





Abstract
Abdominal aortic aneurysm (AAA) is a common, serious vascular disease with no effective pharmacological treatment. The nucleoside adenosine plays an important role in modulating vascular homeostasis, which prompted us to determine whether adenosine kinase (ADK), an adenosine metabolizing enzyme, modulates AAA formation via control of the intracellular adenosine level, and to investigate the underlying mechanisms.
We used a combination of genetic and pharmacological approaches in murine models of AAA induced by calcium chloride (CaCl2) application or angiotensin II (Ang II) infusion to study the role of ADK in the development of AAA. In vitro functional assays were performed by knocking down ADK with adenovirus-short hairpin RNA in human vascular smooth muscle cells (VSMCs), and the molecular mechanisms underlying ADK function were investigated using RNA-sequencing, isotope tracing, and chromatin immunoprecipitation quantitative polymerase chain reaction (ChIP-qPCR). The heterozygous deficiency of ADK protected mice from CaCl2- and Ang II-induced AAA formation. Moreover, specific knockout of ADK in VSMCs prevented Ang II-induced AAA formation, as evidenced by reduced aortic extracellular elastin fragmentation, neovascularization, and aortic inflammation. Mechanistically, ADK knockdown in VSMCs markedly suppressed the expression of inflammatory genes associated with AAA formation, and these effects were independent of adenosine receptors. The metabolic flux and ChIP-qPCR results showed that ADK knockdown in VSMCs decreased S-adenosylmethionine (SAM)-dependent transmethylation, thereby reducing H3K4me3 binding to the promoter regions of the genes that are associated with inflammation, angiogenesis, and extracellular elastin fragmentation. Furthermore, the ADK inhibitor ABT702 protected mice from CaCl2-induced aortic inflammation, extracellular elastin fragmentation, and AAA formation.
Our findings reveal a novel role for ADK inhibition in attenuating AAA via epigenetic modulation of key inflammatory genes linked to AAA pathogenesis.
Time of primary review: 18 days
1. Introduction
Abdominal aortic aneurysms (AAAs), characterized by localized progressive dilatation of the abdominal aorta that exceeds the normal diameter by 50%, are found in up to 8% of men aged >65 years.1 Deaths attributed to AAAs are estimated at more than 150 000 annually worldwide.1,2 Although open or endovascular surgical repair can reduce the risk of life-threatening rupture of large AAAs, few medical therapies have been shown to limit the progressive expansion of AAAs, partially due to an insufficient understanding of the pathogenetic mechanisms of AAA formation.2,3
Studies in animal models and patients with AAA indicate a major role of chronic aortic inflammation in AAA formation and progression.2 As a hallmark of AAA, aortic wall inflammation predicts AAA expansion, rupture, and need for surgical repair in patients.4 In both human AAA tissue and animal models of AAA, microarray-based and RNA-sequencing-based gene expression studies have demonstrated an over-representation of pathways involved in inflammatory responses in aneurysmal aortas.5–8 Vascular smooth muscle cells (VSMCs), as the main cellular component of the aortic wall, promote AAA progression through enhanced inflammatory responses including increased secretion of proinflammatory cytokines and proteolytic enzymes.9–11 Thus, inhibition of aneurysmal wall inflammation, especially VSMC inflammation, may be a useful therapeutic target for AAA.
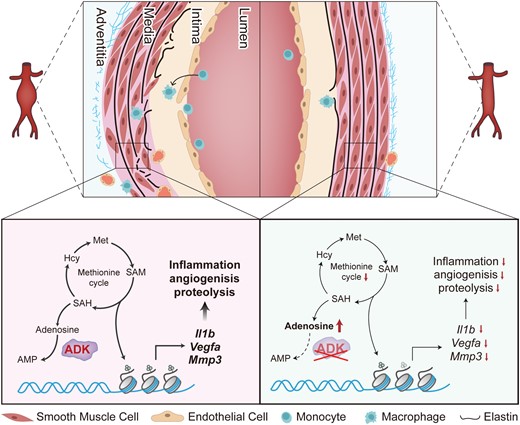
Adenosine, a ubiquitous nucleoside, plays a significant role in protecting against cellular damage in numerous pathological states.12,13 It regulates the function of mammalian cells through the activation of four G-protein-coupled receptors, ADORA1, ADORA2A, ADORA2B, and ADORA3, and through receptor-independent mechanisms.13 Adenosine is generated through the dephosphorylation of adenosine monophosphate (AMP) by an intracellular 5′-nucleotidase or extracellular ecto-5′-nucleotidase.13 In addition to AMP hydrolysis, adenosine can also be produced intracellularly through cleavage of S-adenosylhomocysteine (SAH) by the enzyme SAH hydrolase.13 The bioavailability of adenosine depends upon its transformation to inosine via adenosine deaminase or to AMP via adenosine kinase (ADK).13 ADK is a principal enzyme in maintaining intracellular adenosine homeostasis, and minor changes in ADK activity rapidly translate into major changes in adenosine levels.14 Several studies from our laboratory and others highlight a therapeutic potential of ADK inhibition in various diseases, including arthritis, endotoxemia, and stroke.15–17 However, it remains unclear whether elevated intracellular adenosine in VSMCs via ADK inactivation can inhibit VSMC inflammation and protect mice from angiotensin II (Ang II) or calcium chloride (CaCl2)-induced AAA formation.
In the current study, we utilized a combination of genetic and pharmacological approaches in two murine AAA models to demonstrate the significant protective role of ADK inhibition in VSMCs in the development of AAA. We found that inhibition of VSMC ADK attenuates AAA formation by decreasing the expression of inflammatory genes, including IL1B, IL6, vascular endothelial growth factor A (VEGFA), and matrix metalloproteinase 3 (MMP3), through adenosine receptor-independent epigenetic pathways. These data strongly suggest that inhibition of ADK is an attractive therapeutic strategy for AAA prevention and treatment.
2. Methods
The sources of reagents and detailed methods are provided in the Supplementary Material.
2.1 Animals
Mice were used in accordance with the protocol approved by the Institutional Animal Care and Use Committee of Augusta University. The floxed ADK (ADKF/F) mice were generated by Xenogen Biosciences Corporation (Cranbury, NJ, USA). Global inducible heterozygous ADK-knockout (ADKF/+/Rosa26Cre/ERT2) and VSMC-specific inducible ADK-knockout (ADKF/F/Myh11Cre/ERT2) mice were produced by breeding ADKF/F mice with Rosa26Cre/ERT2 mice (stock no. 008463, The Jackson Laboratory, Bar Harbor, ME, USA) or Myh11Cre/ERT2 transgenic mice (stock no. 019079, The Jackson Laboratory, Bar Harbor, ME, USA). These mice were also crossbred with Apoe−/− mice to obtain ADKF/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/F/Myh11Cre/ERT2/Apoe−/− mice, respectively. All mice were genotyped by PCR amplification with tail DNA samples, and the specific primers were listed in Supplementary material online, Table S1. Tamoxifen (75 mg/kg/day) was given to 8- to 10-week-old male ADKF/+/Rosa26Cre/ERT2, ADKF/+/Rosa26Cre/ERT2/Apoe−/−, and ADKF/F/Myh11Cre/ERT2/Apoe−/− mice and their littermate control mice for five consecutive days by intraperitoneal injection to induce global heterozygous ADK knockout (ADKF/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2/Apoe−/−) or VSMC-specific ADK knockout (ADKΔVSMC/Apoe−/−). We used only male mice in this study according to the recommendation from the Arteriosclerosis, Thrombosis, and Vascular Biology Council.18 All mice were on a C57BL/6J background. The animal procedures within this study conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes or the National Institutes of Health Guide for the Care and Use of Laboratory Animals. In procedures requiring anaesthesia, the mice were anaesthetized with 2% isoflurane. And at the appropriate time, the mice were euthanized with CO2.
2.2 Statistical analysis
Statistical analyses were performed with GraphPad Prism software (GraphPad, La Jolla, CA, USA). Categorical data are shown as percentages, two-sided Fisher’s exact test was used to compare the difference of AAA incidence between two groups, and Mantel–Cox test was used to compare the difference of survival curve between two groups. Continuous data are presented as the means ± standard error of mean (SEM), data normality was assessed via the Shapiro–Wilk test, and F-test or Brown–Forsythe test was used to evaluate the homogeneity of variance. If data were normally distributed, two-tailed unpaired Student’s t-test (for the variances that were equal) or two-tailed unpaired Student’s t-test with Welch’s correction (for the variances that were unequal) was used to compare the difference between two groups, and one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test or two-way ANOVA followed by Tukey post hoc test was performed for comparison among more than two groups when appropriate. For non-normally distributed continuous data and rank data, Mann–Whitney U test was used to compare two groups, and Kruskal–Wallis test followed by Dunn’s post hoc test was used for comparison among more than two groups. For repeated measures data, mixed-effects model followed by Bonferroni’s post hoc test (for when there were missing values) or two-way repeated measures ANOVA followed by Bonferroni’s post hoc test (for when there were no missing values) was used to compare the difference between two groups. Differences were considered statistically significant at P < 0.05, and all results were representative of at least three independent experiments.
3. Results
3.1 ADK deficiency inhibits CaCl2-induced AAA formation
To investigate the role of ADK in AAA development, we generated ADK-deficient mice in a C57BL/6J background. Because homozygous ADK knockout is lethal within 1 month after tamoxifen-induced Cre recombination, we generated heterozygous ADK-inducible knockout (ADKF/+/Rosa26Cre/ERT2) mice by intercrossing ADKF/F mice with Rosa26Cre/ERT2 mice (see Supplementary material online, Figure S1A), and found that there was a nearly 50% reduction of ADK protein in the main organs including the aorta, heart, lung, and kidney (see Supplementary material online, Figure S1B–F), and no significant change in cardiac function or histology of the liver, kidney, adipose tissue, or skeletal muscle of male ADKF/+/Rosa26Cre/ERT2 mice compared with those of control mice 2 weeks after tamoxifen injection (see Supplementary material online, Figure S1G–J).
To determine the role of ADK in CaCl2-induced AAA, male ADKF/+/Rosa26Cre/ERT2 mice and their littermate ADK+/+/Rosa26Cre/ERT2 mice underwent laparotomy followed by CaCl2 application to the infrarenal aorta. Six weeks later, the mice were euthanized and the aortas were collected for analysis (Figure 1A). Morphologically, the aortas of saline-treated ADKF/+/Rosa26Cre/ERT2 mice did not differ from those of saline-treated control mice. The maximal abdominal aortic diameter was significantly larger in CaCl2-treated ADK+/+/Rosa26Cre/ERT2 mice compared with the saline-treated ADK+/+/Rosa26Cre/ERT2 mice. The maximal abdominal aortic diameter was remarkably reduced in CaCl2-treated ADKF/+/Rosa26Cre/ERT2 mice compared with that of CaCl2-treated ADK+/+/Rosa26Cre/ERT2 mice (Figure 1B and C). Medial degeneration and local inflammation are hallmarks of AAA development. Elastin staining showed that CaCl2 caused marked aortic elastin fragmentation in ADK+/+/Rosa26Cre/ERT2 mice, which was significantly blunted in ADKF/+/Rosa26Cre/ERT2 mice (Figure 1D). Moreover, heterozygous ADK gene deletion protected mice against CaCl2-induced aortic inflammation, as evidenced by significantly reduced macrophage infiltration in aortas of ADKF/+/Rosa26Cre/ERT2 mice compared with those of the control mice (Figure 1E). Consistently, the mRNA expression of inflammatory molecules and MMPs was significantly decreased in the aortas of CaCl2-treated ADKF/+/Rosa26Cre/ERT2 mice compared with that of controls (Figure 1F and G). These results demonstrated that heterozygous ADK knockout suppressed CaCl2-induced AAA formation.
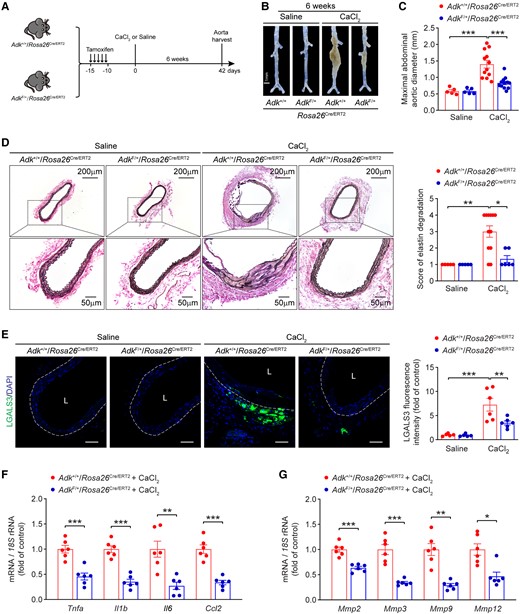
ADK deficiency inhibits CaCl2-induced AAA formation in mice. (A) Experimental schematic representation of CaCl2-induced AAA in male ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice. (B) Representative images of abdominal aortas from ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice at 6 weeks after saline or CaCl2 application. (C) Maximal abdominal aortic diameter in ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice at 6 weeks after saline or CaCl2 application (n = 5–12). (D) Representative staining with elastin (left) and elastin fragmentation score (right) in abdominal aortas from ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice at 6 weeks after saline or CaCl2 application (n = 5–12). (E) Representative images (left) and quantification (right) of LGALS3 immunofluorescent staining of abdominal aortas from ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice at 6 weeks after CaCl2 application (n = 5–6), L: lumen, scale bar = 50 μm. (F, G) qPCR analysis of inflammatory cytokines (F) and MMPs (G) in abdominal aortas from ADK+/+/Rosa26Cre/ERT2 and ADKF/+/Rosa26Cre/ERT2 mice at 6 weeks after CaCl2 application (n = 6). Two-way ANOVA followed by Tukey post hoc test for (C) and (E), Kruskal–Wallis test followed by Dunn’s post hoc test for (D), two-tailed unpaired Student’s t-test for (F) and (G, Mmp2), two-tailed unpaired Student’s t-test with Welch’s correction for (G, Mmp3, and Mmp9), and Mann–Whitney U test for (G, Mmp12). All data are represented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated comparisons.
3.2 ADK deficiency suppresses Ang II-induced AAA formation
We next sought to confirm the aforementioned findings using a distinct AAA model that involved Ang II infusion, a model that recapitulates many pathological aspects of human AAA, including marked inflammation, thrombus formation, and aortic rupture. Male ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice and their littermate ADK+/+/Rosa26Cre/ERT2/Apoe−/− mice were generated, injected with tamoxifen, and infused with saline or Ang II (1 000 ng/kg/min) for 28 days (see Supplementary material online, Figure S1K). Ang II infusion increased systolic blood pressure, but blood pressure did not differ between ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice and control mice (Figure 2A). Ang II infusion resulted in 80% AAA incidence in control ADK+/+/Rosa26Cre/ERT2/Apoe−/− mice, whereas only 30% of ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice developed AAA (Figure 2B). There were no gross differences in aortic morphology between saline-infused ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice and control mice. The maximal suprarenal abdominal aortic diameters of Ang II-infused ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice were significantly smaller than those of the control mice (Figure 2C and D). Meanwhile, the elastic lamina was frequently disrupted and degraded in Ang II-infused ADK+/+/Rosa26Cre/ERT2/Apoe−/− mice but not in ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice (Figure 2E). Moreover, ADK deficiency protected mice against Ang II-induced vascular inflammation, as evidenced by significantly reduced macrophage infiltration in aortas of ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice compared with those of the control mice (Figure 2F). Consistently, the mRNA expression of inflammatory molecules and MMPs was also significantly decreased in the aortas of Ang II-infused ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice compared with that of controls (Figure 2G and H). These changes at protein levels of inflammatory cytokines, including IL1B and IL6, and MMPs, including MMP2 and MMP3, were further confirmed by immunohistochemical staining (see Supplementary material online, Figure S2A–D). Taken together, these data suggest a critical role for ADK in AAA formation induced by Ang II.
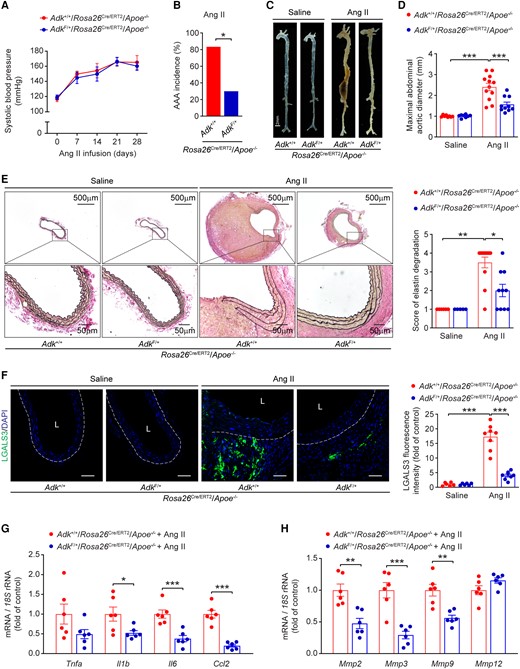
ADK deficiency suppresses Ang II-induced AAA formation in Apoe−/− mice. (A) Systolic blood pressure of male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice during Ang II infusion (n = 6–9). (B) The incidence of Ang II-induced AAA in male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice (n = 10–12). (C) Representative images of whole aortas from male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice following 4 weeks of saline or Ang II infusion. (D) Maximal abdominal aortic diameter in male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice following 4 weeks of saline or Ang II infusion (n = 8–12). (E) Representative staining with elastin (left) and elastin fragmentation score (right) in abdominal aortas from male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice following 4 weeks of saline or Ang II infusion (n = 5–12). (F) Representative images (left) and quantification (right) of LGALS3 immunofluorescent staining of abdominal aortas from male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice following 4 weeks of saline or Ang II infusion (n = 6–8), L: lumen, scale bar = 50 μm. (G, H) qPCR analysis of inflammatory cytokines (G) and MMPs (H) in abdominal aortas from male ADK+/+/Rosa26Cre/ERT2/Apoe−/− and ADKF/+/Rosa26Cre/ERT2/Apoe−/− mice following 4 weeks of Ang II infusion (n = 6). Mixed-effects model followed by Bonferroni’s post hoc test for (A), two-sided Fisher’s exact test for (B), two-way ANOVA followed by Tukey post hoc test for (D) and (F), Kruskal–Wallis test followed by Dunn’s post hoc test for (E), two-tailed unpaired Student’s t-test for (G, Tnfa, Il6, and Ccl2) and (H), and two-tailed unpaired Student’s t-test with Welch’s correction for (G, Il1b). All data are represented as mean ± SEM *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated comparisons.
3.3 ADK deficiency in vascular smooth muscle ameliorates Ang II-induced AAA formation
With the evidence of the involvement of ADK in AAA formation, we examined ADK expression in aortas of mice. Interestingly, after 7 days of Ang II infusion, ADK expression at protein level in the aortas of mice with Ang II infusion did not differ from that of control mice with saline infusion (see Supplementary material online, Figure S3A). In vitro experiments, there was also no significant change in the protein level of ADK in human aortic smooth muscle cells (HASMCs) treated with different stimuli, including Ang II, tumour necrosis factor alpha (TNFA), CaCl2, or H2O2 (see Supplementary material online, Figure S3B and C). Consistently, analysis of genome-wide expression profiling data from human AAA samples (GSE47472) also failed to show a difference in ADK expression at mRNA level (see Supplementary material online, Figure S3D). Then, we investigated the cellular origin of ADK in the formation of AAA. We analysed single-cell RNA-sequencing data (GSE152583) from single cells of aortas from elastase-induced murine AAA, which simulates the denouement of AAA formation (elastin fragmentation), circumventing earlier mechanisms such as changes in VSMC behaviour and function. Compared with many other types of cells, including fibroblasts, endothelial cells, and immune cells, Myh11-positive VSMCs express a high level of ADK transcript (Figure 3A). We further examined ADK expression at the protein level by immunostaining aortas from Ang II-induced murine AAA and human AAA tissues. As shown in Figure 3B and C, ADK was prominently expressed in medial VSMCs in AAA. We then generated VSMC-specific ADK knockout (ADKΔVSMC/Apoe−/−) mice by breeding ADKF/F mice with Myh11Cre/ERT2 and Apoe−/− mice, and treating the consequent mice with tamoxifen (see Supplementary material online, Figure S3E). ADK knockout efficiency in the adventitia-removed aorta was verified by qPCR and western blot (see Supplementary material online, Figure S3F and G). As shown in Supplementary material online, Figure S3H–J, there were no statistical differences in systolic blood pressure, diastolic blood pressure, or heart rate between ADKΔVSMC/Apoe−/− and ADKWT/Apoe−/− mice. The mRNA expression of VSMC markers and inflammatory molecules in adventitia-removed aortas of ADKΔVSMC/Apoe−/− mice was also similar compared with that of ADKWT/Apoe−/− mice (see Supplementary material online, Figure S3K). Next, male ADKΔVSMC/Apoe−/− and ADKWT/Apoe−/− mice were infused with saline or Ang II for 28 days (see Supplementary material online, Figure S4A). No significant differences were found in systolic blood pressure between ADKΔVSMC/Apoe−/− and ADKWT/Apoe−/− mice during Ang II infusion (see Supplementary material online, Figure S4B). However, the survival rate of ADKΔVSMC/Apoe−/− mice was significantly higher than that of ADKWT/Apoe−/− mice after Ang II infusion (Figure 3D), and the AAA incidence was 70% for ADKWT/Apoe−/− mice and only 27% for ADKΔVSMC/Apoe−/ mice (Figure 3E). Ang II infusion increased aortic diameters in ADKWT/Apoe−/− mice in vivo, which was significantly blunted in ADKΔVSMC/Apoe−/− mice (Figure 3F and G). The alterations in the diameters were also confirmed by measuring the maximal suprarenal abdominal aortas ex vivo (Figure 3H and I). Histological analyses showed that the elastic lamina was frequently disrupted and fragmented in Ang II-infused ADKWT/Apoe−/− mice, whereas VSMC ADK deficiency significantly prevented elastic lamina fragmentation (Figure 4A). Moreover, VSMC ADK deficiency protected mice against Ang II-induced pathological changes of abdominal aortas, exhibiting up-regulation of ACTA2 expression in VSMCs (see Supplementary material online, Figure S4C), decreased microvessel formation evidenced with PECAM1 staining (Figure 4B), decreased macrophage infiltration evidenced with CD68 staining (Figure 4C), and decreased expression of MMP2 and MMP3 in vessel wall of ADKΔVSMC/Apoe−/− mice compared with ADKWT/Apoe−/− mice (see Supplementary material online, Figure S4D and Figure 4D). Further, VSMC ADK-specific deficiency significantly decreased the enzymatic activity of MMP3 in abdominal aortas compared with the control group (Figure 4E). Consistently, the mRNA expression of MMPs (Mmp3 and Mmp2, but not Mmp9) and inflammatory molecules (Il1b and Il6, but not Tnfa or Ccl2) was significantly decreased in the aortas of Ang II-infused ADKΔVSMC/Apoe−/− mice compared with controls (Figure 4F–H and Supplementary material online, Figure S4E–H). Taken together, these results indicate that VSMC ADK plays a prominent role in AAA formation in Ang II-infused Apoe−/− mice.
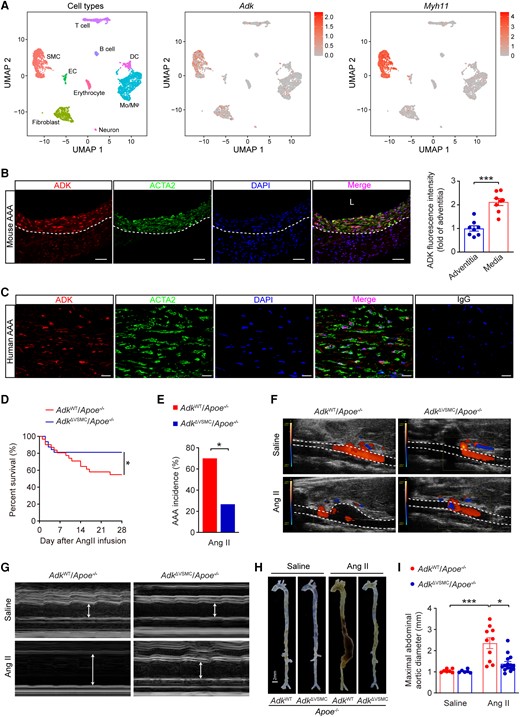
ADK deficiency in VSMCs reduces Ang II-induced AAA formation in Apoe−/− mice. (A) Uniform Manifold Approximation and Projection visualization of cell types present in mouse infrarenal abdominal aortas treated with elastase for 0, 7, and 14 days (left) and the expression of ADK (middle) and Myh11 (right) revealed by scRNA-seq (GSE152583). (B) Representative images (left) of triple immunofluorescence for ADK, ACTA2, and nuclei (DAPI, 4',6-diamidino-2-phenylindole), and quantification (right) of ADK expression in the media and the adventitia of abdominal aortas from male Apoe−/− mice after 4 weeks of Ang II infusion (n = 8), L: lumen, scale bar = 50 μm. (C) Representative images of triple immunofluorescence for ADK, ACTA2, and nuclei (DAPI) on sections of human AAA, scale bar = 20 μm. (D) The survival curve of Ang II-induced AAA in ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice (n = 31–32). (E) The incidence of Ang II-induced AAA in male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice (n = 10–15). (F, G) Representative colour Doppler ultrasound images of aortic flow (F) and M-mode pictures of abdominal aortas (G) from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice following 4 weeks of saline or Ang II infusion. (H) Representative images of whole aortas from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice following 4 weeks of saline or Ang II infusion. (I) Maximal abdominal aortic diameter in male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice following 4 weeks of saline or Ang II infusion (n = 6–15). Two-tailed unpaired Student’s t-test for (B), Mantel–Cox test for (D), two-sided Fisher’s exact test for (E), and Kruskal–Wallis test followed by Dunn’s post hoc test for (I). All data are represented as mean ±SEM *P < 0.05 and ***P < 0.001 for indicated comparisons.
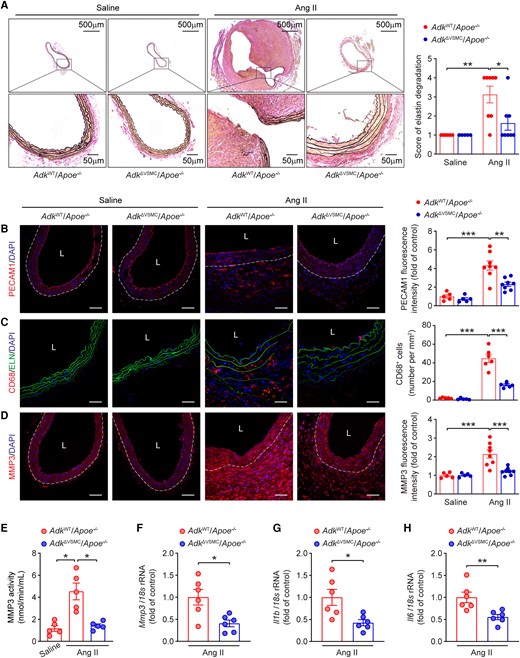
Vascular smooth muscle cell-specific ADK ablation decreases Ang II-induced abdominal aortic pathological changes in Apoe−/− mice. (A) Representative staining with elastin (left) and elastin fragmentation score (right) in abdominal aortas from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice following 4 weeks of saline or Ang II infusion (n = 5–8). (B–D) Representative images (left) and corresponding quantification (right) of PECAM1 (B), CD68 (C), and MMP3 (D) immunofluorescent staining of abdominal aortas from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice after 4 weeks of saline or Ang II infusion (n = 5–8), L: lumen, scale bar = 50 μm. (E) MMP3 catalytic activity in abdominal aortas from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice after 4 weeks of saline or Ang II infusion (n = 5). (F–H) qPCR analysis of Mmp3 (F), Il1b (G), and Il6 (H) in abdominal aortas from male ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice following 4 weeks of Ang II infusion (n = 6). Kruskal–Wallis test followed by Dunn’s post hoc test for (A), two-way ANOVA followed by Tukey post hoc test for (B–D), Brown–Forsythe ANOVA and Welch’s ANOVA test with Dunnett’s T3 multiple comparison for (E), and two-tailed unpaired Student’s t-test for (F–H). All data are represented as mean ±SEM *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated comparisons.
3.4 Adenosine kinase regulates the inflammatory status of VSMCs independent of adenosine receptors
To further explore the molecular mechanisms underlying the function of ADK in AAA development, short hairpin RNA was used to knock down ADK in cultured HASMCs (see Supplementary material online, Figure S5A–C). RNA-Seq was performed to unbiasedly compare the transcriptomic changes in HASMCs transfected with shADK vs. control (shCtrl). A volcano plot showed the distribution of the differentially expressed genes (DEGs) between ADK-knockdown and control HASMCs (Figure 5A). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was used for the identification of key pathways associated with the effects of ADK knockdown on HASMCs, and the top 20 KEGG pathways of all DEGs are shown in Figure 5B. Notably, the mitogen-activated protein kinase (MAPK) signalling pathway was the highest enriched KEGG pathway in HASMCs transfected with shADK. Using western blots, we did not find a decrease in MAPK related signalling pathways (including p-ERK/ERK (extracellular signal-regulated kinase), p-JNK/JNK (c-Jun N-terminal kinase), or p-P38/P38) or NF-κB related pathway (p-P65/P65) in ADK-knockdown HASMCs treated with TNFA (see Supplementary material online, Figure S5D–H). Rather, we did find that proinflammatory cytokines (IL1B and IL1A) and proangiogenic factors [VEGFA and vascular endothelial growth factor B (VEGFB)] were in the top 20 down-regulated genes in the MAPK signalling pathway in ADK-knockdown HASMCs (Figure 5C). Using qPCR, we further validated the decreased mRNA expression of IL1B, IL6, VEGFA, and MMP3 in response to TNFA (Figure 5D–G), a proinflammatory cytokine that is up-regulated in human AAA tissues and has been shown to induce an inflammatory VSMC phenotype.19,20 Moreover, we isolated and cultured smooth muscle cells (SMCs) from the abdominal aortas of ADKF/F/Rosa26Cre/ERT2 and ADK+/+/Rosa26Cre/ERT2 mice (see Supplementary material online, Figure S6A). The purity of abdominal aortic SMCs was excellent, as determined by ACTA2 immunostaining (see Supplementary material online, Figure S6B). We next treated the cultured abdominal aortic SMCs with 4-hydroxytamoxifen to initiate tamoxifen-induced Cre activity. Western blot analysis showed a complete deletion of ADK in ADK-knockout SMCs compared with controls after 4-hydroxytamoxifen treatment (see Supplementary material online, Figure S6C). Consistent with results of the human SMC studies, the mRNA expression of Il1b, Il6, Vegfa, and Mmp3 was significantly suppressed in TNFA-treated ADK-knockout SMCs compared with control cells (Figure 5H–K). Furthermore, the ADK inhibitor ABT702 also significantly decreased TNFA-induced proinflammatory gene expression (including Il1b and Il6) in wild-type abdominal aortic SMCs (see Supplementary material online, Figure S6D and E). In addition, ADK knockdown significantly decreased the expression of several other inflammatory genes (including IL11, CSF1, PTGS2, ICAM1, CCL20, and CXCL8) in HASMCs treated with TNFA (see Supplementary material online, Figure S6F and G). In functional experiments, the number of monocytes adhering to ADK-knockdown HASMCs treated with TNFA was significantly reduced compared with that of control HASMCs (see Supplementary material online, Figure S6H), and the endothelial proliferation assay demonstrated that conditioned medium from ADK-knockdown HASMCs produced significantly fewer EdU+ human umbilical vein endothelial cells as compared with the control group, which was overcome by adding exogenous VEGF (see Supplementary material online, Figure S6I). ADK can regulate levels of both intracellular and extracellular adenosine, and extracellular adenosine alters intracellular cAMP concentration via activation of its receptors. Nevertheless, the content of cAMP in abdominal aortas was comparable between ADKΔVSMC/Apoe−/− mice and ADKWT/Apoe−/− mice after 4 weeks of Ang II infusion (see Supplementary material online, Figure S6J), which suggests that VSMC-specific ADK deficiency has no effect on the activation of adenosine receptors. Meanwhile, as shown in Figure 5L–O, the anti-inflammatory effects of ADK knockdown in HASMCs treated with TNFA were not blunted by DPCPX (8-cyclopentyl-1,3-dipropylxanthine, ADORA1 antagonist), ZM241385 (ADORA2A antagonist), MRS1754 (ADORA2B antagonist), or MRS1523 (ADORA3 antagonist). These results suggest that ADK knockdown inhibits TNFA-induced inflammatory responses through an adenosine receptor-independent mechanism.
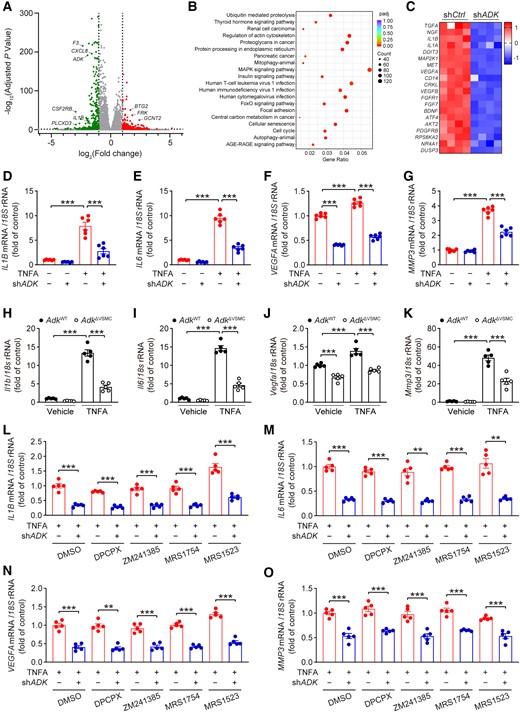
ADK knockdown attenuates inflammatory responses in HASMCs independent of adenosine receptors. (A) Volcano plot with up- or down-regulated genes in ADK-knockdown HASMCs highlighted in red or green, respectively. (B) Scatter plot of the top 20 KEGG pathways of all DEGs in ADK-knockdown HASMCs. The colour of the dot represents the adjusted P value, and the size of the dot represents the number of DEGs. (C) Heatmap analysis of the top 20 down-regulated genes in the MAPK signalling pathway in ADK-knockdown HASMCs. (D) qPCR analysis of mRNA levels of IL1B, (E)IL6, (F) VEGFA, and (G) MMP3 in TNFA-treated (10 ng/mL for 12 h) control and ADK-knockdown HASMCs (n = 6). (H) qPCR analysis of mRNA levels of Il1b, (I) Il6, (J) Vegfa, and (K) Mmp3 in TNFA-treated (10 ng/mL for 24 h) control and Adk-knockout abdominal aortic SMCs (n = 5). (L) qPCR analysis of mRNA levels of IL1B, (M) IL6, (N) VEGFA , and (O) MMP3 in TNFA-treated (10 ng/mL for 12 h) control or ADK-knockdown HASMCs pre-treated for 30 min with 5 μM DPCPX, 5 μM ZM241385, 5 μM MRS1754, or 5 μM MRS1523 (n = 5). Two-way ANOVA followed by Tukey post hoc test for (D–K), two-tailed unpaired Student’s t-test for (L, DPCPX, ZM241385, and MRS1523), (M, DMSO (dimethyl sulfoxide), DPCPX, and MRS1754), (N, DMSO, ZM241385, MRS1754, and MRS1523), and (O, DMSO, ZM241385, and MRS1523), two-tailed unpaired Student’s t-test with Welch’s correction for (L, DMSO, and MRS1754), (M, ZM241385, and MRS1523), and (O, DPCPX, and MRS1754), and Mann–Whitney U test for (N, DPCPX). All data are represented as mean ± SEM **P < 0.01 and ***P < 0.001 for indicated comparisons.
3.5 ADK knockdown decreases inflammatory responses in HASMCs via histone methylation
TNFA significantly increased methionine uptake (Figure 6A), and subsequently increased intracellular S-adenosylmethionine (SAM) and SAH (Figure 6B and C). Accordingly, the SAM:SAH ratio, an index of methylation potential, was also increased (Figure 6D), indicating that SAM-dependent transmethylation reactions are promoted in TNFA-stimulated HASMCs. Adenosine hydrolyzed from SAH is metabolized by ADK to maintain the flux of the methionine cycle.14 In TNFA-treated HASMCs, ADK knockdown significantly increased intracellular adenosine levels (see Supplementary material online, Figure S7A), which may negatively inhibit the flux of the methionine cycle and SAM-dependent transmethylation reactions. SAM is synthesized by methionine and ATP whereas glucose-derived ribose can integrate into the methionine cycle through de novo ATP synthesis (Figure 6E and Supplementary material online, Figure S7B). Therefore, 13C6-glucose tracer is used to monitor the metabolite dynamics of the methionine cycle.21 Within 4 h of 13C6-glucose tracing in cultured VSMCs, m + 5 ATP was the most abundant labelled ATP synthesized de novo from 13C6-glucose (see Supplementary material online, Figure S7C), facilitating the monitor of SAM and SAH. The unlabelled ATP and labelled m + 5 ATP were similar between the control and ADK-knockdown HASMCs (see Supplementary material online, Figure S7D). Furthermore, labelled methionine was extremely low in treated HASMCs (∼0.03‰ of the total methionine pool, Supplementary material online, Figure S7E) and the unlabelled methionine was comparable between the control and ADK-knockdown HASMCs (see Supplementary material online, Figure S7F). Under these conditions, the levels of accumulated m + 5 SAM, m + 5 SAH, and m + 5 adenosine in ADK-knockdown HASMCs were much lower than that of the control HASMCs with TNFA stimulation (see Supplementary material online, Figure S7G and H and Figure 6F), indicating the flux of the methionine cycle was resisted in ADK-knockdown HASMCs. Moreover, the labelled m + 5 SAH even normalized with m + 5 SAM was significantly decreased in ADK-knockdown HASMCs compared with that in control cells (Figure 6G), suggesting that ADK knockdown might inhibit SAM-dependent transmethylation including histone and DNA methylation, which play critical roles in chromatin state and gene transcription. Histone methylation is required for the maintenance of downstream gene transcription and is modulated by the methionine cycle,22 we therefore examined whether a change in the histone modification state occurs in ADK-knockdown HASMCs. Dimethylation or trimethylation at lysines 4, 9, 36, and 79 of histone H3 (H3K4me3, H3K9me2, H3K36me2, and H3K79me3) are known to play a substantial role in defining chromatin states and modulating gene expression. Western blots showed that H3K4me3 but not H3K9me2, H3K36me2, and H3K79me3 was reduced in ADK-knockdown HASMCs treated with TNFA (Figure 6H–L). Consistently, methionine restriction also led to a rapid drop in the level of H3K4me3, but not H3K79me3, in HASMCs (see Supplementary material online, Figure S7I), indicating H3K4 methylation is more sensitive to the methionine cycle than other histone methylation in HASMCs. The decreased H3K4me3 in ADK-knockdown HASMCs was further confirmed by immunofluorescent staining (see Supplementary material online, Figure S7J). Moreover, the level of H3K4me3 was also significantly reduced in TNFA-treated ADK-knockdown abdominal aortic SMCs compared with control cells (see Supplementary material online, Figure S7K). Given that H3K4me3 has been widely described to be associated with active chromatin and gene transcription, and the total H3K4me3 level was decreased in ADK-knockdown HASMCs treated with TNFA, we used chromatin immunoprecipitation coupled with qPCR (ChIP-qPCR) to investigate H3K4me3 chromatin occupancy of selective genes whose expression is down-regulated in ADK-knockdown HASMCs. As shown in Figure 6M–P, H3K4me3 enrichment in the promoters of IL1B, IL6, VEGFA, and MMP3 was significantly decreased in ADK-knockdown HASMCs treated with TNFA compared with the control group. Moreover, 7 days of Ang II infusion produced lower levels of H3K4me3 in medial VSMCs of ADKΔVSMC/Apoe−/− mice compared with those of ADKWT/Apoe−/− mice (Figure 6Q). Altogether, these data suggest that the anti-inflammatory effects of intracellular adenosine induced by ADK inhibition in VSMCs are mediated through inhibition of H3K4 trimethylation.
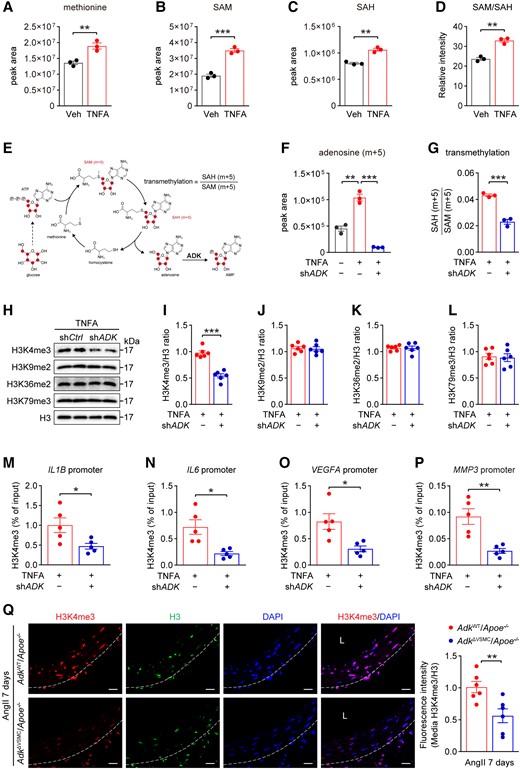
ADK knockdown decreases inflammatory responses in HASMCs via histone methylation. (A) Liquid chromatography–mass spectrometry (LC–MS) analysis of intracellular methionine, (B) SAM, (C) SAH, and (D) SAM/SAH in HASMCs treated with TNFA (10 ng/mL) for 4 h (n = 3). (E) Schematic diagram of the expected isotopologues of methionine cycle metabolites produced during 13C6-glucose tracing. The transmethylation of SAM can be monitored with the ratio of labelled SAH (m + 5) to labelled SAM (m + 5). (F–G) Mass isotopomer distribution of adenosine (m + 5) (F) and the SAH (m + 5):SAM (m + 5) ratio (G) in control or ADK-knockdown HASMCs treated with or without TNFA (10 ng/mL) for 4 h in a medium containing 13C6-glucose (22 mM) (n = 3). (H–L) Representative western blot results of H3K4me3, H3K9me2, H3K36me2, H3K79me3, and total H3 (H) in TNFA-treated (10 ng/mL for 4 h) control or ADK-knockdown HASMCs and relative ratio of H3K4me3/H3 (I), H3K9me2/H3 (J), H3K36me2/H3 (K), and H3K79me3/H3 (L) were quantitated by densitometric analysis of the corresponding western blots (n = 6). (M–P) ChIP analysis of H3K4me3 on promoter of IL1B (M), IL6 (N), VEGFA (O), and MMP3 (P) in TNFA-treated (10 ng/mL for 4 h) control or ADK-knockdown HASMCs. Enrichment of the indicated gene promoters was assessed by qPCR (n = 5). (Q) Representative images (left) and quantification (right) of H3K4me3 immunofluorescent staining of abdominal aortas from ADKWT/Apoe−/− and ADKΔVSMC/Apoe−/− mice after 7 days of Ang II infusion (n = 5), L: lumen, scale bar = 50 μm. Two-tailed unpaired Student’s t-test for (A–D), (G), (I–M), (O), and (Q), one-way ANOVA with Bonferroni’s post hoc test for (F), and two-tailed unpaired Student’s t-test with Welch’s correction for (N) and (P). All data are represented as mean ± SEM *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated comparisons.
3.6 Adenosine kinase inhibitor ABT702 suppresses CaCl2-induced AAA formation in mice
To examine the effect of pharmacological inhibition of ADK in suppression of AAA development, ABT702, a specific ADK inhibitor exhibiting anti-inflammatory effects,23,24 was administered intraperitoneally to wild-type male at a dose of 1.5 mg/kg twice a week following CaCl2 induction (Figure 7A). Six weeks later, the mice were euthanized and their aortas were collected for analysis. No morphological change was observed in the aortas of ABT702-treated mice compared with those of vehicle-treated mice. Treatment with ABT702 significantly abrogated the ability of CaCl2 to induce AAA in the mice (Figure 7B). The maximal abdominal aortic diameter was significantly reduced in ABT702-treated mice compared with vehicle-treated mice 6 weeks after CaCl2 induction (Figure 7C). In line with data from ADK-deficient mice, histology of murine aortas with Verhoeff’s elastic staining showed that CaCl2 caused marked aortic elastin fragmentation in vehicle-treated mice, whereas this was significantly blunted in ABT702-treated mice (Figure 7D). Moreover, immunofluorescent and immunohistochemical staining showed that ABT702 treatment protected mice against CaCl2-induced aortic inflammation, as evidenced by markedly reduced macrophage infiltration (Figure 7E and Supplementary material online, Figure S8A), and decreased expression of IL1B, IL6, MMP2, and MMP3 in aortas of ABT702-treated mice compared with those of vehicle-treated mice (see Supplementary material online, Figure S8B–E). Consistently, the mRNA expression of inflammatory molecules and MMPs was also significantly reduced in the aortas of ABT702-treated mice compared with control mice 6 weeks after CaCl2 induction (Figure 7F and G). These results demonstrate that administration of the ADK inhibitor ABT702 attenuates CaCl2-induced AAA formation via suppressing vascular inflammation.
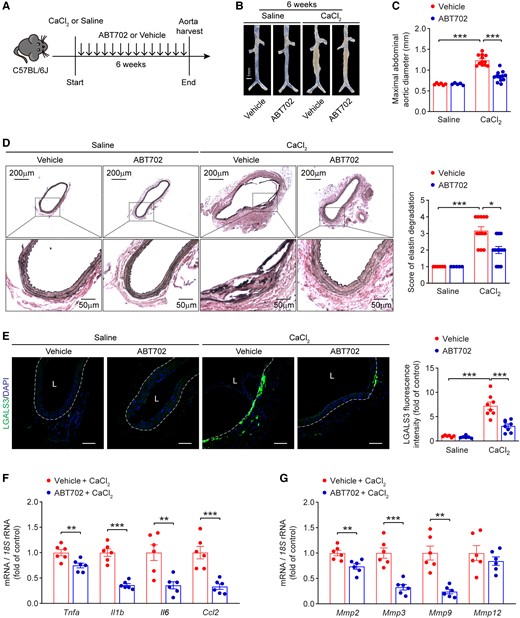
Adenosine kinase inhibitor ABT702 reduces CaCl2-induced AAA in mice. (A) Experimental schematic representation of CaCl2-induced AAA in wild-type male mice treated with vehicle or ABT702. (B) Representative images of abdominal aortas from vehicle- and ABT702-treated mice at 6 weeks after saline or CaCl2 application. (C) Maximal abdominal aortic diameter in vehicle- and ABT702-treated mice at 6 weeks after saline or CaCl2 application (n = 5–12). (D) Representative staining with elastin (left) and elastin fragmentation score (right) in abdominal aortas from vehicle- and ABT702-treated mice at 6 weeks after saline or CaCl2 application (n = 5–12). (E) Representative images of LGALS3 immunofluorescent staining (left) and quantification of LGALS3+ cells (right) in abdominal aortas from vehicle- and ABT702-treated mice at 6 weeks after saline or CaCl2 application (n = 6–8), L: lumen, scale bar = 50 μm. (F) qPCR analysis of inflammatory cytokines and (G) MMPs in abdominal aortas from vehicle- and ABT702-treated mice at 6 weeks after CaCl2 application (n = 6). Two-way ANOVA followed by Tukey post hoc test for (C) and (E), Kruskal–Wallis test followed by Dunn’s post hoc test for (D), two-tailed unpaired Student’s t-test for (F) and (G, Mmp2, Mmp3, and Mmp12), and two-tailed unpaired Student’s t-test with Welch’s correction for (G, Mmp9). All data are represented as mean ± SEM *P < 0.05, **P < 0.01, and ***P < 0.001 for indicated comparisons.
4. Discussion
In the present study, we demonstrated a key role for ADK in the pathogenesis of AAA formation in mice. Heterozygous and VSMC-specific ADK deficiency reduces CaCl2- or Ang II-induced inflammation and AAA formation. Moreover, deficiency of ADK dramatically improves key pathological aspects of AAA, including elastin fragmentation, inflammatory gene expression, macrophage infiltration, and neovascularization. Mechanistically, ADK deficiency leads to decreased levels of H3K4me3, an activating histone mark on the promoters of inflammatory, angiogenesis, and matrix metalloproteinase genes in VSMCs. Consistent with results from genetic approaches, pharmacological inhibition of ADK with ABT702 also rescued the AAA phenotype. However, it is noteworthy that clinical development of ABT702 was halted owing to the side effects, which do not appear related to the mechanism of ADK inhibition.25 Thus, it is an ongoing subject to develop novel, safe, and potent ADK inhibitors, especially targeting the nuclear ADK.26
AAA formation is a complex process including multiple molecular and cellular events involved in interactive immune and vascular cell responses. ADK in VSMCs appears to be required for AAA formation because ADK was predominantly expressed in aortic media in mouse and human AAA lesions, and deletion of ADK in VSMCs attenuated aortic dilation, decreased elastin fragmentation, and reduced vascular inflammation, demonstrating that VSMC ADK is essential for AAA formation. In addition to VSMCs, ADK has been shown to regulate endothelial function and inflammation.17,27 It is possible that endothelial ADK is also important for AAA formation. However, since the majority of ADK-positive cells were VSMCs, ADK is likely to promote aortic aneurysms in large part by regulating VSMC phenotype (inflammation and proliferation, but not dedifferentiation or migration, Supplementary material online, Figures S9 and S10). Future studies are necessary to determine whether ADK in endothelial cells or other cell types is important for AAA formation.
VSMC ADK deficiency decreases the expression of inflammatory molecules and MMP3, and protects against Ang II-induced AAA formation. Inhibition of VSMC inflammation has been demonstrated to protect mice from AAA formation.28,29 For example, VSMC-specific Smad4-knockout mice developed aortic aneurysms in association with induction of proinflammatory IL1B, and therapeutically neutralizing IL1B significantly attenuated aneurysm progression.28 Moreover, aortic IL1B expression showed a positive correlation with aortic expansion in Ang II-infused mice.7 Another proinflammatory cytokine, IL6, was higher in patients with AAA than in controls, and its receptor IL6R was identified as a genetic susceptibility locus for AAA.30,31 In VSMCs of EP4-Tg mice, Il6 was the most strongly up-regulated cytokine, and inhibition of IL6 prevented Ang II-induced AAA formation.29 MMPs, including MMP3, also play a key role in the formation of AAA. The knockout of Mmp3 reduced Ang II-induced AAA formation in mice, and a bone-marrow transplantation experiment confirmed that MMP3 expression in vascular cells, but not haematopoietic cells, was required to promote AAA.32 In the present study, the expression of Il1b, Il6, and Mmp3 was reduced in the aortas of Ang II-infused ADKΔVSMC/Apoe−/− mice. Moreover, in response to inflammatory stimulation, the expression of IL1B, IL6, and MMP3 was reduced by ADK knockdown in HASMCs. Therefore, the decreased AAA formation in Ang II-infused ADKΔVSMC/Apoe−/− mice is likely mediated, at least in part, by decreased expression of Il1b, Il6, and Mmp3. In addition, AAA formation is also associated with induction of proangiogenic cytokines and increased adventitial neovascularization, and pharmacological inhibition of VEGFA or its receptor suppressed AAA formation.33–35 Thus, decreased VEGFA expression in ADK-knockdown HASMCs, and decreased vascularization in the aortic wall, may also contribute to the reduced aortic inflammation and AAA formation in Ang II-infused ADKΔVSMC/Apoe−/− mice. Besides, one early study has shown that SMC proliferation is enhanced in human AAAs.36 Recently, single-cell transcriptome analysis also revealed that among the four types of SMC clusters in human aortic aneurysmal tissue, two types of SMCs exhibited proliferative properties and highly expressed synthetic SMC marker genes; however, unlike traditional synthetic SMCs, these proliferating SMCs did not show low expression of contractile genes.37 Therefore, ADK inactivation may protect against AAA via reduced inflammatory response and pathological proliferation of SMCs without effects on the expression of SMC contractile genes.
In vitro studies in VSMCs demonstrated that knockdown of ADK decreased TNFA-induced proinflammatory gene expression in association with reduced H3K4me3 promoter occupancy of inflammatory genes including IL1B, IL6, VEGFA, and MMP3. In human AAA, inflammatory pathways are broadly activated, the most significant of which was reported to be TNFA signalling via NF-κB pathway.8 Indeed, TNFA promoted the expression of inflammatory genes, including IL1B, IL6, VEGFA, and MMP3, in both human and murine aortic VSMCs. Moreover, an enhanced SAM:SAH ratio induced by TNFA may result in increased levels of H3K4me3 in the early state of Ang II-induced AAA.38 In ADK-knockdown VSMCs, increased intracellular adenosine did not affect the phosphorylation of P65, ERK, JNK, or P38, but inhibited the flux of the methionine cycle and SAM-dependent transmethylation reactions, resulting in low level of H3K4me3 on the promoters of inflammatory genes and reduced gene expression.39–42 The decreased H3K4me3 observed in this study is in line with previous studies showing decreased levels of H3K4me3 in Ado1-deficient yeast and ADK-deficient endothelial cells with proinflammatory stimulation.17,43 Moreover, the inhibition of H3K4 trimethyltransferase SMYD3 decreases the expression of inflammatory genes and MMPs through decreased binding of H3K4me3 to the promoter regions of the target genes in VSMCs.44 Our findings are also in agreement with recent studies that showed that blocking the methionine cycle in immune cells decreases proinflammatory responses through down-regulation of histone methylation.21,22 Interestingly, excessive methionine supplementation exacerbates the development of AAA,45 which may result from an increased methionine cycle flux that drives proinflammatory gene expression through the hypermethylation of H3K4me3.
Other possible mechanisms, such as the down-regulation of homocysteine in ADK-deficient VSMCs,46 may also contribute to the alleviation of vascular inflammation and AAA formation in Ang II-infused ADKΔVSMC/Apoe−/− mice. Previous studies have shown that homocysteine directly interacts and activates the Ang II type I receptor to promote vascular injury and aneurysm formation.47–49 Therefore, VSMC ADK deficiency may decrease intracellular homocysteine levels, which could in turn contribute to decreased inflammation in aortas observed in the present study. In addition, although our data suggest that the anti-inflammatory effects of ADK knockdown in VSMCs are independent of adenosine receptors in vitro, we cannot rule out the possibility that VSMC adenosine receptors are involved in vivo, or that increased adenosine in ADK-deficient VSMCs can activate adenosine receptors on other cell types in the aorta through paracrine effects in vivo. Our recent study showed that ADK knockdown in VSMCs increased the levels of extracellular adenosine.50 Using the elastase-induced AAA model, Bhamidipati et al.51 reported that Adora2a knockout or inhibition exacerbated aneurysm formation in mice, while ADORA2A agonist attenuated AAA formation in part by inhibiting immune cell recruitment. Hence, it remains possible that in ADKΔVSMC/Apoe−/− mice, adenosine released from VSMCs into the extracellular milieu activates adenosine receptors on immune cells to inhibit AAA formation.
The expression of ADK in mouse arterial aneurysms remains relatively unchanged. Recent research focusing on human calcified arteries has revealed a connection between ADK and vascular calcification.52 Consequently, future investigations should aim at comparing ADK expression and activity in VSMCs from normal human arteries and arterial aneurysms. Despite the potential similarity in expression/activity levels observed in the aforementioned VSMCs, the substantial anti-aneurysm effect should not dismiss the therapeutic potential of ADK inhibition. In the field of medicine, therapeutic targets may not always align precisely with pathogenic genes or molecules, as illustrated by the example of SGLT2 inhibitors in the treatment of type 2 diabetes.53 Therefore, further exploration is warranted to uncover the potential efficacy of ADK inhibition in mitigating arterial aneurysms.
Abdominal aortic aneurysm (AAA) is associated with marked aortic inflammation leading to progressive aortic expansion. Here, we report that increased intracellular adenosine, through adenosine kinase (ADK) inhibition, reduces aortic inflammation, elastin fragmentation, and neovascularization, thereby modulating AAA formation. These effects are independent of adenosine receptors and are associated with decreased S-adenosylmethionine-dependent trimethylation of histone H3K4. Pharmacological augmentation of intracellular adenosine with an ADK inhibitor could be an effective medical therapy for AAA.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
J.X., Z.L., Q.Y., K.O., M.H., H.K., R.M., N.W., and Y.H. designed the research. J.X., Z.L., Q.Y., Q.M., Y.Z., Y.C., D.Z., and T.L. performed experiments. J.X., Z.L., Q.Y., G.Z., H.S., J.Z., and Y.H. analysed data. J.X., D.F., C.M., R.M., N.W., and Y.H. wrote and revised the article. J.X., M.H., H.K., N.W. and Y.H. provided the reagents or materials and participated in experimental design. Y.H. had primary responsibility for the final content. All authors read and approved the final article.
Acknowledgements
We are grateful for the support of the Metabolomics Facility in the National Protein Science Technology Center of Tsinghua University, Beijing, China.
Funding
This work was supported in part or in whole by grants from the National Natural Science Foundation of China (82100506 and 82070461); the Guangdong Basic and Applied Basic Research Foundation (2023A1515010816 and 2020A1515010010); the China Postdoctoral Science Foundation (2020M680003); the Shenzhen Fundamental Research Program (JCYJ20190808155801648 and GXWD20201231165807007-20200818123312001); the Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2023SHIBS0004); the American Heart Association (19TPA34910043 and 22TPA968801); and the National Institutes of Health (R01HL142097, R01EY030500, R01EY033369, and R01EY033737).
Data availability
The data relating to this article are incorporated into the article and its Supplementary material.
References
Author notes
Jiean Xu, Zhiping Liu and Qiuhua Yang have contributed equally to this article.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}