





This editorial refers to ‘Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart’ by J. Nah et al.,https://doi.org/10.1093/cvr/cvac003.
The quintessential response of the myocardium to increased work load is heart hypertrophy. Heart hypertrophy is an adaptive response mechanism that promotes growth of the heart muscle in attempt to normalize diastolic filling pressure.1 Cardiac hypertrophy can result from an increase in peripheral resistance, valvular defects, or cardiac injury following myocardial infarction. These changes set into motion a cascade of signalling events that promote cardiac myocyte growth. The physiological increase in cardiac wall mass off-sets the increased ventricular loading conditions and maintains cardiac function according to the Laws of Laplace, reviewed in.2,3 In general, the heart is among the most metabolically active organs in the body with a high energy demand for maintaining normal cardiac functioning, which rises exponentially in response to hypertrophic growth. For this reason, cardiac myocytes rely heavily on mitochondrial oxidative metabolism as the primary adenosine triphosphate (ATP) source and is reflected by the fact that more than 30% of the cardiac myocyte volume is comprised of mitochondria. However, with the continuous loss of functional mitochondria and consequential decline in ATP, the adaptive hypertrophic response can readily transition into pathological hypertrophy resulting in heart failure.4
The mitochondrial cristae on the inner mitochondrial membrane serve as a site for electron transport chain complexes, important in oxidation of Nicotinamide adenine dinucleotide (NADH), Flavin adenine dinucleotide (FADH), and ATP energy production. Dysfunction of mitochondria is often been associated with cardiomyopathies and heart failure.5 Stress conditions that lead to ionic imbalance and calcium overload results in increased reactive oxygen species, mitochondrial permeability transition pore opening, loss of membrane potential, and impaired ATP production. Moreover, the leakage of mitochondrial DNA from damaged mitochondria can trigger an immune damage associated molecular patterns response leading to pathological cardiac remodeling and dysfunction.5,6
In contrast to cells that readily divide, the dysfunctional damaged mitochondrial pool is distributed to each of the newly formed daughter cells upon each cell division, thereby diluting the potential impact of dysfunctional mitochondria on cell viability. In the context of the post-mitotic adult heart in which cardiac myocytes do not readily undergo cell division, alternative mitochondrial quality control mechanisms have evolved that selectively remove damaged or irreparable mitochondria from the cell via specialized process known as mitophagy.7
The process of mitophagy is an evolutionary conserved from yeasts to humans and plays an important role in several biological processes including development, differentiation, inflammation, and cell death.8 Mitochondria can reportedly be discarded by multiple processes including the canonical Pink1/Parkin mitophagy pathway, conventional ATG7/Atg5/LC3 mitochondrial autophagy pathway and more recently recognized Unc51-like-kinase 1 (ULK1)/Rab9-mediated alternative mitophagy pathway. These pathways differ from each other with respect to their respective initiating protein complexes but have similar final step where engulfed mitochondria are degraded by the lysosome. The Pink1/Parkin canonical mitophagy pathway is among the most studied mitochondrial clearance pathway. Pink1 (PTEN)-induced putative kinase 1 is a mitochondrial targeted kinase, which is stabilized and accumulates on the outer mitochondrial membrane in response to loss of mitochondrial membrane potential. Pink1 subsequently recruits the E3-ligase Parkin to the outer mitochondrial membrane which ubiquitinates a number of mitochondrial proteins essential for removal of the defective mitochondria. Pink/Parkin mediated mitophagy has been shown to be important for mitochondrial clearance in aging, myocardial infarction, and other cardiovascular diseases.7,9
Autophagy is well characterized and widely studied process implicated in degrading and recycling of defective proteins and organelles including mitochondria. It involves a key autophagy protein, ATG7 which in concert with ATG5 and LC3II proteins initiate the autophagy process. Recent studies showing diminished mitochondrial clearance in the absence of ATG7 suggest an important role for ATG7 in mitophagy.10 Acute activation of ATG7 mediated mitophagy was shown to be cardioprotective in response to high fat diet.11 Notably, the common feature among classical autophagy clearance pathways is the requirement for ATG7 or ATG5 to initiate the process. However, a recently described alternative autophagy pathway independent of ATG7/ATG5 has been described.
Alternative mitophagy, which involves ULK1 and the endosomal protein Rab9 protein, has been shown to protect cardiac myocytes by increasing clearance of damaged mitochondria during high fat diet, diabetes, and ischaemia/reperfusion.11,12 ULK1-mediated phosphorylation of Rab9 at serine 179 is an essential step for initiation of alternative mitophagy. Further, in the alternative autophagy pathway, the phagophore membrane that would otherwise engulf depolarized damaged mitochondria is derived from the trans-Golgi membrane and not from the endoplasmic reticulum as would be the case in the canonical pathway.12
Hence, defects in mitophagy lead to accumulation of dysfunctional mitochondria and cell death which ultimately, leads to adverse remodeling and heart failure. Notably restoration or induction of mitophagy has been shown to limit the cardiomyopathies. Despite of the increasing awareness of mitophagy forms, the exact role of each mitophagy pathway is unknown.
Nah et al.13 provide new evidence that both conventional and alternative mitophagy occur following pressure overload hypertrophy; however, the authors show that these mitochondrial clearance mechanisms are spatially and temporally separated which result in different functional outcomes (Figure 1).
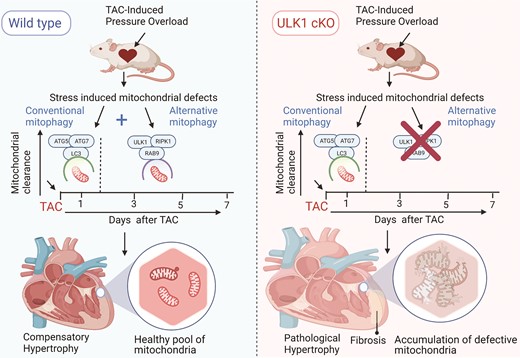
ULK1-induced alternative mitophagy prevents the development of pathological cardiac hypertrophy under pressure overload. The study by Nah et al.13 provides evidence to show the cardioprotective effect of ULK1-mediated alternative mitophagy against pressure overload stress (TAC mouse model). In response to pressure overload, ATG7/ATG5-dependent mitophagy is acutely activated but rapidly diminished after 1 day. Conversely, ULK1/Rab9-mediated alternative mitophagy is activated and sustained for several days following TAC, actively clearing defective mitochondria. Moreover, the myocardium of the mice deficient for ULK1 (ULK1cKO) exhibited large dysfunctional mitochondria with poorly defined cristae. Indices of pathological cardiac remodelling such as increased fibrosis, loss of mitochondrial integrity, and cardiac dysfunction were evident. These findings point towards the importance of ULK1-mediated alternative mitophagy in mitochondria clearance and attenuating pressure overload induced cardiac dysfunction.
Herein, Nah et al.13 identified Ulk1/Rab9-mediated alternative mitophagy as the predominant form of mitophagy in the heart in response to pressure overload, with a major cardioprotective role. Nah et al.13 systematically showed the activation of the two mitochondrial clearance pathways in the heart following transverse aortic constriction (TAC) induced pressure overload, an early At5/Atg7/LC3 mediated mitochondrial autophagy that peaked at day 1 and then rapidly declined. In contrast, ULK1/Rab9 mediated alternative mitophagy peaked between 3 and 5 days and lasted to a greater extent compared to At5/Atg7/LC3 mediated mitophagy. To test the functional significance of ULK1-mediated alternative mitophagy and distinguish it from Atg5/7 mediated mitophagy, the authors conducted a parallel pressure overload study in wild type (WT) mice and cardiomyocyte specific deletion of ULK1 (cULK1 KO) and Atg7 (Atg7cKO) mice. Through this study, the authors provide compelling new evidence to prove that ULK1/Rab9-mediated mitochondrial clearance is crucial for clearing damaged mitochondria and preventing decline in cardiac function. Interestingly, both WT mice as well as Atg7cKO mice defective for conventional autophagy exhibited normal clearance of dysfunctional mitochondria via ULK1/Rab9 pathway and displayed healthy pool of mitochondria. However, ULK1cKO mice with intact conventional autophagy but defective for alternative mitophagy displayed accumulation of dysfunctional large sized mitochondria with diffused compromised cristae and disrupted oxidative metabolism. The authors further investigated the functional changes in the hearts of WT and ULK1cKO mice for up to 30 days following TAC. Interestingly, in contrast to WT mice that exhibited adaptive and mild hypertrophy, TAC induced severe hypertrophy, fibrosis, and cardiac dysfunction in ULK1cKO mice, suggesting the importance of ULK1-mediated alternative mitophagy in eliminating dysfunctional mitochondria and preventing decline in cardiac performance (Figure 1). Interestingly, use of TAT-Beclin restored mitophagy levels and improved cardiac function in ULK1cKO mice in response to stress. All together, the reduced bioenergetics from loss of functional mitochondria combined with defects in mitochondrial clearance triggers the molecular events leading to development of severe pathophysiological condition of heart which could be reversed by restoration of mitochondrial clearance process.
Although this study by Nah et al.13 elegantly shows the importance of mitochondrial clearance by UlK1/Rab9 in preventing pathological signalling leading to severe maladaptive hypertrophy, several questions remain unanswered. First, that mitochondrial and cardiac dysfunction observed in ULK1cKO mice could be rescued significantly by use of an autophagy inducer compound, TAT Beclin 1, raises the question as to whether ULK1 specific mitophagy is really critical for maintaining mitochondrial and cardiac function or has some alternate function in the cell beyond its ascribed role in autophagy. TAT Beclin 1 mediated rescue of mitophagy even in the absence of ULK1 would suggest that restoration of mitophagy is important for cardio protection, regardless of the nature of the mode of mitophagy, i.e. whether it be through Atg7/5 or ULK1. The second question that remains unanswered is what are the factors that cause termination of the ATG5/7 conventional pathway peak after 1 day of TAC. Furthermore, it would be interesting to test whether stabilizing ATG5/7-mediated autophagy would rescue cardiac dysfunction in the absence of ULK1. Third, in this study, the authors did not assess the activation status of Pink1/Parkin-mediated classical mitophagy in response to TAC raising the possibility that TAT-Beclin conferred mitochondrial clearance might occur through PINK/Parkin pathway. Therefore, further investigation to dissect the mechanisms of TAT Beclin1 induced mitophagy in the absence of ULK1 would shed light on the importance of this pathway under physiological and pathological conditions in relation to other modes of autophagy.
Nevertheless, the finding of this study strongly advocates the importance of mitochondria quality control process for protecting the heart against the detrimental effects of accumulated damaged mitochondria under pressure overload conditions. Interventions that stimulate mitophagy under stress conditions may prove beneficial in protecting the myocardium from adverse pathological remodelling during stress.
Funding
This work was supported by a foundation grant to L.A.K from the CIHR and funds from the St. Boniface Hospital Research Foundation. L.A.K. holds a Canada Research Chair in Molecular Cardiology. Figure was created with BioRender.com.
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.
Conflict of interest: None declared.

{kind=link}