of the University of Heidelberg (Germany), and the Else-Kröner-Fresenius Stiftung.
Data availability
The data underlying this article are available in the article itself or its Supplementary material, or from the authors upon reasonable request.
References
Author notes
Xiaojun Liu and Haobo Li Co-first authors.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}