





Abstract
Calcification of the arterial wall and valves is an important part of the pathophysiological process of peripheral and coronary atherosclerosis, aortic stenosis, ageing, diabetes, and chronic kidney disease. This review aims to better understand how extracellular phosphates and their ability to be retained as calcium phosphates on the extracellular matrix initiate the mineralization process of arteries and valves. In this context, the physiological process of bone mineralization remains a human model for pathological soft tissue mineralization. Soluble (ionized) calcium precipitation occurs on extracellular phosphates; either with inorganic or on exposed organic phosphates. Organic phosphates are classified as either structural (phospholipids, nucleic acids) or energetic (corresponding to phosphoryl transfer activities). Extracellular phosphates promote a phenotypic shift in vascular smooth muscle and valvular interstitial cells towards an osteoblast gene expression pattern, which provokes the active phase of mineralization. A line of defense systems protects arterial and valvular tissue calcifications. Given the major roles of phosphate in soft tissue calcification, phosphate mimetics, and/or prevention of phosphate dissipation represent novel potential therapeutic approaches for arterial and valvular calcification.
Introduction
Physiological mineralization of bone is essentially an extracellular process in which calcium and phosphates precipitate on the extracellular matrix (ECM). Due to the extracellular prevalence of calcium and the intracellular predominance of phosphates, extracellular inorganic or exposed tissue phosphates are subject to rate limitation during the initiation and propagation of mineralization.
Phosphates and phosphoryl transfers are essential components of prokaryotic and eukaryotic life, being used for both structural (nucleic acids1 and phospholipid membranes2) and functional storage and release of chemical energy3 in nucleoside triphosphates purposes (Figure 1). This cellular and tissue ubiquity of organic phosphates in initial life and progress is directly related to the basic chemical properties of phosphates, involving their thermodynamic instability, facilitating transfers and kinetic stability, thereby giving their structural solidity (e.g. DNA structure).4 The physicochemical properties of organic phosphates associated with their negative charges are fundamental for nucleophilic transfer and confinement of their components and metabolites to subcellular and cellular boundaries.5
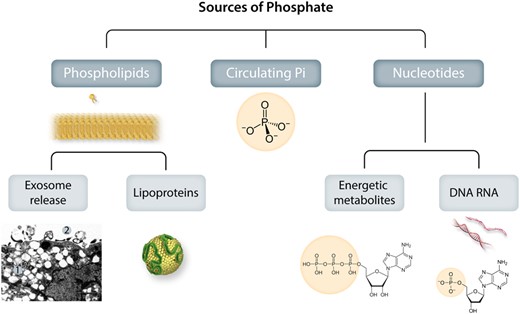
Sources of phosphate for valvular and vascular mineralization. Phosphates exist in the body both in the chemical form of minerals (inorganic phosphate, Pi) and in biologically active forms integrated in molecules for structural and energy metabolism (organic phosphate). Extracellularly exposed phosphate groups in organic molecules will become hot sports for calcium precipitation and hydroxyapatite crystallization. The organic integration of inorganic phosphate can also take the reverse form, where Pi is released extracellularly from organic sources to directly form mineralization. Photo inset shows: A: Electron microscopic view of SMC phagocytic consequences: 1. Cytosolic microvesiculation, 2. Exosome release.
As a result, phosphates change from a mineral chemical form (inorganic phosphate, Pi) to a biologically active and integrated form in organic molecules (nucleic acids, phospholipids; Table 1). Phosphates can also be exocytosed in different forms as a result of endoplasmic reticulum stress, lysosomal microvesiculation, exocytosis, and cell death. Furthermore, extracellular nucleotides (mainly ATP and ADP) not only act via purinoreceptors to signal cellular responses but are also metabolized by ecto-enzymes to generate extracellular phosphates. In response to different environmental conditions (such as ageing) or associated diseases [such as chronic kidney disease (CKD), diabetes, homocysteinaemia,6 or osteoporosis7,8] phosphates could take the reverse course from organic integration to an inorganic form, and from an intracellular to an extracellular anion, thereby directly promoting mineralization.
Sources of phosphate for valvular and arterial mineralization
| Circulating inorganic phosphate (Pi) |
| Membranous phospholipids |
| Phospholipid-transporting lipoproteins |
| Nucleic acids |
| Purinergic system and pyrophosphate metabolism |
| Circulating inorganic phosphate (Pi) |
| Membranous phospholipids |
| Phospholipid-transporting lipoproteins |
| Nucleic acids |
| Purinergic system and pyrophosphate metabolism |
Sources of phosphate for valvular and arterial mineralization
| Circulating inorganic phosphate (Pi) |
| Membranous phospholipids |
| Phospholipid-transporting lipoproteins |
| Nucleic acids |
| Purinergic system and pyrophosphate metabolism |
| Circulating inorganic phosphate (Pi) |
| Membranous phospholipids |
| Phospholipid-transporting lipoproteins |
| Nucleic acids |
| Purinergic system and pyrophosphate metabolism |
Soft tissue calcification is a double-edged sword. As a common healing response to injury, vascular calcifications could be beneficial responses in the circulation. However, more frequently, dysregulated calcification is maladaptive and participates in cardiovascular pathologies. Cardiovascular mineralization includes micro and macro precipitations of calcium phosphate (CaP) on the ECM. This may be associated with a dysregulation of systemic phosphocalcic metabolism, either in CKD (metastatic calcification) or associated with diabetes and ageing (degenerative calcification). Here, cardiovascular tissue calcification in the different pathologies of the arterial wall (atherosclerosis, Mönckeberg disease, aneurysms) and heart valves will be addressed.
Despite this environmental biodiversity, the common denominator of mineralization initiation in soft vascular tissues is, as in bone, the bioavailability of extracellular phosphates exposed as initial calcium-binding substrates and CaP precipitation on ECM. Therefore, understanding the role of phosphates is crucial to understanding the pathophysiology of calcification and how to therapeutically target phosphate biology for preventing pathological cardiovascular consequences of calcifications, particularly with the challenge of maintaining bone homeostasis. The aim of this review is to focus on the diversity of phosphate sources (both in its organic and inorganic forms) and its role in the mineralization initiation of arterial and valvular tissues. These local roles interfere with systemic phosphocalcic homeostasis, as well as local cellular exocytosis that will transform organic phosphate-containing cellular molecules into extracellular Pi or exposed phosphates.
Bone growth and mineralization
Two phosphorus and three calcium molecules are required to form a crystal molecule of calcium phosphate Ca3(PO4)2, which polymerizes to initiate physiological mineralization of calcium in teeth and bones and pathological mineralization in soft tissues. The mineralization process of soft tissues, observed in arterial and valvular pathologies, shares numerous similarities with physiological bone mineralization.
Bioavailable extracellular phosphates form the initial substrate for calcium binding, followed by CaP precipitation in the organic matrix, preceding formation of hydroxyapatite crystals containing tricalcium phosphate (mineral form of calcium), polymerization and growth, leading to osteogenesis (ossification), and bone tissue formation as an endoskeleton of vertebrates. The collagen organization of bone matrix (essentially collagen type I), is axially staggered and compacted forming a hierarchical organized scaffold for mineralization. This physiological process is highly specific to bone tissue through the initial release of phosphates, as well as the activities of differentiated cells (osteoblast) and specific proteins (e.g. bone morphogenetic proteins, BMPs).9 During phylogeny, calcium phosphate biomineralization appeared in the fishbone (endoskeleton) and develops largely with the appearance of sarcopterygians (first joint) and terrestrialization (endoskeleton) .10 Bone calcium accounts for 90% of the total body calcium in mammals.
Similar dynamics and chronology are present in vascular and valvular calcifications involving first CaP precipitation, polymerization, and formation of hydroxyapatite crystals, and finally extension to tissue mineralization. The collagen ECM of vascular and valvular tissues being primarily devoted to other functions, however constitutes a less organized scaffold. Ectopic mineralization is a result of amorphous CaP forming solid mineral crystals incorporated into the ECM.
Calcifying cardiovascular pathologies
Atherosclerosis
Atherosclerotic calcifications predominate in the abdominal aorta and femoral arteries, presenting calcified protrusions towards the lumen. This site-specific calcification may be related to the specific haemodynamics of the legs, involving oscillatory flow with retrograde shear stress in protodiastole, thereby impairing endothelial function.11–13 This phenomenon is modulated by hydrostatic pressure (gravitational force),14 and prolonged sitting impedes shear stress-dependent vasodilatation and promotes peripheral arterial disease.15 In this haemodynamic context, endothelial dysfunction promotes subjacent mineralization. Endothelial cells are an important source of microvesicles.16 In particular, hyperphosphataemia associated with CKD is toxic to endothelial cells,17 and this effect can be prevented by autophagy.18 Therefore, endothelial dysfunction and reduction of shear stress are involved in the initiation and progression of vascular calcifications. In this context, a recent study using single-cell transcriptomics in young versus old primates reported endothelial erosion and increased signalling pathways of calcification with ageing.19
The development of atherosclerosis is initiated by the unidirectional wall transfer of plasma low-density lipoprotein (LDL)20 and apolipoprotein B (apoB) to interact with intimal glycosaminoglycans.21 Further initiating processes include foam cell formation,22 followed by an inward migration and subendothelial proliferation of smooth muscle cells (SMCs), and the synthesis of ECM. In this atherosclerotic environment, phosphates accumulate in the form of phospholipids through the release of SMC exosomes23 and the transport of plasma apolipoprotein24,25 (Figure 2). Furthermore, extracellular DNA retention within the shoulder of the plaque serves as a locus to precipitate CaP,26 as will be further outlined below (Figure 2).
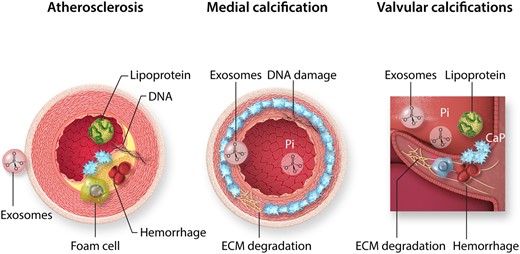
Calcifying cardiovascular pathologies. Cardiovascular mineralization occurs as atherosclerosis, medial calcification, and valve calcifications. Atherosclerotic plaque calcification is characterized by lipoprotein infiltration, foam cell formation and exosome release from smooth muscle cells, as well as intraplaque haemorrhage, cell death, and DNA retention. In contrast, medial calcification develops in the absence of inflammatory cells and involves degradation of the internal elastic lamina in the extracellular matrix (ECM), DNA damage, exosome release, and calcium phosphate precipitation from circulating inorganic phosphate (Pi). Valve calcification is highly dependent on biomechanical and haemodynamic factors, and has the characteristics of atherosclerotic and medial calcification, with a strong inflammatory component. The initial localizations of calcifications are indicated in blue.
In coronary artery disease, coronary artery calcium scoring is one of the most powerful predictors of acute coronary syndromes.27 The initial coronary fatty streaks and fibroatheroma are usually associated with microcalcifications (0.5–15 µm) that, as a result of the calcification mechanisms discussed below, progress to macrocalcification (>3 mm) in association with atherosclerosis progression.28 Spotty calcifications have been suggested to be associated with plaque instability, whereas extensive calcifications may be prone to stabilizing the lesion. The instability associated with spotty lesions may be due to the distortion forces caused by the density mismatch between the solid hydroxyapatite and the smooth elastic wall (see below, von Misses stress). Nevertheless, the source and role of phosphates in the progression from micro- to macrocalcification remains to be explored.
Medial calcification
Diabetes and CKD are major risk factors for medial calcification, also referred to as Mönckeberg disease,29 and are associated with the degradation of the elastic lamina30 (Figure 2). This disease is primarily a SMC pathology, which is related to the activation of SMC endosomes, exosome release, precipitation of hydroxyapatite in the degraded internal elastic lamina, and an absence of immune cells.31
The genetic Hutchinson-Gilford syndrome, due to progerin accumulation in the nuclear envelope, is characterized by a defect in nuclear mechanotransduction with SMC endosomal stress,32 DNA damage and SMC disappearance, leading to intense calcifications.33 These abnormalities are at least partially reproduced in CKD and physiological ageing.34 In diabetic patients, medial calcifications are frequently associated with neuropathy.35,36 Moreover, sympathectomy increases the frequency of vascular calcification in limbs37 and hand in systemic sclerosis.38 These observations are particularly interesting because they suggest that the role of SMCs is predominant in the development of medial calcifications, and that the release of extracellular vesicle (exosomes, microvesicles, apoptotic bodies) is potentially the main phenomenon linking SMC to ECM mineralization.
Valvular calcifications
The mechanotransduction of the left heart valve is highly haemodynamically related to the cardiac cycle.39 The aortic valve consists of three layers with different biomechanical properties and tissue composition; the collagen-rich fibrosa, the proteoglycan-rich spongiosa, and the elastin-rich ventricularis.40 The aortic valve interstitial stromal cells (VICs) are devoid of myosin (high ECM synthesis, low contractility), but express a high level of tensegrity,10 thereby transducing the ability of VIC to resist mechanical stress and fatigue (peak stress intensity multiplied by frequency). Recently, layered tissue and VIC differentiation were confirmed by multi-omics mapping.41 Fibrosis and calcification pathways predominate in fibrosa. Fibrosa-derived VICs in primary culture conserve these phenotypes, suggesting that the tissue microenvironment has an epigenetic imprint in VIC biology. This spatio-temporal tissue environment in relation to mechanosensitivity and biomechanical function can be summarized in that the fibrosa is mainly sensitive to tensile stress during diastole, whereas ventricularis is mainly sensitive to shear stress on the aortic valve during systole. It is worth noting that calcific aortic valve disease (CAVD) initiates and progresses from the fibrosa, whereas, the rare calcified mitral valve disease initiates and progresses from the ventricularis, which is exposed to high tensile stress during systole. Mitral calcifications usually predominate in the mitral annulus at the junction of the small mitral leaflet with the posterior annulus.42 In relation to local haemodynamics, degenerative mitral disease begins with lipid deposition, which manifests as yellow fatty streaks in the ventricularis. In this disease, plasma hypertriglyceridaemia was identified as an independent risk factor, including genetic imprinting43 and apo(E) transport by a very LDL.44
Why is the arterial system the main target for extraosseous calcium mineralization?
Soft-tissue calcification
Inorganic Pi and calcium concentrations at or near saturation concentrations and a nidus for crystal nucleation represent the two prerequisites for the initiation of calcium phosphate precipitation and crystallization. In healthy soft living tissue, extracellular mineralization can be prevented physiologically through different mechanisms: unavailability of Pi, phosphate and calcium chelators, and loose connective tissue. In addition, inhibitors (e.g. fetuin, Matrix Gla protein, osteopontin, and pyrophosphate) that are systemically present and/or actively produced locally prevent soft tissue calcification and mineralization.45 However, in different microenvironments, such protections either disappear or become insufficient to prevent calcium binding to extracellular phosphates, thereby initiating mineralization through precipitation, crystallization and polymerization on soft matrices. Under pathological conditions, soft tissue calcifications can be observed in tumours,46 skin, connective tissue disorders such as scleroderma,47 muscles,48 tendons,49 abscess,50 and venous wall (phleboliths).51 These soft tissue localizations are usually dystrophic, related to tissue injuries, and occur in association with a physiological systemic phosphorous metabolism. Sometimes, they may be metastatic, associated with systemic phosphocalcic disorders or abnormal ECM for precipitation, such as calciphylaxis in end-stage CKD.52
Mass transport
The arterial system that is subject to high blood pressure is particularly exposed to this non-physiological mineralization process. This is reflected in a high prevalence of calcifications in the arterial wall and the aortic valve, though these preferential localizations are not exclusive. A major physiological consequence of the acquisition of blood pressure in the arterial system is the unidirectional outward mass transport of soluble plasma components through the wall. This means that in addition to energetic substrates, oxygen, glucose, and lipoproteins, ionic molecules (e.g. phosphate and calcium) and circulating calcification inhibitors (e.g. fetuin synthesized by the liver) will also reach the arterial wall from the circulation. The consequences of numerous bifurcations, associated with the in-parallel evolution of circulation, are the frequent collision of the circulating cells with the arterial wall.10 Both aspects are involved in the process of arterial calcification by directly influencing (i) the transport through and retention within the wall of certain plasma lipoproteins, and (ii) creating privileged sites of haemodynamic injuries for the development of atherosclerosis.53 In response, SMCs acquired a phenotypic plasticity,54 involving their ability to promote endocytosis of plasma soluble components and phagocytosis of wall-penetrating circulating cells. In order to maintain cell homeostasis, such input clearance abilities are associated with important exocytosis capacities, including molecular exocytosis of phosphate-rich exosomes. The exocytosis capacities are directly related to endosomal activity; they consume biochemical energies.55
In the aortic valve, the diastolic transvalvular pressure gradient between the aorta and the left ventricular cavity (90 mmHg) is the driving force for the advective plasma soluble components, including lipoproteins such as LDL and Lp(a), which leads to lipid and phospholipid retention within the leaflet base, particularly in the hinge region between the leaflets and the aortic wall of the sinus of Valsalva.56 This initial stage of CAVD largely resembles the fatty streaks observed in atherosclerosis, including exposed phosphates from various sources (exosomes, lipoproteins, and nucleic acids). In CAVD, Lp(a) seems to play a specific role, which will be discussed further below.
ECM in CaP nucleation, precipitation, and crystallization
In the arterial wall, fragmented elastin, for example, serves as an ECM substrate for calcium phosphate precipitation.57 Similar to vascular calcification, valvular calcium phosphate precipitation, and crystallization require a suitable ECM as retention and nucleation determinants of mineralization. For example, calcifications are the major limit to the durability of valve bioprotheses, which are composed of a xenogenic matrix (porcine or bovine) neutralized by glutaraldehyde cross-linking. Since glutaraldehyde pre-treatment of aortic graft in rats promotes calcification,58 it is likely that the ECM cross-linking compacts the matrix fibrillar network and facilitates Pi retention. This is further supported by the crosslinking strategy used to develop new scaffolds for bone neo-formation and solidity,59 as well as glycation crosslinking of ECM in diabetes that promotes mineralization.60
In contrast, the expression of certain noncollagenous matrix proteins serves to protect the vascular and valvular tissues from calcium phosphate (CaP) precipitation. The vitamin K-dependent matrix Gla-protein is synthesized by SMCs and VICs and incorporated into their ECMs.61 MGP contains five γ-carboxyglutamic acid residues capable of binding metal ions and preventing Pi and Ca precipitation and crystallization on ECM.62 MGP-deficient mice exhibited extensive vascular calcifications, leading to reduced life span as a result of vascular rupture.63,64 Vitamin K is required for the post-translational γ-carboxylation of glutamic acids in MGP, which may constitute the link between the observed vascular and valvular calcifications induced by anticoagulant vitamin K antagonists, thereby raising the notion of the therapeutic possibilities of vitamin K supplementation to prevent vascular and valvular calcification.
Other mineralization regulators present in ECM include the SIBLING (Small Integrin-Binding Ligand N-linked Glycoprotein) family, whose member osteopontin (OPN) forms a bridge that limits CaP aggregation and inhibits its crystallization. Unlike MGP, OPN is not expressed in normal vessels and, therefore, has been suggested as an inducible inhibitor of CaP deposition in the vascular wall.45 In support of the latter, OPN deletion does not induce spontaneous murine vascular calcification, but exacerbates the vascular calcification observed in MGP-deficient mice.65
Osteoblast phenotype switching of SMC and VIC
As previously indicated, the initiating process of valvular and vascular mineralization is the physiochemical precipitation of ionized calcium on exposed or inorganic phosphates. Although calcification inhibitors produced by the liver, SMCs, and VICs serve to limit precipitation, the structural vascular and valvular cells may not actively participate in the actual CaP precipitation. This has been demonstrated in vitro, where either lyzed or fixed SMCs exhibit faster calcification compared to living cells in the presence of a high content of phosphate and calcium in the cell culture medium.66 However, in the latter study, the nanostructure of the crystals was qualitatively different between living and dead cells, and the authors concluded that despite the initial vascular CaP deposition appearing to be cell-independent, living SMCs participated in the process of hydroxyapatite crystallization.66 Observations of spherical CaP deposits, present in both calcified on non-calcified human aortic valves, have supported that CaP precipitation indeed precedes the calcification of aortic valves.67
Vascular and valvular CaP deposition induces the expression of osteoblastic markers in SMCs 66,68 and VICs, respectively, which in turn optimize the ECM to facilitate hydroxyapatite crystallization in the arterial wall and valves. Hence, the osteoblast-like SMC and VIC phenotypes promote the mineralization of ECM with a more or less osteoid morphology.69 This response to the local extracellular phosphate microenvironment, endosomal stress (including oxidative stress and increased endosomal/exosomal turnover) corresponds to specific changes in the gene expression pattern associated with epigenetic phenomena.70 Nevertheless, the simple addition of β-glycerophosphate in SMC culture media also promotes this phenotypic change.71 The induction of osteochondrogenic morphogens, e.g. the BMP and Wnt, as well as the osteoblast transcription factor Runx2, represents the hallmark of the phenotypic transition of SMC and VIC during vascular and valvular calcification.9 This procalcifying gene pattern also includes the appearance of MSX2, SOX9, osterix, ALP and NFkB, following a slow reduction in contractile proteins and partial repression of myocardin72 due to changes in transcription factors.73 However, it should be noted that despite an induction during calcification, the SMC osteogenic gene expression levels remain up to 40 times lower compared to bone-forming osteoblasts.74
Intravascular, intraplaque, and intravalvular haemorrhages
The relationship between valvular and arterial haemorrhages and calcifications75 is bidirectional: calcifications promote haemorrhages and red blood cells (RBC), while haemoglobin and iron promote calcification of these elastic vascular tissues (Figure 3). In the context of the soft viscoelastic arterial and valvular tissues, calcifications promote a mechanical mismatch between the high strain of elastic tissue (measured by finite element analysis) and the solid crystals, provoking a distortion energy at the interface between solid calcifications and elastic tissues (von Mises stress).78 This distortion promotes fatigue-like repeated micro-damages, such as microscopic tears or macroscopic haemorrhages at the interface between the calcifications and elastic tissue, where the distortion force is maximal. When calcifications develop in the arterial intima, they can cause breaches that are sensitive to plaque rupture.79,80 If calcifications develop deeper in the media layer, the distorting forces could incite tears of the neovascularization (see above) and lead to peri-calcification haemorrhages.81 In this context, phagocytosis of small calcium phosphate crystals could induce cell death via the intracellular release of ionized calcium.82 Like healthy arterial media, healthy valves are avascular tissue.83 Similarly, and in line with the early stages of atherosclerosis, lipid accumulation in the fibrosa promotes the development of angiogenesis within the valves, a process always associated with aortic valve disease. In this situation, microcalcifications develop in the fibrosa and can cause neovascularization tears and haemorrhages, which accelerate the calcification process.84,85 Haemoglobin and its derivates (heme, ferrous iron, free radicals, and NF-κB activation) promote the osteoblastic differentiation of valvular interstitial cells,86 as well as SMC in the arterial wall.87 Therefore, there is a vicious circle between calcifications and RBC/ferrous iron, which promote both valvular85 and vascular calcifications in association with classical cardiovascular risk factors; ageing, tobacco, dyslipidaemia, etc. Moreover, RBC could be a source of vesicles and exposed phosphates from phospholipids.88 Importantly, phagocytosis of RBC releases numerous microvesicles and exosomes that expose the amphiphilic pole of phospholipids, as will be discussed further below.
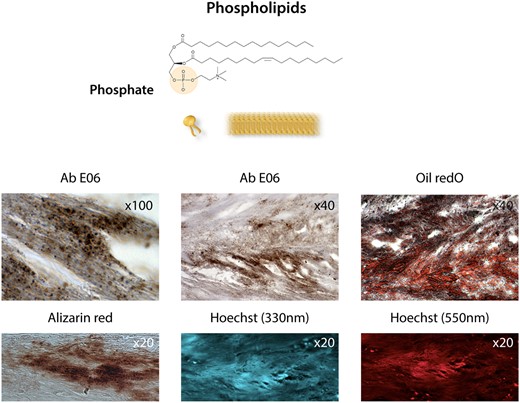
Membranous phospholipids. Phosphate at the polar head of phospholipids in cell membranes, i.e. the plasma membrane, mitochondrial membranes, and the nuclear envelope, as well as in exosomes. Bottom photo panel: The co-localization of oxidized phospholipids stained with the antibody Ab E06 (Sigma) and lipids (oil red O staining), calcification (Alizarin red staining after EDTA chelation of Ca++), Hoechst staining at 330 nm (UV), and at 550 nm (red) indicates the presence of phosphates (see text76,77) in a section of an aortic valve leaflet in the initial calcification step.
Sources of phosphate
Circulating phosphate
Extracellular ionized phosphate and calcium are present in plasma and interstitial fluids. The physiological concentration of ionized calcium (2.2–2.6 mmol/L) in interstitial fluids is twice the phosphate concentration (0.8–1.45 mmol/L). Extracellular calcium is 103 to 104 fold higher than intracellular calcium. These extracellular concentrations of anion and cation are both controlled by systemic calcium/phosphate homeostasis and metabolism. This involves a pattern of endocrine molecules and cellular receptors, including vitamin D and parathyroid hormone (for recent reviews, see refs89,90). Since Pi levels in the blood are close to the saturation level,91 prevention of crystal initiation on nucleating structures and inhibition of crystal growth require the presence of active calcification inhibitors.45 This systemic extracellular phosphocalcic metabolism, homeostatic or not, directly or indirectly influences soft tissue mineralization.
When calcium phosphate product (Ca x Pi) reaches a critical point, the CaP will physicochemically precipitate. Therefore, hyperphosphataemia, due to CKD and/or hyperparathyroidism for instance, may increase the propensity for spontaneous vascular and valvular hydroxyapatite deposition on nucleating structures. Therefore, preventing an excess of systemic Pi levels is an important strategy to limit cardiovascular mineralization. One of these endogenous Pi regulators is Klotho, a co-receptor for fibroblast growth factor 23 (FGF23), which decreases renal phosphate reabsorption.92 Klotho-deficient mice exhibit hyperphosphataemia, as well as vascular and valvular calcifications,93 illustrating the importance of this regulatory mechanism in retaining physiological Pi and preventing soft tissue mineralization.
Another potential means of limiting CaP precipitation is by generating colloidal calcium-phosphate complexes, such as calciprotein particles (CPPs) formed with the glycoprotein fetuin A.94 Since fetuin-A deficiency increases murine atherosclerotic calcification only in the presence of hyperphosphataemia,95 and since fetuin A levels decrease in CKD,92 CPP may serve to limit mineralization as a result of a disturbed Pi—Ca balance.96 However, the effective span of this pathway may be limited, since primary CPPs can transit to larger CPPs. These so-called secondary CPPs induce vascular inflammation and SMC osteogenic differentiation,94 which eventually counteracts the potential beneficial effects of plasma-derived colloidal calcium phosphate with fetuin-A.96
Membranous phospholipids
In soft tissues, the main localization of phosphates is at the polar head of phospholipids in cell membranes, i.e. the plasma membrane, mitochondrial membranes, and the nuclear envelope (Figure 3). As membrane components, phospholipids are made up of membranes that bud in the form of microparticles, vesicles, and exosomes released by activated cells, as well as during cell suffering and death with the release of apoptotic bodies. The accumulation of such structures on the collagen matrix in the epiphyseal growth plates represents the mechanism by which mineralization progresses during bone growth.97 Matrix vesicles, produced by chondrocytes and osteoblasts, are specific microparticles that provide an initial site for mineralization (apatite polymerization) during bone formation.55 In this physiological context of bone mineralization, matrix vesicles share a high level of structural and functional similarities with exosome synthesis and release. The cytosolic process of microvesiculation is common to all eukaryotic cells and is specifically defined by endosomal biogenesis (10–100 nm). Given the homeostatic nature of endosomal activity, there is a highly regulated balance between endosomal entry (endocytosis, phagocytosis) and exit of exocytosis, secretion and exosome release55 as illustrated in Figure 4. It is interesting to note that such microstructures are selectively packaged and highly enriched in microRNA, also containing phosphates.98 In analogy with these physiological micro-vesiculations in bone formation, a similar pathological mechanism of cellular budding, release of apoptotic bodies99 or exosomes,100 mainly generated by SMC membranous phospholipids, could drive the development of arterial calcifications101,102 (Figure 4). In this phospholipid context, sphingomyelin and phosphatidylserine have the privileged ability to precipitate Ca++ on exposed phosphates. In particular, palmitoyl-oleoyl phosphatidylserine has a greater affinity for ionized Ca++ compared to other calcium-binding phospholipids.102,103 In this context, the phosphatase encoded by the PHOSPHO1 gene plays a role in bone mineralization due to phosphocholine as its specific target.104,105 Different tissue isoforms obtained by alternative splicing have been characterized. PHOSPHO1 exists in the microvesicles of the bone formation matrix, where it plays an important role in Pi intra-vesicular release.106 PHOSPHO1 also exists in SMC microvesicles,107 and its inhibition suppresses SMC calcifications in vitro.108
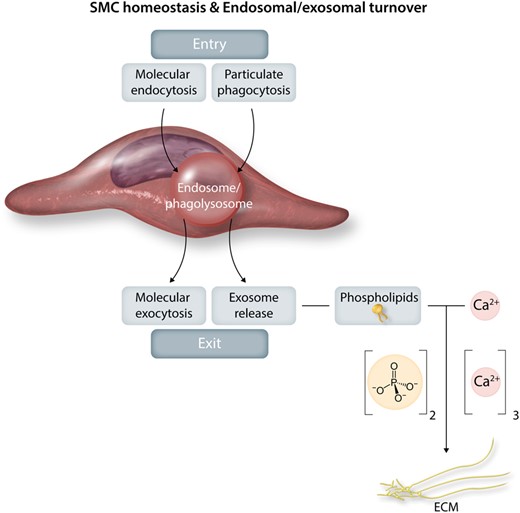
Smooth muscle cell homeostasis and endosomal/exosomal turnover. Homeostatic endosomal activity is a highly regulated balance between endosomal entry (phagocytosis, endocytosis, autophagy, heterophagy) and exit by molecular exocytosis and secretion and exosome release. Such release generated by smooth muscle cell membranous phospholipids can drive the development of arterial calcifications.
Phospholipid-transporting lipoproteins
As for cholesterol, phospholipids are transported on the surface of all lipoproteins and actively participate in their packaging. Although HDL is the main transporter of ingested phospholipids,109 Lp(a) is primarily involved in the transport of oxidized phospholipids.110 Lp(a) is a particular lipoprotein similar to LDL particles, but differentiated by the apolipoprotein (apo) constitution. Lp(a) is composed of apo B100 covalently linked to apo(a), which is a highly differentiated protein that was recently derived from plasminogen (Plg) in humans and great apes through gene duplication. This homology is characterized by the presence of kringle domains, which provides a unique ability for both apo(a) and Plg in binding lysine residues. This binding is prevented by lysine mimetics, for example, tranexamic acid or α-aminocaproic acid. The number of kringle IV type 2 (KIV-2) repeats is genetically determined and defines several circulating plasma apo(a) isoforms of different lengths, which is the dominant cause of large interindividual variability of Lp(a) plasma concentrations.111 The plasma level of apo(a) is inversely proportional to its length (number of KIV-2).112 Due to the presence of apo B in Lp(a), the behaviour Lp(a) is similar to that of LDL, but the presence of kringles in apo(a) further provides Lp(a) binding specificities that are similar to Plg.113 In phylogenetic mice devoid of Lp(a), positive transgenesis of human Lp(a) promotes atherosclerosis and aortic valve calcifications.114 Repeated intravenous injections of human Lp(a) similarly incited valve calcification in rabbits.114 Oxidized phospholipids may represent a specific link between Lp(a) and valvular and arterial pathologies, including calcifications115 (Figure 2). Lp(a) is also a transporter of PCSK9 and co-localizes with retention of oxidized phospholipid.116
So far, t-PA is regarded as the main activator of Plg in the arterial tissue. A new Plg Receptor [PlgR lysine (k) terminal(t), PlgRkt] has been described.117 PlgRkt is linked to urokinase (u-PA) and binds to UPAR on the cell membrane. Clustering of PlgRkt and u-PA/UPAR on cell membranes promotes Plg activation in tissues. There are numerous data showing that u-PA activation occurs in arterial aneurysms118 and human aortic valves.119 PlgRkt has the activities of both promoting Plg activation117 by u-PA in the tissue, as well as engulfing Lp(a).120 The specific linkage to Lp(a), via phospholipids,115 was recently confirmed,121 but the specific role of apo(a) and its specific binding to PlgRkt has up till now not been explored. This point is important from a therapeutic point of view, since lysine mimetics (tranexamic acid or α-aminocaproic acid, cf. supra) inhibit the binding of apo(a) to the lysine residue and, therefore, the endocytosis of apo(a).122 However, the therapeutic implications of these antifibrinolytics for soft tissue calcification is yet to be established.
The phospholipase (PL) and alkaline phosphatase (ALP) enzymatic families are directly and indirectly involved in the exposure and release of phosphate from phospholipids to promote mineralization. ALPs are conserved enzymes, already present in prokaryotes, particularly in Escherichia coli. In humans, there are four isozymes, intestinal, placental, bacterial ALP, and tissue-non-specific ALP (TNAP). TNAP is a GPI-anchored membrane protein that exists on cell membranes, but also in cell-derived matrix vesicles in bone formation, and exosomes released from SMC during pathological processes.123 Membranous ALP participates in the release of Pi from phospholipids, but predominantly in the release of Pi from pyrophosphates (PPi),124 which will be further outlined below. Therefore, ALP participates in the initiation of the extracellular formation of calcium phosphate crystals and mineralization.125 Loss‐of‐function mutations in tissue ALP (hypophosphatasia) lead to extracellular accumulation of pyrophosphate, rickets, osteomalacia and teeth hypomineralization in children, but less severe forms (such as repeated fractures) in adulthood.126 ALP is present on arterial cells, including SMC127 and endothelial cells.128 Elevated plasma ALP activity is an independent prognostic marker for cardiovascular disease (CVD) in humans,129 and TNAP inhibition can effectively prevent ectopic calcification in vitro and ex vivo.130
Phospholipases (PL A1, A2, B, C, D) are enzymes that hydrolyze phospholipids, which release fatty acids and different forms of the polar head containing phosphates. Secretory PLA2 is highly involved in the phospholipid metabolism of vascular SMC membranes. Extracellular PLA2 is transported in association with lipoproteins (LP-PLA2), mainly LDL and Lp(a).131 LP-PLA2 is a cardiovascular risk biomarker,132 but clinical trials of PLA2 inhibitors did not reveal a positive impact on CVD.133 However, specific effects on calcifications were not explored in those studies. A systems biology approach was recently applied to propose PLA2 inhibition as a powerful anti-calcification agent in human SMC culture and an in vivo mouse model.24 In agreement with these findings, PLA2 is positively correlated with the expression of BMP, TNAP, and osteopontin.134 PLD releases lysophosphatidic acid, an important signalling molecule capable of enhancing procalcifying genes,135 and also promotes mineralization.136
Taken together, these observations support the proposal that phospholipase diversity and activity directly or indirectly promote mineralization by facilitating Pi production, calcium phosphate precipitation, and procalcifying gene expression.137
Nucleic acids
Nucleic acids are biopolymers constituted on the backbone of phosphate and sugar (ribose or deoxyribose), on which sequential base pairs are anchored (Figure 5). Free nucleic acids are powerful triggers of the innate immune response, induction of interferon, secretion of IL-1β and activation of different cell death pathways.138 In addition to representing an important source of Pi, the polyanionic nature of DNA will interact strongly with cationic calcium. It is worth noting that hydroxyapatite columns were initially used to purify DNA,139 while calcium phosphate nanoparticles were used as vectors for cell DNA transfection.140 Therefore, extracellular free tissue DNA (and RNA) are potential ‘hot spots’ for ionized calcium precipitation on exposed nucleic acid phosphates. The release of free DNA is directly related to cell death and possibly to tissue extracellular traps. Indeed, the co-localization of vascular calcifications with extracellular free DNA within the arterial wall is associated with cell disappearance in the core of human atherosclerotic plaques26 and the calcified aortic valves (Figure 5). Moreover, injections of free DNA into the aortic wall in rodents can cause calcification.26 In this context, it is important to note that 4′,6-Diamino-2-Phenylindole (DAPI, Hoechst 33420), which is commonly used to stain nucleic acids, is not highly specific to DNA. In addition to forming hydrogen bonds with guanine-cytosine in double-stranded DNA and with adenine-uracil in RNA, DAPI/Hoechst also forms complexes with anionic phosphates through electrostatic interactions with positively charged end-groups.76 This property of DAPI/Hoechst has been used for the detection of inorganic polyphosphates (poly-Pi) in bacteria.77,141 Although the DAPI-DNA complex fluoresces at an excitation wavelength of 360 nm, the DAPI/Hoechst interactions with poly-Pi cause a red-shift of the fluorescence at 520–550 nm.76,Figures 3 and 5 depict phosphates detected by this technology, as a background for valve calcification after decalcifying treatment with EDTA (a chelator of divalent cations); nevertheless, this fluorescence is not visible on non-decalcified controls. Inorganic polyphosphates are also present in tissues, cells, organelles, and plasma,142,143 particularly in human osteoblasts.144 However, to the best of our knowledge, the electrostatic interactions of DAPI with Pi have not yet been applied for phosphate detection in the context of soft tissue calcification.

Nucleic acids. The phosphate group in nucleic acids is attached to a main chain of sugars (ribose or deoxyribose), in which sequential base pairs are anchored. Bottom photo panel of different staining of an aortic valve calcification: (i) Alizarin red staining of calcium before EDTA chelation; (ii) higher magnification; (iii) Hoechst fluorescent staining (DAPI, U.V. wave length: 330 nm) after EDTA chelation of calcium. (iv) Hoechst fluorescent staining (red, wave length: 550 nm). Hoechst binds to the guanine-cytosine pair of intact DNA, but also to phosphate (see text). (v) Anti-phosphatidylcholine antibody (Sigma) stained by E06 Calcification. (vi) TUNEL binds to DNA breakages of fragmented DNA through an enzymatic terminal deoxynucleotidyl transfer of a tagged oligonucleotide. Therefore, this staining shows the presence of damaged DNA in the calcification background after treatment with EDTA. (vii) Immunostaining of calcification by DNA (ab27156, abcam) and histone antibodies (ab AE-4, Santa Cruz Biotech). (viii) Scanning Electron Microscopy of intact calcification.
Another source of free nucleic acids is the polymeric ADP-ribose (PAR) chain, constituted by polymerization of monomer [ribose-pyrophosphate (diphosphate)-Adenine (nitrogen purine base), PAR] under the control of Poly(ADP-ribose) polymerase-1 (PARP-1). The polymer is devoid of membrane, forming non-microvesicular condensates and capable of being conjugated with targeted proteins, which is known as parylation.145 This system is directly involved in the initial step of DNA repair. PARP activity increases in response to DNA damage and oxidative stress. Furthermore, PAR deposition has been detected in the ECM of human arteries with either medial or atherosclerotic intimal calcification, whereas either normal arteries or non-calcified areas in diseased arteries exclusively exhibited nuclear PAR positivity.146 Importantly, PARP1 is up-regulated in calcified aortic valves.147 Therefore, DNA damage leading to PARP activation and subsequent PAR-chain synthesis and extracellularization may physiochemically stimulate ECM calcification,146 suggesting that DNA damage may serve as a starting point for arterial and valvular calcification though repair responses.146,147 In this process, ionized Ca precipitates in the diphosphate moiety of ADP-ribose. Indeed, conditional aortic PARP1 knockdown in a rodent model of adenine-induced CKD reduces medial calcification.148
Finally, a single-nucleotide polymorphism in the histone deacetylase HDAC9 gene was associated with abdominal aortic calcifications, suggesting also the involvement of chromatin acetylation/deacetylation in vascular mineralization.149 Among the inherited syndromes150 associated with diffuse calcifications of the arterial system, Singleton-Merten Syndrome (SMS) is interesting in the nucleotide context. Gain-of-function mutation in retinoic acid-inducible gene-I (RIG-I), which is a helicase capable of binding to double-stranded (ds) RNA in response to interferon-stimulated gene, triggers SMS that is potentially related to exposure and/or abnormal metabolism of nucleotide phosphates.151 SMS is characterized by dental dysplasia, osteoporosis, and diffuse arterial calcifications associated with phosphate release, which could be reproduced experimentally in mice.152 In this monogenic context of rare inherited arterial calcification syndromes [generalized arterial calcifications of infancy (GACI), pseudoxanthoma elasticum, calcifications of joints and arteries, idiopathic basal ganglia calcifications], all observed gene mutations directly affect phosphate metabolism.150 Once more, these genetic observations provide evidence both for the predominant initial role of extracellular phosphates in arterial calcifications and for the diversity of the phosphate process (enzymes, transporters) capable of promoting arterial calcifications.
Purinergic system as phosphate source and pyrophosphate metabolism
Besides exposed nucleic acids, intracellular phosphoryl transfers and extracellular purinergic signalling also represent potential sources of phosphates. Purinergic extracellular signalling, including nucleosides and nucleotides as ligands, as well as P1 and P2 receptors respectively, is a ubiquitous regulatory pathway in the CV system.153 Intracellular ATP is regenerated from AMP through the AMP-activated protein kinase (AMPK),154 thereby maintaining the energy homeostatic balance. Although intracellular energetic signalling may not provide a phosphate source for extracellular ectopic calcification, activation of AMPK by metformin prevents vascular calcification,155 suggesting that maintaining the intracellular energetic balance is protective. Conversely, the suppression of AMPK activity promotes atherosclerosis and vascular calcification in mice.156
In contrast, the metabolism of extracellular ATP involving ectoenzymes is crucial for the generation of Pi and inorganic pyrophosphates (PPi). In plasma, the minimum Pi and Ca product necessary to induce hydroxyapatite crystallization on collagen is much higher than in other fluids, which led to the discovery of PPi as a critical systemic mineralization inhibitor.157 PPi consists of two ester-linked Pi molecules and owes its name to PPi preparation from heating Pi (pyro meaning fire in Greek).158 In addition to physicochemically preventing calcium phosphate crystallization, PPi also competes with Pi for binding to established hydroxyapatite surfaces, thereby slowing down or terminating CaP mineralization processes.
PPi is generated by ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), through the hydrolysis of ATP into PPi and ADP. However, the metabolism of ATP and ADP can also contribute to Pi when hydrolyzed by ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1; Figure 6). Consequently, the ENPP1/ENTPD1 ratio has been suggested as a key regulator of the Pi and PPi synthesis from ATP in SMCs159 (Figure 6). As mentioned above, monogenetic disorders affecting ENPP1 and the ATP transporter ABCC6 have provided critical evidence for the decisive role of extracellular ATP availability and PPi biosynthesis for soft tissue mineralization.45 Likewise, the genetic targeting of these components in mice leads to spontaneous cardiovascular mineralization, as recently reviewed.45 The pathways are summarized in Figure 6.
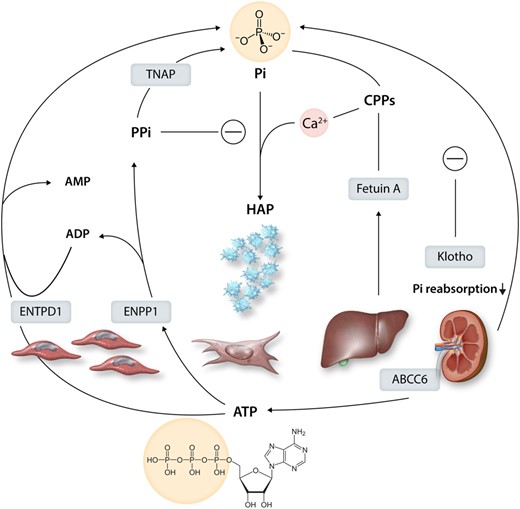
Purinergic and pyrophosphate metabolism into inorganic phosphate (Pi) and its regulation. Intracellular ATP is transported to the extracellular space by the transporter ABCC6. The hydrolysis of ATP by ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) generates inorganic pyrophosphate (PPi), which inhibits the formation of hydroxyapatite (HAP). In contrast, metabolism of ADP to by ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1) and PPi metabolism by tissue non-specific alkaline phosphatase (TNAP) increase Pi and HAP formation. Pi levels are regulated by calcification inhibitors, including Fetuin-A produced by the liver, which forms colloidal calcium-phosphate complexes called calciprotein particles (CPPs). It also shows the endogenous Pi regulator klotho, a co-receptor for fibroblast growth-factor 23 (FGF23), which decreases renal phosphate reabsorption.
Hydrolysis of PPi into Pi is catalyzed by ALP activity, which when produced by mineralized tissues, consumes local PPi levels and releases this physiological brake on hydroxyapatite deposition and growth. Tissue ALP was up-regulated in SMCs cultured under procalcifying conditions,74,159 and either genetic knockdown or pharmacological ALP inhibition could decrease the in vitro calcification of SMCs74 and VICs.160
The Pi/PPi161 balance emerges as a key regulator for soft tissue calcification in vessels and valves. In this balance, the regulation of Pi levels, for example, by fetuin-A and Klotho (cf. supra; Figure 6) also plays a major role. Restoring PPi by oral administration reverses the soft tissue calcification caused by the genetic targeting of systemic and local PPi in mice.162 In addition, further optimization of PPi accumulation, by means of ATP replacement therapy in combination with TNAP and ENTPD inhibitors, reduces vascular calcification in a murine model of Hutchinson-Gilford progeria syndrome.159 It should also be noted that PPi can crystalize with calcium, referred to as calcium PPi deposition (CPPD), and presents as chondrocalcinosis in the joints.163 Although articular chondrocalcinosis is associated with vascular calcification,163 direct vascular CPPD has not been reported so far.
Similar to PPi, inorganic poly-Pi prevents calcium precipitation on orthophosphates and inhibits hydoxyapatite formation.142 Inorganic poly-Pis are phosphate polymers assembled at different chain lengths by phosphoanhydride bonds. First described in bacteria, poly-Pi is a marker of protozoa acidocalcisomes, conserved from bacteria to man in organelles derived from common evolutionary steps.77 Several polyphosphate kinases have been identified in bacteria, but not in humans.164 The most important source of Poly-Pi in mammals is platelets.143 PolyPs are involved in numerous steps of coagulation164 and are also actively involved in calcium homeostasis within mitochondria.165 Poly-Pi is degraded by TNAP, but the possible preventive role of poly-Pi in the soft tissue mineralization process remains to be explored.
Therapeutic potential of phosphate inhibitors for cardiovascular mineralization
In summary, based on the concepts discussed above, phosphate emerges as a key regulator of vascular and valvular calcification and, as such, an attractive therapeutic target. Although there is a rationale for reducing phosphate in hyperphosphataemic CKD, the effects of phosphate reduction in normophosphataemia are largely unknown. Targeting the Pi and PPi balance to promote the anti-calcifying effects of PPi and its analogues would be an alternative therapeutic strategy within the phosphate pathways. Recently, phosphomimetics that replace Pi in the hydoxyapatite mineralization process to inhibit crystal growth are being evaluated in clinical trials for their efficacy in inhibiting cardiovascular calcification. These different phosphate-targeted therapeutic approaches are summarized in Figure 7 and further discussed below.
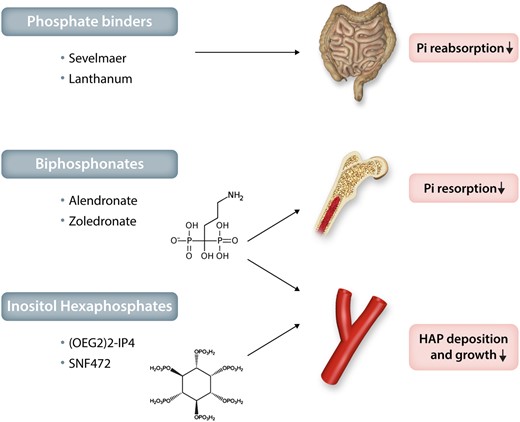
Therapeutic potential of phosphate inhibitors for cardiovascular mineralization. Examples of phosphate binders, bisphosphonates and inositol hexaphosphates and their effects on the absorption and resorption of inorganic phosphate (Pi), as well as on hydroxyapatite (HAP) deposition and growth during valvular and vascular mineralization.
Phosphate binders
Sevelamer is a non-absorbed phosphate-binding polymer, which decreases circulating phosphate levels in CKD by reducing gastrointestinal uptake (Figure 7). Randomized trials in haemodialysis patients have shown that controlling serum Pi levels with sevelamer prevents the progression of coronary and aortic calcifications, which was observed in the arm treated with calcium-containing phosphate binders.166,167 Although these data suggest favouring non-calcium-containing phosphate binders in haemodialysis, it is not yet certain whether an increase in oral calcium intake in the control group may have accelerated the vascular calcification in that study. Sevelamer and lanthanum (both non-calcium-based phosphate binders) decreased atherosclerotic calcification in a murine model by combining hyperlipidaemia and renal failure.168
Bisphosphonates
Bisphosphonates are PPi analogues that cannot be hydrolyzed by ALPs. The clinical bisphosphonate use in the treatment of osteoporosis is based on the bone-conserving actions mediated through inhibition of bone resorption. These include direct effects on bone-resorbing osteoclasts in terms of osteoclast apoptosis and interaction with specific cellular processes, including the mevalonate pathway for nitrogen-containing bisphosphonates.169 Therefore, the potential beneficial effects of bisphosphonates on vascular and valvular mineralization can be anticipated both by a decrease in Pi release from bone resorption (Figure 7) and by mimicking PPi’s protection against dystrophic calcification without being limited by TNAP degradation (Figure 6). Indeed, in a murine model of GACI through mutations in the gene encoding ENPP1 (cf. supra) the bisphosphonate etidronate both reduced aortic calcification and restored bone architecture.170
A systematic review of clinical studies evaluating the effects of bisphosphonates on vascular mineralization concluded that etidronate and nitrogen-containing bisphosphonates, such as alendronate (Figure 7), may reduce vascular calcification in haemodialysis patients. In contrast, the existing data on the effects of oral bisphosphonates on vascular calcification in non-CKD populations were either inconclusive or showed moderate effects.8 Likewise, a recent post hoc analysis of a randomized trial of zoledronic acid administered annually to postmenopausal women with osteoporosis found no significant effect on abdominal aortic calcification over 3 years.171
Some of the available observational studies have supported an association between bisphosphonate use and the progression of aortic stenosis and valve calcification.172 Randomized studies are ongoing on alendronate treatment in non-osteoporotic patients with moderate aortic stenosis.173
Inositol hexaphosphates
Phytate (myo-inositol hexaphosphate; InsP6) is a naturally occurring product in legumes, seeds and nuts, which inhibits vascular calcification in rodent models.174 In analogy to PPi, InsP6 binds to hydroxyapatite, so as to prevent crystal growth. Oligo(ethylene glycol) (OEG) conjugates of InsP6 increase its bioavailability after oral administration and decrease degradation through enzymatic hydrolysis compared to unconjugated InsP6.175 In the latter study, OEG-conjugated InsP6 reduced the uptake of the calcium-binding radiotracer 8F-NaF in ex vivo incubations of calcified femoral artery and aortic valve samples.175 The intravenous formulation of InsP6 (SNF472) administered during haemodialysis sessions reduced the progression of coronary and aortic valve calcium volume score compared to placebo in a recently reported phase 2b study in patients with end-stage renal disease.176
Summary and conclusion
Extracellular phosphates and exposed tissue phosphates, on which ionized calcium can precipitate, are the key limiting factor for the initiation and progression of calcification processes in the arterial wall and heart valves. Sources of extracellular phosphates are numerous, including circulating Pi, phospholipids (from cell membranes, microvesiculation, and lipoproteins), nucleic acids, purinergic, and pyrophosphate metabolism.
While several pathways and regulatory processes for phosphate-mediated mineralization are shared with bone homeostasis, the ectopic mineralization of the arterial wall and valves have distinct features in terms of ECM structure and cellular actions and activation. In particular, for vascular and valvular structural cells to remain in a homeostatic state, a high exosomal activity must be retained to keep the clearance function intact within the arterial wall and valves. This, along with energetic phosphoryl transfers and the need for phosphate as part of the membrane phospholipid structure, further illustrates the need of phosphate for the structure and function of the vascular wall.
Possible effects on bone mineralization should also be considered when interfering with extracellular phosphate availability and pyrophosphate metabolism. The occurrence of side effects on bone homeostasis must therefore be closely monitored when evaluating treatments targeting the calcium phosphate balance should also consider.
The initial clinical assessment of phosphate binders, bisphosphonates and inositol hexaphosphates supports the potential of targeting phosphates to slow down valvular and vascular calcification. Other potential therapeutic targets that are yet to be evaluated for their effects on cardiovascular calcification include lysine mimetics (tranexamic acid or α-aminocaproic acid) and phospholipase inhibitors to limit phospholipids as exposed phosphates for calcium precipitation and mineralization. Therefore, understanding the major role of exposed phosphates may open up new therapeutic avenues to prevent cardiovascular calcification.
Funding
M.B. is an awardee of the Gutenberg Chair of Excellence from the Région Grand Est and the Eurométropole de Strasbourg (France) and supported by the Swedish Research Council (Grant number 2019-01486). J.-B.M. was supported by the French Society of Cardiology (Alain Castaigne Award) and Avenir Foundation (Major prize 2019).
Conflict of interest: M.B. has acted as a consultant for Inositec AG.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}