





Abstract
Well-controlled mitochondrial homeostasis, including a mitochondria-specific form of autophagy (hereafter referred to as mitophagy), is essential for maintaining cardiac function. The molecular mechanism mediating mitophagy during pressure overload (PO) is poorly understood. We have shown previously that mitophagy in the heart is mediated primarily by Atg5/Atg7-independent mechanisms, including Unc-51-like kinase 1 (Ulk1)-dependent alternative mitophagy, during myocardial ischaemia. Here, we investigated the role of alternative mitophagy in the heart during PO-induced hypertrophy.
Mitophagy was observed in the heart in response to transverse aortic constriction (TAC), peaking at 3–5 days. Whereas mitophagy is transiently up-regulated by TAC through an Atg7-dependent mechanism in the heart, peaking at 1 day, it is also activated more strongly and with a delayed time course through an Ulk1-dependent mechanism. TAC induced more severe cardiac dysfunction, hypertrophy, and fibrosis in ulk1 cardiac-specific knock-out (cKO) mice than in wild-type mice. Delayed activation of mitophagy was characterized by the co-localization of Rab9 dots and mitochondria and phosphorylation of Rab9 at Ser179, major features of alternative mitophagy. Furthermore, TAC-induced decreases in the mitochondrial aspect ratio were abolished and the irregularity of mitochondrial cristae was exacerbated, suggesting that mitochondrial quality control mechanisms are impaired in ulk1 cKO mice in response to TAC. TAT-Beclin 1 activates mitophagy even in Ulk1-deficient conditions. TAT-Beclin 1 treatment rescued mitochondrial dysfunction and cardiac dysfunction in ulk1 cKO mice during PO.
Ulk1-mediated alternative mitophagy is a major mechanism mediating mitophagy in response to PO and plays an important role in mediating mitochondrial quality control mechanisms and protecting the heart against cardiac dysfunction.
1. Introduction
Heart failure is a major health issue world-wide. Despite the recent development of medical treatments targeting distinct mechanisms contributing to the progression of heart failure, the 5-year survival rate of heart failure patients remains modest.1 At the cellular level, cardiomyocytes (CMs) in end-stage heart failure patients commonly exhibit mitochondrial dysfunction, with a reduced rate of ATP synthesis and increases in oxidative stress, that can lead to cell death.2
Autophagy is an important mechanism of cellular degradation characterized by the presence of double membrane vesicles, and a major contributor to the cellular protein and organelle quality control mechanisms.3 Previous studies have shown that autophagy is a homeostatic mechanism that is essential for maintaining cardiac function at baseline and in response to stress4: down-regulation of autophagy induces baseline cardiac dysfunction and exacerbates heart failure in response to pressure overload (PO)5 and volume overload.6 Moreover, activation of mitophagy, a mitochondria-specific form of autophagy, is critical for maintaining mitochondrial function and cardiac performance in the presence of PO.7 Importantly, stress-induced activation of mitophagy is often transient and the heart rapidly goes into failure once mitophagy is inactivated.7 Increasing lines of evidence suggest, however, that reactivation of mitophagy is a promising modality to restore cellular function of CMs and retard the progression of heart failure in response to stress in the heart.7 Thus, understanding the molecular mechanism through which mitophagy is activated should provide us with an important clue to controlling the level of mitophagy in the heart during stress, including PO.
Previous studies of mitophagy have focused almost exclusively on the Atg5–Atg7–LC3- and Pink1–Parkin-dependent conventional mechanism of mitophagy.8,9 However, increasing lines of evidence suggest that mitophagy is mediated through multiple mechanisms,10 including Atg5–Atg7–LC3- or Pink1–Parkin-independent mechanisms.11,12 Furthermore, mitophagy in Mouse Embryonic Fibroblast (MEF) cells is predominantly activated though a Rab9-dependent alternative pathway.13 In fact, we have shown that alternative mitophagy activated through an Atg7-independent and Unc-51-like kinase 1 (Ulk1)-, Rab9-, Rip1-, and Drp1-dependent mechanism, rather than Atg7-, LC3-, or Parkin-dependent mitophagy, is the major mechanism of mitophagy in the heart in response to myocardial ischaemia.12,14 Furthermore, mitophagy is activated in the heart during PO even after conventional autophagy is down-regulated,7 suggesting that mitophagy may be activated through unconventional mechanisms, including alternative mitophagy.
The present study thus aimed to clarify the molecular mechanism of mitophagy that is activated during PO, using ulk1 cardiac-specific knock-out (cKO) mice with transverse aortic constriction (TAC) as a model of PO in the heart. The goals in this study were to (i) demonstrate activation of alternative mitophagy during PO, and (ii) clarify the functional significance of Ulk1-dependent alternative mitophagy in mitochondrial function and cardiac function in the presence of PO.
2. Methods
2.1 Mouse models
Cardiac-specific Atg7 or Ulk1 knockout (KO) mice were generated by crossing Atg7flox/flox or Ulk1flox/flox mice with Myh6-Cre transgenic mice.14 Transgenic mice expressing Myh6-driven Mito-Keima were used for mitophagy assays. Pathogen-free mice were housed in a temperature-controlled environment with 12-h light/12-h dark cycles and were given food and water ad libitum. We used both male and female mice in this study. Animal studies were performed according to the guidelines from the NIH Guide for the Care and Use of Laboratory Animals. All experiments involving animals were approved by the Rutgers New Jersey Medical School’s Institutional Animal Care and Use Committee (protocol number: 201900133).
2.2 CM cultures
Primary cultures of CMs were prepared from 1-day-old Charles River Laboratories/Wistar Institute BR-Wistar rats (Harlan Laboratories, Somerville, NJ, USA) as described previously.6 A CM-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Isolation and culture of adult mouse CMs (AMCMs) were conducted using a Langendorff-free method according to a protocol described previously.15 This method utilizes direct needle perfusion of the left ventricle (LV) ex vivo, which allows isolation, separation, and culture of AMCMs. These myocytes were used for mitophagy assays within 1 day after isolation.
2.3 Transverse aortic constriction
The TAC model was used to assess the effect of PO on the development of heart failure in mice.16 Mice at 10–16 weeks of age (body weight: 25–35 g) and fed a normal chow diet were randomly divided into two groups, PO with TAC or sham operation. Age- and sex-matched mice were used for the control group. The background of all mice was C57BL/6J. Backcross was carried out more than five generations. Mice were anaesthetized with pentobarbital sodium (60 mg/kg) or isoflurane inhalation. For isoflurane inhalation, mice were put into an induction chamber connected to an isoflurane vaporizer and anaesthetized with 4–5% isoflurane. When the animal was deeply anaesthetized, it was intubated, connected to a ventilator, and mechanically ventilated while the surgical plane of anaesthesia was maintained by continuous inhalation of 1–2% isoflurane during surgery. The left side of the chest was opened at the second intercostal space. Aortic constriction was performed by ligation of the transverse thoracic aorta between the innominate artery and left common carotid artery with a 27-gauge needle using a 7-0 prolene suture. Sham operation was performed without constricting the aorta. We followed the development of cardiac hypertrophy, heart failure, and changes in histology and signalling mechanisms at multiple time points (from 1 day to 8 weeks). Successful application of TAC was confirmed by transverse aortic velocity above 4 m/s, as evaluated with Doppler echocardiography. Mice that died due to heart failure were included in the survival analysis but were excluded from the assessment of cardiac function and histology. There were no unexpected adverse events during the procedure. All operations and analyses were performed with randomization in a blinded manner with regard to mouse genotype.
2.4 Echocardiography
Mice were anaesthetized using 12 μL/g body weight of 2.5% Avertin (Sigma-Aldrich), since isoflurane occasionally lowers blood pressure and induces irregular heartbeat. Echocardiography was performed using ultrasonography (Visualsonics Vevo 770, Toronto, Canada) with a 30-MHz ultrasound transducer. Two-dimensional guided M-mode measurements of LV internal diameter were obtained from at least three beats and then averaged. LV end-diastolic dimension was measured at the time of the apparent maximal LV diastolic dimension, and LV end-systolic dimension was measured at the time of the most anterior systolic excursion of the posterior wall. All analyses were performed in a blinded manner with regard to mouse genotype.
2.5 Histological analysis
The heart was isolated, fixed with 4% phosphate-buffered paraformaldehyde (PFA), embedded in paraffin, and cut into 10 μm-thick sections. Serial sections of the heart were stained with wheat germ agglutinin (WGA) for analysis of CM cross-sectional area (CSA) and Picrosirius Red for analysis of myocardial fibrosis. CSA was obtained by tracing the outlines of 100–200 random CMs with a clear nuclear image from the LV. These analyses were performed in a blinded manner using a fluorescence microscope (Eclipse E800, Nikon) and Image J Software (NIH, Bethesda, MD, USA).
2.6 Immunostaining assay
CMs were seeded in gelatin-coated four-well chambers and treated with Ad-shControl or Ad-shUlk1. After 5 days, cells were subjected to 6 h of glucose deprivation and then incubated with 1 μM MitoTracker Red CMXRos (CST, #9082) for 30 min. Cells were washed in phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde for 10 min and then permeabilized for 10 min with 0.1% Triton X-100. Blocking was performed with 3% bovine serum albumin in PBS for 1 h. Subsequently, cells were incubated with anti-p62 antibody (Abnova, H00008878-M01) overnight at 4°C. After washing with PBS/T, they were incubated with Alexa Fluor 488 (Invitrogen, #A-11034) for 1 h at room temperature in a dark chamber. Cells were washed with PBS/T, mounted on slides, and examined by confocal microscopy.
2.7 Evaluation of mitochondria inside Rab9-positive vesicles
In order to evaluate the engulfing of mitochondria by Rab9-positive vesicles, we stained heart sections with anti-Rab9 (Cell Signaling Technology, #5118) and anti-TOMM20 (Sigma-Aldrich, WH0009804M1) antibodies. Rab9-positive vacuoles that colocalized with mitochondria (TOMM20) in each field were counted and divided by the number of DAPI-positive nuclei using ImageJ software.
2.8 Keima with mitochondrial targeting signal
For observation of mitophagy in the heart, transgenic mice with CM-specific expression of Mito-Keima (Tg-Mito-Keima) were used.14 For the detection of Mito-Keima signals in the heart, hearts were excised, washed with PBS, and fixed with 4% PFA (pH 9.5) for 30 min, as described previously.14 Immediately after the fixation, the heart sections were prepared by vibratome and subjected to confocal imaging. All images were acquired using living cells at 37°C by Nikon A1RSI confocal microscopy. Images were acquired by an unbiased observer blinded to mouse genotype and experimental conditions.
2.9 TAT-Beclin 1
The L17 type of TAT-Beclin 1 amino acid sequence was YGRKKRRQRRGGVWNATFHIWHD. The control peptide, TAT-Scrambled (TS), consisted of the TAT protein transduction domain, a GG linker, and a scrambled version of the C-terminal 11 amino acids of TAT-Beclin 1 (YGRKKRRQRRGGWNHADHTFVWI). For in vivo experiments, D-amino acid peptides were dissolved in H2O and stored at –80°C until use. For induction of autophagy in vitro, CMs were washed with PBS (–) and treated with peptides dissolved in OPTI-MEM acidified with (0.15% v/v) 6 N HCl.17 For in vivo induction, mice were injected with TS or TAT-Beclin 1 at 5 mg/kg through the jugular vein as described previously.7
2.10 Electron microscopy
Electron microscopy (EM) was performed in a blinded manner as described previously.14 Heart tissue was removed from the animal and quickly rinsed in PBS. The tissue was placed in a Petri dish with 0.5% glutaraldehyde and 0.2% tannic acid in PBS, diced into 2 mm cubes and then transferred to modified Karnovsky’s fixative [4% formaldehyde and 2.5% glutaraldehyde containing 8 mmol/L CaCl2 in 0.1 mol/L sodium cacodylate buffer (pH 7.4)]. Samples were washed with PBS and post-fixed in 1% osmium tetroxide in 0.1 mol/L sodium cacodylate buffer (pH 7.4) for 1 h to produce osmium black. Samples were then dehydrated through a graded series of ethanol washes and embedded in Epon/SPURR resin (EM Science) that was polymerized at 65°C overnight. Sections of heart tissues were prepared with a diamond knife on a Reichert-Jung Ultracut-E Ultramicrotome and stained with UrAc (20 min) followed by 0.2% lead citrate (2.5 min). Images were photographed with a Jeol JEM-1200EX electron microscope.
2.11 Real-time quantitative polymerase chain reaction
Total RNA was extracted from mouse hearts using the RNeasy Plus Universal Kit (QIAGEN). Total RNA was converted to cDNA using PrimeScript RT Master Mix (Takara). Primer sequences are shown below.
Atrial natriuretic peptide (ANP)-For: TCGGAGCCTACGAAGATCC; ANP-Rev: TTCGGTACCGGAAGCTGTT; brain natriuretic peptide (BNP)-For: GGTCCAGCAGAGACCTCAAA; BNP-Rev: CAACTTCAGTGCGTTACAGCC.
2.12 Immunoblot analyses
The methods used for preparation of cell lysates from in vitro and in vivo samples and for immunoblot analyses have been described previously.14 The antibodies used include LC3 (Novus, NB100-2220), p62 (Abnova, H00008878-M01), GAPDH (Cell Signaling, 2118S), αTubulin (Sigma-Aldrich, T6199), Atg7 (Cell Signaling Technology, #8558), RIP1 (Cell Signaling Technology, #3493), RIP3 (Cell Signaling Technology, #95702), Rab9 (Cell Signaling Technology, #5118), Ulk1 (Millipore Sigma, A7481), Drp1 (Cell Signaling Technology, #5391), and COXIV (Cell Signaling Technology, #4850). Phospho-Rab9(S179) antibody has been described previously.14
2.13 Immunoprecipitation
CMs were lysed with IGEPAL CA-630 buffer [50 mmol/L Tris-HCl (pH 7.4), 1% IGEPAL CA-630, 10 mmol/L EDTA, 150 mmol/L NaCl, 50 mmol/L NaF, 1 μmol/L leupeptin, and 0.1 μmol/L aprotinin] containing phosphatase inhibitor. For IP of YFP-Rab9, the GFP-Trap-A Kit (Chromotek) was used according to the manufacturer’s instructions. For IP of HA-Drp1, the Pierce HA Tag IP Kit (Thermo Fisher Scientific) was used according to the manufacturer’s instructions.
2.14 Scientific justification
Avertin Tribromoethanol (Avertin) is appropriate for short procedures in mice. Because Avertin has few side effects and the haemodynamic analysis and echocardiography do not require a long time,18,19 we have applied Avertin to anaesthetize mice for haemodynamic measurement and echocardiology in this study. Avertin is known to have much less of a cardio-suppressive effect, but Ketamine/Xylazine or Pentobarbital is known to suppress cardiac function and heart rate. Due to the cardiovascular focus of this study, it is important to keep cardiac function as close to the physiological condition as possible during experiment.
2.15 Statistics
Statistical analyses were conducted using the GraphPad Prism 9.0 software programme. Data are presented as line graphs expressing mean ± standard deviation for the indicated number of experiments or as dot-plots expressing mean ± standard error of the mean for the indicated number of mice. The difference in means between two columns or multiple columns was evaluated using the two-tailed unpaired Student’s t-test (for two columns), one-way ANOVA with Tukey’s post hoc tests (for multiple comparisons), or one-way ANOVA with Dunnett’s post hoc test (for multiple comparisons to control). The difference in means between more than two groups was evaluated using two-way ANOVA with Tukey’s or Šídák’s post hoc test, as appropriate. A P value of less than 0.05 was considered statistically significant. The reported significance values are always two-sided. Normal distribution of data was confirmed by the Shapiro–Wilk’s test. Non-normally distributed data were evaluated by the non-parametric Mann–Whitney U test after confirming equality of ranks variances.
3. Results
3.1 Mitophagy is acutely up-regulated by both Atg7- and Ulk1-dependent pathways during PO in the heart
We have shown previously that, although general autophagy is activated transiently within 6 h after TAC and inactivated within 24 h, mitophagy, evaluated with electron microscopic (EM) analyses, is activated with a distinct time course in the heart, peaking at 3–7 days after TAC.7 Activation of mitophagy after inactivation of conventional autophagy suggests that mitophagy may be activated by unconventional mechanisms, including alternative mitophagy.14 To better understand the underlying molecular mechanism mediating mitophagy in the heart during PO, we examined the involvement of conventional and alternative mitophagy.12 To this end, we used mice with cardiac-specific knockout of Atg7 (atg7cKO), in which the conventional mechanism of autophagy and mitophagy is fully abolished, and mice with cardiac-specific knockout of Ulk1 (ulk1cKO), in which conventional mitophagy is little affected, whereas alternative mitophagy is almost fully abolished.7 EM analyses showed that mitophagy/autophagy was observed after 1 day of TAC in both control and ulk1cKO but not in atg7cKO mice, suggesting that mitophagy/autophagy at this time point is activated through the conventional Atg7-dependent mechanism of autophagy (Figure 1A and B). The activity of mitophagy/autophagy was calculated by counting the number of autophagosomes containing mitochondria, indicated by arrows, and autolysosomes, indicated by arrowheads, under EM observation (Figure 1A). The activity of mitophagy/autophagy was further increased 3 days after TAC in both control and atg7cKO mice, but not in ulk1cKO mice, suggesting that Ulk1-dependent mitophagy/autophagy is predominantly activated 3 days after TAC (Figure 1A and B). To specifically examine mitophagy activity, we also evaluated the level of mitophagy using cardiac-specific Mito-Keima mice crossed with atg7cKO (Keima–atg7cKO), ulk1cKO (Keima–ulk1cKO), or wild-type (WT) (Keima) mice.14 These mice underwent TAC and the area of dots with high 560 nm/440 nm ratios (acidic dots), reflecting autophagosomes containing mitochondria fused with lysosomes,14 were analysed. The area of acidic dots was slightly increased in both WT and ulk1cKO but not in atg7cKO mice 1 day after TAC, suggesting that modest mitophagy activation 1 day after TAC occurs predominantly through an Atg7-dependent mechanism. However, the area of acidic dots was further increased in WT and atg7cKO mice but less so in ulk1cKO mice 3 and 5 days after TAC (Figure 1C and D). The differences in % of mitophagy/autophagy activity in Figure 1A and the area of acidic dots in Figure 1C between WT and atg7cKO mice, reflecting Atg7-dependent mitophagy, were higher than the difference between WT and ulk1cKO mice 1 day after TAC. However, the difference between WT and ulk1cKO mice became significantly greater than the difference between WT and atg7cKO mice 3 and 5 days after TAC, indicating that Ulk1-dependent alternative mitophagy is the major form of mitophagy at 3–5 days (Supplementary material online, Figure S1A and B). On the other hand, as we previously reported,7 LC3-II was rapidly and transiently up-regulated in response to TAC in WT and ulk1cKO mouse hearts but not in atg7cKO mouse hearts. Interestingly, LC3-II was increased in a more sustained manner in ulk1cKO mouse hearts (Supplementary material online, Figure S1C). Taken together, these observations suggest that mitophagy is induced within 1 day after TAC through conventional autophagy but is induced via an alternative mechanism, namely the Ulk1-dependent pathway, thereafter in response to PO.
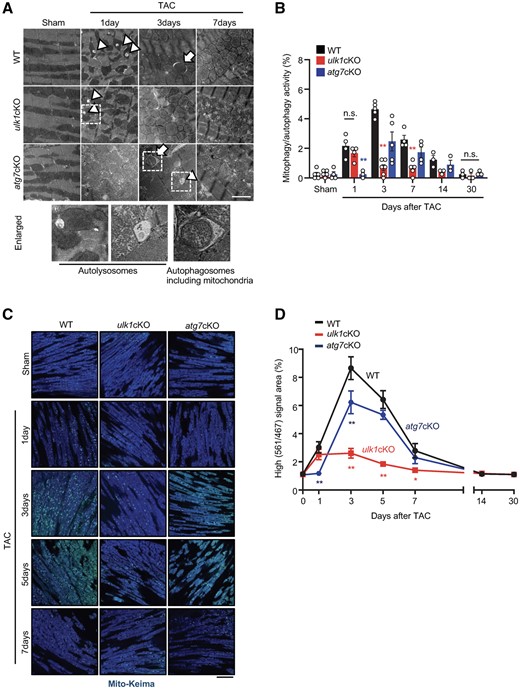
Ulk1 plays a major role in mediating mitophagy in response to pressure overload. (A and B): 3–4-month-old WT, atg7cKO, and ulk1cKO mice were subjected to either sham operation or TAC for 1, 3, 7, 14, and 30 days. Representative EM images are shown in (A) (scale bar, 2 µm). Mitophagy was quantified. Mean values ± S.E., n = 3–6; *P < 0.05, **P < 0.01 vs. WT for each time course, each value was the average of nine different images from each mouse, n.s., no significance. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (B). Arrows indicate autophagosomes containing mitochondria; arrowheads indicate autolysosomes. % of mitophagy/autophagy activity was calculated by dividing the number of autophagosomes containing mitochondria + the number of autolysosomes by areas (1000 µm2). (C and D): 3–4-month-old WT/Mito-Keima, atg7cKO/Mito-Keima, and ulk1cKO/Mito-Keima mice were subjected to either sham operation or TAC for 1, 3, 5, 7, 14, and 30 days. Lysosomal degradation of mitochondria was detected with Mito-Keima. The bright signal corresponding to a high 560 nm/440 nm ratio indicates lysosomal localization of Mito-Keima, representing mitophagy. Representative images are shown in (C) (scale bar, 100 μm). High (561 nm/467 nm) signal areas of Mito-Keima dots/total microscopic areas are shown in (D). Mean values ± S.E., *P < 0.05, **P < 0.01 vs. WT for each time course. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test.
3.2 ulk1cKO mice exhibited severe cardiac dysfunction in response to TAC
The results presented above suggest that Ulk1 plays an important role in mediating mitophagy more prominently than Atg7 in response to PO. In order to investigate the role of endogenous Ulk1 in the heart during PO, control and ulk1cKO mice were subjected to either TAC or sham operation and cardiac function was evaluated 1–30 days after TAC. Although TAC decreased echocardiographically evaluated left ventricular ejection fraction (LVEF) 14 and 30 days after TAC in WT mice, TAC significantly decreased LVEF as early as 3 days after TAC and thereafter compared to sham operation in ulk1cKO mice (Figure 2A and B). LVEF was significantly smaller in ulk1cKO mice than in WT mice 3 days after TAC and thereafter (Figure 2A and B). Heart weight/tibia length (HW/TL) was significantly greater in ulk1cKO mice than in WT mice 7 days after TAC and thereafter (Figure 2C and D). Lung weight/tibia length (LW/TL), an index of lung congestion, was also slightly increased 3 and 7 days after TAC and was drastically greater 30 days after TAC in ulk1cKO mice than in WT mice (Supplementary material online, Figure S2A). LV end-diastolic pressure was significantly increased 3 days after TAC in ulk1cKO mice compared to in WT mice (Supplementary material online, Figure S2B). These results suggest that endogenous Ulk1 plays an essential role in maintaining cardiac function as early as 3 days after TAC, when Ulk1-dependent mitophagy reaches a peak. CM CSA, evaluated with WGA staining, in ulk1cKO mice was similar to that in control mice under basal conditions but was significantly greater in ulk1cKO mice than in control mice after 7 days of TAC (Figure 2E and F). The mRNA levels of ANP and BNP were significantly higher in ulk1cKO hearts than in control mouse hearts 1 day after TAC and thereafter (Supplementary material online, Figure S2C and D). The percentage of myocardial fibrosis was significantly greater in ulk1cKO than in control mice 7 days after TAC (Figure 2G and H). These results suggest that TAC-induced cardiac hypertrophy and cardiac dysfunction are exacerbated in ulk1cKO compared to WT mice.
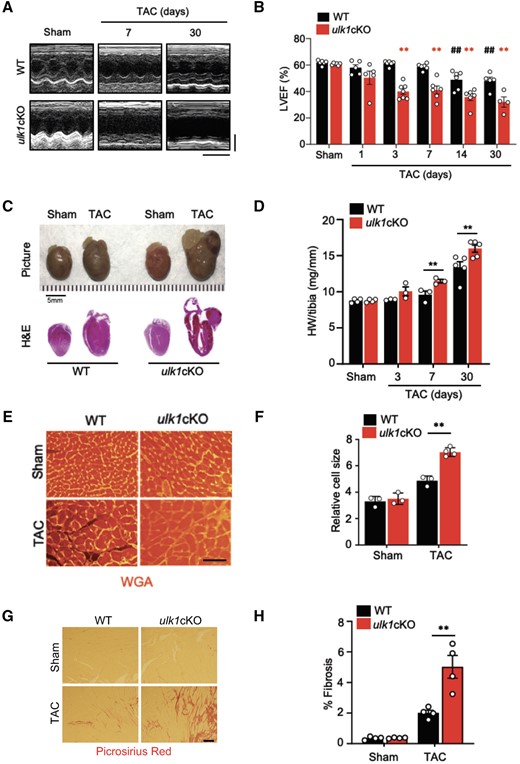
Down-regulation of Ulk1 exacerbates cardiac dysfunction after TAC. Three- to 4-month-old WT and ulk1cKO mice were subjected to either sham operation or TAC for 1, 3, 7, 14, and 30 days. (A) Representative echocardiographs. Scale bars, vertical 2.5 mm and horizontal 250 ms. (B) % of LVEF was evaluated by echocardiography. **P < 0.01 vs. ulk1cKO sham; ##P < 0.01 vs. WT sham. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test. (C) Gross morphology and heart sections of WT and ulk1cKO mice stained with haematoxylin and eosin (H&E) staining. Scale bars, 5.0 mm. (D) Heart weight to tibia length (HW/tibia) ratio. *P < 0.05 vs. WT at each time point. Mean values ± S.E. are shown. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test. (E and F) Left ventricles were fixed, sectioned, and stained with Wheat Germ Agglutinin (WGA). Scale bars, 500 µm (E). Relative cell size was measured (F). Mean values ± S.E., **P < 0.01. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test. (G and H) Left ventricles were fixed, sectioned, and stained with Picrosirius red. Scale bars, 200 µm (G). Picrosirius red positive areas were measured (H). Mean values ± S.E., n = 4 each; **P < 0.01. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test.
3.3 Ulk1/Rab9-dependent alternative mitophagy is dominantly activated during PO
We have shown previously that LC3-dependent autophagy is inactivated within a day after PO. Thus, the Ulk1-dependent Atg7-independent mitophagy observed 3–7 days after PO should be LC3-independent. We hypothesized that mitophagy activated 3–7 days after PO is mediated through alternative mitophagy, which is mediated through an LC3-independent mechanism.14 One of the important characteristics of alternative mitophagy is association of Rab9 with autophagosomes containing mitochondria. Confocal microscopic analyses showed that there was a significant increase in Rab9 puncta co-localized with mitochondria stained with anti-TOMM20 antibody 3 days after TAC compared to sham operation (Figure 3A and B). As we previously reported, LC3-dependent conventional autophagy was activated within 1 day after TAC but it was decreased thereafter (Supplementary material online, Figure S3A).7 Alternative mitophagy is accompanied by increased phosphorylation of Rab9 at Ser179, which is mediated through Rip1.14 Although the level of Rab9 was not significantly altered in response to PO, phosphorylation of Rab9 at S179 was induced 3 and 7 days after TAC compared to sham operation (Figure 3C–E). The levels of Rip1 and Rip3 were also increased in the heart, peaking around 7 days after TAC (Figure 3C, F, and G). Rip3 interacts with Ulk1 to regulate Ulk1-dependent alternative autophagy in mouse embryonic fibroblasts.20 We have shown previously that Rip1 interacts with Rab9 in an Ulk1-dependent manner in CMs. Here we found that Rip3, but not p38αMPAK or p42/44MAPK, also interacts with Rab9 in cultured CMs, suggesting that Rip1/3 is involved in alternative mitophagy in CMs (Supplementary material online, Figure S3B). These results are consistent with the notion that Ulk1/Rab9-dependent alternative mitophagy is activated at 3–7 days of PO in the heart.
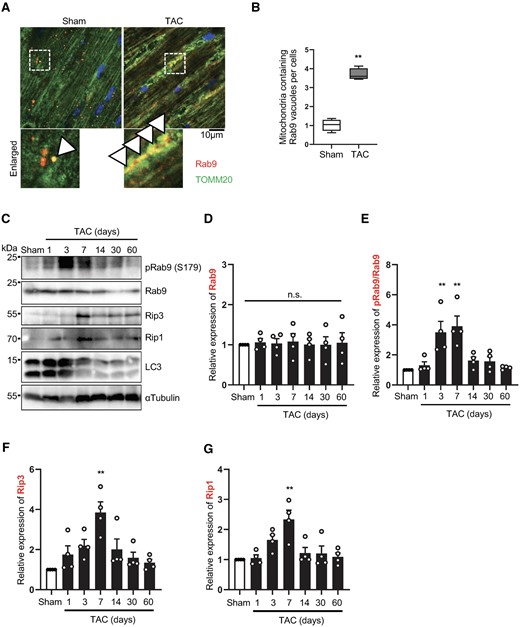
Alternative autophagy is induced during pressure overload in the heart. Three-month-old WT mice were subjected to either sham operation or TAC for 1, 3, 7, 14, 30, and 60 days. Hearts were analysed by confocal microscopy (A and B) or Western blotting (C–G) to assess alternative mitophagy. Representative fluorescence images of sham and 3-day TAC heart sections stained with anti-Rab9 antibody (red), anti-TOMM20 antibody (green), and DAPI (blue) (A). Arrowhead indicates co-localization of Rab9 and mitochondria. The graph shows quantification of double positive Rab9 and mitochondria dots (B). Mean values ± min to max., n = 4. Values were measured from four different slides per sample. **P < 0.01. Statistical analyses were conducted with unpaired Student’s t-test. Heart lysates were analysed by Western blotting using anti-Rip1, anti-Rip3, anti-phospho-Rab9 (S179), anti-Rab9, anti-LC3, and anti-αTubulin antibodies (C). The relative ratios of Rab9 to αTubulin (D), phospho-Rab9 to Rab9 (E), Rip1 to αTubulin (F), and Rip3 to αTubulin (G) were quantified. Mean values ± S.E. n = 4; **P < 0.01 vs. sham. Statistical analyses were conducted with one-way ANOVA followed by Dunnett’s multiple comparisons test.
3.4 LC3-dependent conventional mitophagy is preserved but Rab9-dependent alternative mitophagy is defective in ulk1cKO during PO
To determine the molecular mechanism of early cardiac dysfunction in ulk1cKO during PO, we first evaluated the level of conventional autophagy during TAC in ulk1cKO mice. To this end, we compared the levels of LC3-dependent autophagy 12 h after TAC, the time point when conventional autophagy reaches a peak,7 in control and ulk1cKO mice. The level of LC3-II was significantly increased after TAC in both control and ulk1cKO mice (Figure 4A and B). The level of p62, a protein known to be degraded by autophagy, was significantly decreased by TAC in both control and ulk1cKO hearts (Figure 4A and C). EM analysis showed that the number of autolysosomes is significantly increased in both control and ulk1cKO mice 12 h after TAC (Figure 4D and E). These results suggest that conventional autophagic flux is activated in response to TAC at this time point in both control and ulk1cKO mice and that down-regulation of Ulk1 does not affect conventional autophagy during PO.
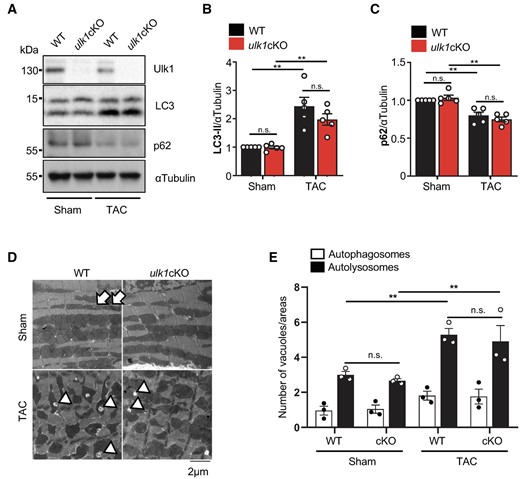
Conventional autophagy is preserved in ulk1cKO during the early stage of TAC. (A–E) 3–4-month-old WT and ulk1cKO mice were subjected to either sham operation or TAC for 12 h. Heart lysates were analysed by Western blotting using anti-Ulk1, anti-LC3, anti-p62, and anti-αTubulin antibodies (A). The relative ratios of LC3-II to αTubulin (B) and p62 to αTubulin (C) were quantified. Mean values ± S.E., n = 5; **P < 0.05. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test. (D and E) Samples from hearts of mice subjected to TAC for 12 h were subjected to electron microscopic analysis. Autophagosomes (APs, arrows) and autolysosomes (ALs, arrowheads) are indicated (D). The number of cytoplasmic APs and ALs was counted. Mean values ±S.E., n = 3; each value was obtained from 10 different pictures. *P < 0.05. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (E).
We next evaluated alternative mitophagy in ulk1cKO mice during PO. Although the protein levels of Rip1 and Rip3 were increased in response to TAC in control hearts, they were not increased in ulk1cKO hearts (Figure 5A; Supplementary material online, Figure S4A and B). Although the ratio of S179 phosphorylated Rab9/total Rab9 was increased in response to TAC in control hearts, it was not increased in ulk1cKO hearts (Figure 5A and B). We have shown previously that the Ulk1/Rab9/Rip1 complex phosphorylates Drp1 at Ser616, thereby inducing mitochondrial translocation of Drp1 and alternative mitophagy in response to ischaemic stress in the heart.14 In order to test whether mitochondrial translocation of Drp1 is affected in ulk1cKO mice, control and ulk1cKO heart lysates were subjected to subcellular fractionation. In control hearts, the level of mitochondrial Drp1 was significantly increased 3 days after TAC, the time point when mitophagy reaches a peak. However, the increase in mitochondrial Drp1 in response to TAC was significantly attenuated in ulk1cKO hearts (Supplementary material online, Figure S5A and B). Since Ulk1-dependent phosphorylation of Rab9 at S179 and mitochondrial translocation of Drp1 are important features of alternative mitophagy,14 these results suggest that the mitophagy activated 3 days after TAC is Ulk1/Rab9-dependent alternative mitophagy. Next, we examined whether Ulk1 deficiency affects the localization of mitophagy receptors, such as p62. We evaluated mitochondrial localization of p62 following glucose deprivation, an intervention known to induce mitophagy in CMs. Colocalization of p62 and Mito-Red was increased by starvation, not only in control but also in Ulk1 knockdown conditions, in CMs (Supplementary material online, Figure S5D and E). In addition, the level of p62 in the mitochondrial fraction was also increased by TAC in both WT and ulk1cKO hearts (Supplementary material online, Figure S5C). These results suggest that the loss of Ulk1 does not affect the localization of mitophagy receptors during mitophagy in the heart.
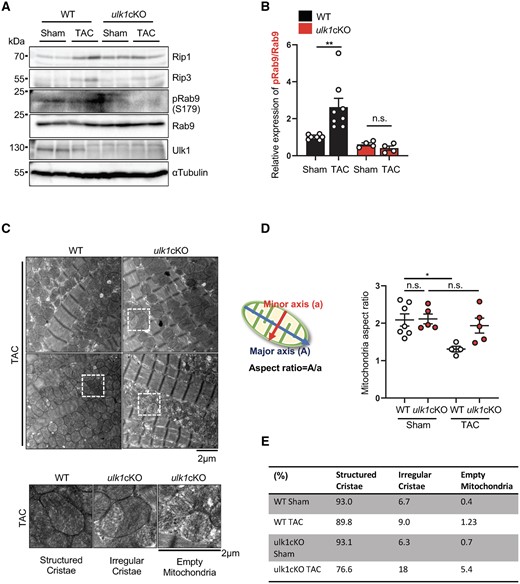
Alternative mitophagy is defective in ulk1cKO mice during TAC. (A–E) 3–4-month-old WT and ulk1cKO mice were subjected to either sham operation or TAC for 3 days. Heart lysates were analysed by Western blotting using anti-Rip1, anti-Rip3, anti-phospho-Rab9 (S179), anti-Rab9, anti-Ulk1, and anti-αTubulin antibodies (A). The relative ratios of phospho-Rab9 to Rab9 (B) were quantified. Mean values ± S.E., **P < 0.01. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test. (C–E) Samples from hearts of mice subjected to 3-day TAC were subjected to electron microscopic analyses. Representative EM images are shown in upper panel of (C). Enlarged mitochondria are shown in lower panel of (C). Mitochondria aspect ratio was quantified. Mean values ±S.E., *P < 0.05. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test (D). Mitochondria were categorized by the morphology of cristae and quantified. The percentage of structured or irregular cristae and empty mitochondria was quantified (n = 5 each group) (E).
Since mitochondrial Drp1 mediates fission, we evaluated whether mitochondrial fission is altered in ulk1cKO mice in response to TAC. We evaluated the aspect ratio of individual mitochondria by TEM analysis. The fused form of mitochondria has a greater mitochondria aspect ratio, whereas mitochondrial fission causes mitochondria to take on a round shape with a smaller aspect ratio.21 Although the average aspect ratio of a single mitochondrion was significantly decreased in control mice in response to TAC, it was not decreased in ulk1cKO mice (Figure 5C and D). Furthermore, the morphology of mitochondrial cristae was preserved in the WT mouse heart 3 days after TAC, but exhibited increased irregularity and reduced density in the ulk1cKO heart (Figure 5C and E). In addition, although dysfunctional mitochondria were increased, mitochondrial biogenesis, as indicated by Transcription Factor A, Mitochondrial (TFAM) expression, was not markedly changed in ulk1cKO heart during TAC (Supplementary material online, Figure S5F). These results are consistent with the notion that alternative mitophagy plays an essential role in maintaining mitochondrial quality during PO.
3.5 TAT-Beclin 1 activates both conventional and alternative mitophagy in the heart
To elucidate whether defective alternative mitophagy contributes to the early development of cardiac dysfunction in ulk1cKO mice, we conducted a rescue experiment using the well-established autophagy inducer TAT-Beclin 1. TAT-Beclin 1 stimulates autophagy by mobilizing endogenous Beclin 1 from the Golgi apparatus.17 We have shown previously that TAT-Beclin 1 induces autophagy and mitophagy in CMs.7 Furthermore, TAT-Beclin 1 induces mitophagy in the atg7cKO mouse heart,9 suggesting that it can also activate unconventional forms of mitophagy. We, therefore, first tested whether TAT-Beclin 1 activates alternative mitophagy. We confirmed that TAT-Beclin 1 stimulates autophagic flux in cultured CMs (Supplementary material online, Figure S6A and B). We found that TAT-Beclin 1 treatment also increases the level of S179 phosphorylated Rab9 in CMs in vitro (Supplementary material online, Figure S6C). In addition, TAT-Beclin 1 treatment significantly increased the number of fragmented mitochondria engulfed by GFP-Rab9-positive vacuoles in cultured CMs (Supplementary material online, Figure S6D and E). These results suggest that TAT-Beclin 1 activates alternative mitophagy in CMs.
Since TAT-Beclin 1 mobilizes endogenous Beclin 1, which, in turn, induces both conventional and alternative autophagy,22 we reasoned that TAT-Beclin 1 should be able to induce both conventional and alternative mitophagy in vivo. Thus, we next evaluated whether TAT-Beclin 1 activates mitophagy in the ulk1cKO heart. We first tested the effect of TAT-Beclin 1 in AMCMs isolated from Mito-Keima and Mito-Keima-ulk1cKO hearts. TAT-Beclin 1 treatment increased acidic dots of Mito-Keima not only in AMCMs isolated from Mito-Keima mice (Figure 6A and C) but also in AMCMs isolated from Mito-Keima-ulk1cKO mice, albeit to a lesser extent than in those from Mito-Keima mice (Figure 6B and C). TAT-Beclin 1 (5 µmol/L) activated mitophagy in AMCMs isolated from Mito-Keima-ulk1cKO mice, reaching a peak at 24 h (Figure 6D). To evaluate the effect of TAT-Beclin 1 in vivo, TAT-Beclin 1 (20 mg/kg) was intra-peritoneally injected into Mito-Keima and Mito-Keima-ulk1cKO mice. One day after TAT-Beclin 1 injection, heart slices were subjected to microscopic analyses for observation of Mito-Keima as described previously.14 The area of dots with high 560 nm/440 nm ratio was significantly increased by TAT-Beclin 1 in both control mouse and ulk1cKO mouse hearts (Figure 6E and F). These results suggest that TAT-Beclin 1 can activate mitophagy in ulk1cKO mouse hearts. Together with our previous results,7,9 these data show that TAT-Beclin 1 activates both conventional and alternative mitophagy. Thus, TAT-Beclin 1 may be able to rescue cardiac dysfunction in ulk1cKO mice in response to TAC by reactivating mitophagy if the primary cause of heart failure exacerbation in ulk1cKO mice is suppression of mitophagy.
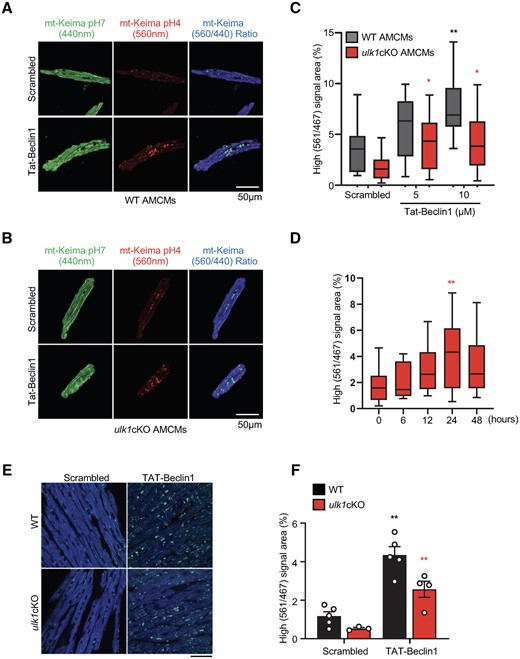
TAT-Beclin 1 peptide activates mitophagy in CMs and mouse heart. (A–D) Adult cardiomyocytes (CMs) were isolated from WT/Mito-Keima or ulk1cKO/Mito-Keima mice as described in Methods. Representative fluorescence images of adult CMs from WT/Mito-Keima (A) or ulk1cKO/Mito-Keima mice (B) (scale bar, 50 μm). The graph shows quantification of high (561 nm/467 nm) Keima signal area after TAT-Beclin 1 treatment (C and D). Mean values ± S.D., n = more than 10 cells per group; *P < 0.05, **P < 0.01 vs. scrambled of each group. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (C) or one-way ANOVA followed by Dunnett’s multiple comparisons test (D). (E and F) 3-month old WT/Mito-Keima and ulk1cKO/Mito-Keima mice were injected with TAT-Beclin 1 for 24 h. Representative images are shown in (E) (scale bar, 100 μm). High (561 nm/467 nm) signal areas of Mito-Keima dots/total microscopic areas are shown in (D) and (F). Mean values ± S.E., n = 3–5 each group; *P < 0.05, **P < 0.01 vs. scrambled of each group. Statistical analyses were conducted with two-way ANOVA followed by Šídák’s multiple comparisons test.
3.6 Mitophagy can be reactivated by TAT-Beclin1 in ulk1cKO mice during PO
We next evaluated whether TAT-Beclin 1 treatment rescues cardiac dysfunction in ulk1cKO mice during PO. Since TAT-Beclin 1 actively promotes mitophagy 24 h after injection,9 we injected TAT-Beclin 1 every 24 h starting 1 day before the TAC surgery (Figure 7A). Although the LV ejection fraction (LVEF) did not change in WT mice, it was decreased significantly in ulk1cKO mice 3 and 7 days after TAC. However, TAT-Beclin 1 injection completely normalized LVEF in ulk1cKO mice 3 and 7 days after TAC (Figure 7B and C; Supplementary material online, Figure S7A). Although TAC for 7 days increased both HW/TL and LW/TL in ulk1cKO mice, both were significantly reduced by TAT-Beclin 1 injection after 7 days of TAC (Supplementary material online, Figure S7B and C). CM CSA was not altered by TAT-Beclin 1 injection after 7 days of TAC in control mice but was significantly reduced by TAT-Beclin 1 injection in ulk1cKO mice after 7 days of TAC (Supplementary material online, Figure S7D and E). TAC-induced increases in the percentage of myocardial fibrosis were also significantly decreased by TAT-Beclin 1 injection in ulk1cKO but not in control mice 7 days after TAC (Supplementary material online, Figure S7F and G). In addition, TAT-Beclin 1 injection restored mitophagy/autophagy activity in ulk1cKO mice after 3 days of TAC, evaluated by EM analyses of the numbers of autophagosomes containing mitochondria and autolysosomes (Figure 7D and E). TAC-induced mitochondrial dysfunction, evidenced by increased cristae disorganization, was reversed in ulk1cKO hearts in the presence of TAT-Beclin 1 injection (Figure 7F and G). Thus, re-activation of mitophagy by TAT-Beclin 1 injection restores cardiac function in the ulk1cKO heart during PO. Furthermore, exacerbation of cardiac dysfunction in ulk1cKO mice in response to PO is at least in part mediated through suppression of mitophagy. Our results further suggest that, regardless of the molecular mechanisms of mitophagy, mitophagy acts as an adaptive mechanism against PO and reactivation of either conventional or alternative mitophagy by TAT-Beclin 1 is an effective modality to delay the progression of heart failure.
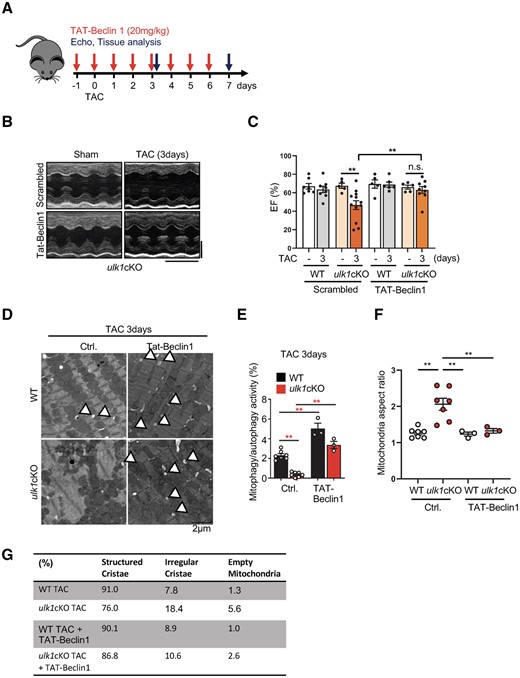
TAT-Beclin 1-induced mitophagy alleviates cardiac dysfunction in response to TAC in ulk1cKO. (A–G) 3- to 4-month-old WT and ulk1cKO mice were subjected to either sham operation or TAC for 7 days. TAT-Beclin 1 peptide was injected on a daily basis from 1 day before TAC surgery until 7 days after TAC, as described in (A). Representative echocardiographs. Scale bars, vertical 2.5 mm and horizontal 250 ms (B). % of LVEF was evaluated by echocardiography. Mean values ± S.E., *P < 0.05. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (C). (D–G) Heart samples from mice subjected to TAC for 3 days were subjected to electron microscopic analyses. Representative EM images are shown. Arrowheads indicate autolysosomes (D) (Scale bar, 2 μm). Mitophagy/autophagy activity was quantified. Mean values ± S.E., **P < 0.01. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (E). Mitochondria aspect ratios were quantified. Mean values ± S.E.; **P < 0.01. Statistical analyses were conducted with two-way ANOVA followed by Tukey’s post hoc test (F). Mitochondria were categorized by the morphology of cristae and quantified. The percentage of structured or irregular cristae and empty mitochondria were quantified (n = 3–6 each group) (E).
4. Discussion
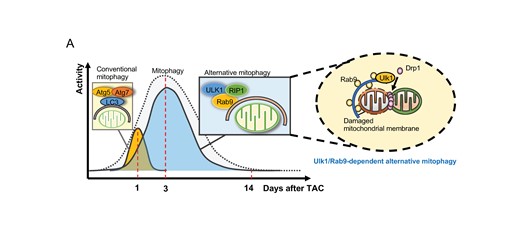
We have shown previously that conventional autophagy and mitophagy are activated with distinct time courses in response to TAC.7 Conventional autophagy is activated rapidly and transiently and returns to baseline within 1 day in response to TAC. On the other hand, mitophagy reaches a peak at 3 days and returns to baseline around 7 days after TAC.7 We here show that the mitophagy activated 3 days after TAC is primarily mediated through an Ulk1-dependent mechanism, including alternative mitophagy (Graphical Abstract).
Mitophagy activation 3 days after TAC was accompanied by increased co-localization between Rab9 puncta and TOMM20 and increased phosphorylation of Rab9 at Ser 179, important features of alternative mitophagy.14 We have shown previously that Drp1 is translocated to mitochondria 1–5 days after TAC and that activation of mitophagy after TAC is mediated by a Drp1-dependent mechanism.7 Since both Ulk1 and Drp1 play an essential role in mediating alternative mitophagy, as shown in this study and the previous study,14 these results together strongly support the hypothesis that alternative mitophagy is a major mechanism mediating mitophagy in the heart in response to PO.
Cardiac-specific down-regulation of Ulk1 did not affect the baseline phenotype in the heart. However, it exacerbated TAC-induced decreases in LVEF and increases in lung congestion and promoted cardiac hypertrophy after 7 days of TAC. Loss of Ulk1 function in the heart did not significantly affect conventional autophagy, whereas it dramatically reduced mitophagy in the heart after TAC. Loss of Ulk1 function also completely inhibited phosphorylation of Rab9 at Ser179, a key feature of alternative mitophagy.14 Although the exacerbation of heart failure in ulk1cKO mice in response to TAC could be mediated by multiple mechanisms, the fact that ulk1cKO mice exhibit prominent mitochondrial dysfunction and that a rescue of mitophagy by TAT-Beclin 1 alleviated TAC-induced heart failure in ulk1cKO mice suggests that Ulk1-dependent mitophagy, including alternative mitophagy, plays an important role in maintaining mitochondrial quality and cardiac function in the presence of PO.
We have shown previously that Ulk1-induced phosphorylation of Rab9 at Ser179 mediates Rip1-induced phosphorylation of Drp1 at Ser616 and alternative mitophagy in response to glucose deprivation in CMs.14 Here we show that TAC-induced Ser179 Rab9 phosphorylation is also Ulk1 dependent. However, expression of Rab9 (S179D), a Rab9 Ser179 phospho-mimetic mutant, in ulk1cKO mouse hearts did not exhibit a cardioprotective effect (data not shown), suggesting that Ulk1 may require additional mechanisms besides phosphorylation of Rab9 at Ser179 to induce mitophagy. Interestingly, TAC up-regulated both Rip1 and Rip3 in the heart in an Ulk1-dependent manner. The underlying molecular mechanism through which Ulk1 affects the levels of Rip1 and Rip3 is currently unknown. We have shown previously that Ulk1-induced phosphorylation of Rab9 at Ser179 facilitates Rab9–Rip1 interaction and consequent formation of a large protein complex consisting of Ulk1, Rab9, Rip1, and Drp1.14 Thus, it is possible that Ulk1-dependent formation of the large protein complex may stabilize both Rip1 and Rip3. Further investigation is needed to test this hypothesis.
We have shown previously that TAT-Beclin 1 can stimulate mitophagy in CMs at baseline.7 TAT-Beclin 1 can stimulate mitophagy in the heart in the presence of PO even after the conventional form of autophagy is down-regulated7 and in atg7cKO mouse hearts in the presence of high fat diet consumption.9 Here we show that TAT-Beclin 1 can promote mitophagy in CMs in which Ulk1 is down-regulated. In addition, we show that TAT-Beclin 1 activates both LC3- and Rab9-dependent autophagy in CMs. Our results suggest that TAT-Beclin 1 is able to reactivate mitophagy even when one (or more) mechanism(s) of mitophagy is inactivated. As a key regulator of class III PI3K, Beclin 1 plays an essential role in both the conventional and alternative forms of mitophagy through generation of autophagic vacuoles.23 For this reason, unlike Atg7 or Ulk1, knockdown of Beclin 1 inhibits both conventional and alternative mitophagy.13 Although the detailed molecular mechanism through which Beclin 1 activates alternative mitophagy is not yet clear, the ability of TAT-Beclin1 to activate mitophagy in various conditions is remarkable and, thus, it may be useful in a wide variety of cardiac conditions in which mitochondrial quality control mechanisms are impaired.
Currently, we do not know the mechanism through which alternative mitophagy is inactivated by 2 weeks after TAC. Since phosphorylation of Rab9 at Ser179, the critical mechanism mediating alternative mitophagy,12 is down-regulated after reaching a peak around 7 days after TAC, investigating the molecular pathways contributing to de-phosphorylation of Rab9 at Ser179, including down-regulation of Rip1/Rip3, should provide useful information to elucidate the mechanism mediating the inactivation of alternative mitophagy.
In summary, we here demonstrate that Ulk1-dependent mitophagy, including alternative mitophagy, is the major mechanism of mitophagy after TAC and plays an important role in maintaining mitochondrial quality and cardiac function in the presence of PO. Despite its importance, alternative mitophagy is inactivated by 14 days after TAC, which leads to the development of heart failure. We propose that interventions to reactivate either conventional or alternative mitophagy may alleviate or delay the development of heart failure in patients with PO.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
J.N., A.S., and J.S. conceptualized the study. J.N. and J.S. developed methodology and wrote the original draft. J.N. validated and engaged in formal analysis. J.N., R.M., P.Z., E.-A.S., A.I., W.M., Y.N., and C.H. performed investigations. T.S., Y.-K.J., and J.S. provided resources. J.N. and J.S. reviewed and edited the manuscript. J.S. supervised and administered the project and acquired funding.
Acknowledgements
We thank Daniela Zablocki for critical reading of the manuscript.
Funding
This work was supported by the Brain Pool programme funded by the Ministry of Science and ICT through the National Research Foundation of Korea 2021H1D3A2A02039802 (J.N.), the American Heart Association (AHA) Postdoctoral Fellowship 18POST34050036 (J.N.) and Merit Award 20Merit35120374 (J.S.), Foundation Leducq Transatlantic Networks 15CVD04 (J.S.), and US Public Health Service Grants HL067724, HL091469, HL112330, HL138720, HL144626, HL150881, and AG23039 (JS).
Data Availability
The data underlying this article are all available within the article and in its online supplementary materials.
Translational perspective
Heart failure is often accompanied by mitochondrial dysfunction in cardiomyocytes. Elimination of dysfunctional mitochondria by mitochondria-specific forms of autophagy, termed mitophagy, is a crucial mechanism for maintaining mitochondrial function in the stressed heart. We discovered that an unconventional form of mitophagy mediated through an Atg7-independent and Ulk1- and Rab9-dependent mechanism is a predominant form of mitophagy in the heart in response to pressure overload. Interventions to restore mitophagy by stimulating the signalling mechanism of the Ulk1-Rab9-dependent mitophagy should delay the development of heart failure in patients with increased afterload.
References
Author notes
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}