




Abstract
Autosomal dominant cerebellar ataxias have a marked heterogeneous genetic background, with mutations in 34 genes identified so far. This large amount of implicated genes accounts for heterogeneous clinical presentations, making genotype–phenotype correlations a major challenge in the field. While polyglutamine ataxias, linked to CAG repeat expansions in genes such as ATXN1, ATXN2, ATXN3, ATXN7, CACNA1A and TBP, have been extensively characterized in large cohorts, there is a need for comprehensive assessment of frequency and phenotype of more ‘conventional’ ataxias. After exclusion of CAG/polyglutamine expansions in spinocerebellar ataxia genes in 412 index cases with dominantly inherited cerebellar ataxias, we aimed to establish the relative frequencies of mutations in other genes, with an approach combining panel sequencing and TaqMan® polymerase chain reaction assay. We found relevant genetic variants in 59 patients (14.3%). The most frequently mutated were channel genes [CACNA1A (n = 16), KCND3 (n = 4), KCNC3 (n = 2) and KCNA1 (n = 2)]. Deletions in ITPR1 (n = 11) were followed by biallelic variants in SPG7 (n = 9). Variants in AFG3L2 (n = 7) came next in frequency, and variants were rarely found in STBN2 (n = 2), ELOVL5, FGF14, STUB1 and TTBK2 (n = 1 each). Interestingly, possible risk factor variants were detected in SPG7 and POLG. Clinical comparisons showed that ataxias due to channelopathies had a significantly earlier age at onset with an average of 24.6 years, versus 40.9 years for polyglutamine expansion spinocerebellar ataxias and 37.8 years for SPG7-related forms (P = 0.001). In contrast, disease duration was significantly longer in the former (20.5 years versus 9.3 and 13.7, P=0.001), though for similar functional stages, indicating slower progression of the disease. Of interest, intellectual deficiency was more frequent in channel spinocerebellar ataxias, while cognitive impairment in adulthood was similar among the three groups. Similar differences were found among a single gene group, comparing 23 patients with CACNA1A expansions (spinocerebellar ataxia 6) to 22 patients with CACNA1A point mutations, which had lower average age at onset (25.2 versus 47.3 years) with longer disease duration (18.7 versus 10.9), but lower severity indexes (0.39 versus 0.44), indicating slower progression of the disease. In conclusion, we identified relevant genetic variations in up to 15% of cases after exclusion of polyglutamine expansion spinocerebellar ataxias, and confirmed CACNA1A and SPG7 as major ataxia genes. We could delineate firm genotype–phenotype correlations that are important for genetic counselling and of possible prognostic value.
Introduction
Hereditary cerebellar ataxias are a heterogeneous group of neurological diseases presenting as a cerebellar syndrome, with a combination of gait alteration, limb incoordination, dysarthria, and eye movement anomalies (Schols et al., 2004). Additional neurological (e.g. pyramidal signs, seizures, cognitive impairment, extrapyramidal symptoms such as dystonia) or extraneurological (e.g. cardiomyopathy, diabetes, chorioretinal dystrophy) signs often complete the clinical picture along with evolution (Durr, 2010). Progressive cerebellar syndromes can be of various origins, but many forms are genetically inherited. They can be transmitted on all described inheritance modes, including autosomal dominant.
The genetic background of autosomal dominant cerebellar ataxias, so-called spinocerebellar ataxias (SCAs), is heterogeneous, with at least 34 genes identified. The most frequent forms are due to CAG trinucleotide repeat expansions in seven genes leading to polyglutamine stretch elongation in their respective proteins: ATXN1 (MIM 601556)/SCA1 (MIM 164400) (Orr et al., 1993), ATXN2 (MIM 601517)/SCA2 (MIM 183090) (Pulst et al., 1996), ATXN3 (MIM 607047)/SCA3 – Machado-Joseph disease (MIM 109150) (Kawaguchi et al., 1994), CACNA1A (MIM 601011)/SCA6 (MIM 183086) (Zhuchenko et al., 1997), ATXN7 (MIM 607640)/SCA7 (MIM 164500) (David et al., 1997), TBP (MIM 600075)/SCA17 (MIM 607136) (Koide et al., 1999), ATN1 (MIM 607462)/dentatorubral-pallidoluysian atrophy (MIM 125370) (Koide et al., 1994). In SCA8, CTG/CAG trinucleotide repeat expansions were described in patients (Koob et al., 1999), then also in controls (Stevanin et al., 2000; Worth et al., 2000), leading to controversy as to their pathogenic role. In transgenic mice, they induce a progressive neurological phenotype, and encode almost pure polyglutamine stretches from the ATXN8 transcript, and an RNA with non-coding CUG expansion from the opposite strand transcript ATXN8OS (Moseley et al., 2006).
Intronic expansions have also been described in SCA10 (Matsuura et al., 2004) (MIM 603516) with ATTCT repeats in intron 9 of ATXN10 (MIM 611150), SCA12 (Holmes et al., 1999) (MIM 604326) with CAG repeats in the 5’UTR region of PPP2R2B (MIM 604325), SCA31 (Sato et al., 2009) (MIM 117210) with TGGAA pentanucleotide repeats in a common intron of genes BEAN (MIM 612051) and TK2 (MIM 188250), and SCA36 (Kobayashi et al., 2011) (MIM 614153) with GGCCTG hexanucleotide repeats in the first intron of NOP56 (MIM 614154).
Conventional mutations have been described in 22 genes so far (Supplementary Table 1): SPTBN2/SCA5 (Ikeda et al., 2006), TTBK2/SCA11 (Houlden et al., 2007), KCNC3/SCA13 (Waters et al., 2006), PRKCG/SCA14 (Chen et al., 2003), ITPR1/SCA15-16 (van de Leemput et al., 2007), KCND3/SCA19-22 (Duarri et al., 2012; Lee et al., 2012), TMEM240/SCA21 (Delplanque et al., 2014), PDYN/SCA23 (Bakalkin et al., 2010), EEF2/SCA26 (Hekman et al., 2012) (one family), FGF14/SCA27 (van Swieten et al., 2003), AFG3L2/SCA28 (Di Bella et al., 2010), ELOVL4/SCA34 (Cadieux-Dion et al., 2014) (one family), TGM6/SCA35 (Wang et al., 2010a), ELOVL5/SCA38 (Di Gregorio et al., 2014), CCDC88C/SCA40 (Tsoi et al., 2014) (one family), TRPC3/SCA41 (Fogel et al., 2015) (one patient), CACNA1A (Yue et al., 1997), DNMT1 (Winkelmann et al., 2012), OPA1 (Amati-Bonneau et al., 2008; Hudson et al., 2008), VAMP1/SPAX1 (Bourassa et al., 2012) (SPAstic ataXia type 1, one family), and GRID2 (Coutelier et al., 2015b). The involvement of IFRD1 in SCA18 is debated (Brkanac et al., 2009), as the only reported mutation is rather frequent in the ExAC database (www.exac.broadinstitute.org, 58 heterozygous patients/60 655); and as the affected amino acid is not conserved in distant mammals such as the elephant. Biallelic mutations in POLG are responsible for a broad clinical spectrum, including sensitive or cerebellar ataxia in some instances (Van Goethem et al., 2001; Luoma et al., 2005). If autosomal dominant transmission has been well documented in progressive external ophtalmoplegia (Van Goethem et al., 2001; Lamantea et al., 2002), it is still unclear whether heterozygous variant can cause cerebellar ataxia (Schulte et al., 2009).
Compared to polyglutamine ataxias, these conventional forms are more slowly progressive (Durr, 2010), with a longer survival and a less severe disability stage (Monin et al., 2015). The cerebellar component is more important than additional signs, which are prominent in polyglutamine SCAs, where intrafamilial presentation varies more (Marelli et al., 2011a). Mutations in DNMT1, OPA1 and POLG account for more complex phenotypes, with deafness and narcolepsy in the first case, and mitochondria-related symptoms in both others (optic atrophy, spasticity). VAMP1 variants are responsible for a markedly spastic ataxia.
Until recently, the classical genetic work-up in dominant cerebellar ataxias was to genotype patients for CAG repeats in polyglutamine SCA genes. Mutations in other genes were only looked for in case of clinical suspicion, which was not frequent because phenotype–genotype correlations are non-univocal (Coutelier et al., 2015c), and few specific symptoms exist for most conventional SCAs. Hence, many patients were never screened for most of these genes, and diagnostic yield remained low, with around 40% of individuals without diagnostic (Durr, 2010; Ruano et al., 2014). We thus aimed to (i) assess the efficiency of a panel sequencing approach in dominant cases of cerebellar ataxias; and (ii) study the frequency and phenotype of a large cohort of index cases for mutations in conventional SCA genes.
Material and methods
Patient recruitment, clinical evaluation and initial genetic work-up
Four hundred and twelve index patients were recruited as part of the SPATAX cohort of patients with Spastic Paraplegia and Ataxia (https://spatax.wordpress.com/). They were examined by at least one member of the SPATAX network, and clinically assessed with a standardized evaluation form (https://spatax.files.wordpress.com/2013/09/fichecliniquespatax-eurospa-2011.pdf). Functional stages were evaluated as follows: 0, no functional handicap; 1, no functional handicap but signs at examination; 2, mild functional handicap, able to run; 3, moderate functional handicap, unable to run, limited walking without aid; 4, severe functional handicap, walking with one stick; 5, walking with two sticks; 6, unable to walk, requiring wheelchair; 7, confined to bed. Autosomal dominant inheritance was assumed based on positive first-degree familial history in more than 95% of patients. However, in many cases, DNA was only available for the index patient. Five patients were sporadic, three had unclear familial history due to premature death of one parent, and four had first or second degree relatives with another neurological disorder (Parkinson’s disease, tremor, supranuclear palsy or uncategorized motor impairment). Patients gave informed consent, and blood samples were collected in accordance with local French regulations [Paris Necker ethics committee approval (RBM 01-29 and RBM 03-48) to A.B. and A.D.]. DNA was extracted using standard procedures. Polyglutamine expansions in ATXN1, ATXN2, ATXN3, ATXN7, CACNA1A and TBP were excluded in all patients. In previous research settings, several genes have been screened for in a subset of patients and positive cases therefore excluded (including four SCA5, one SCA11, one SCA12, six SCA13, 11 SCA14, one SCA22, three SCA23, nine SCA28, 12 SCA36). During the course of the study, causative mutations were nevertheless identified in six patients of the cohort (one HTT, one FA2H, one SCA1, two SCA2, one SCA3). ITPR1 deletions, responsible for the SCA15 subtype of SCA, were looked for in all index cases, with a TaqMan® PCR assay designed by Obayashi et al. (2012), in parallel with panel sequencing. Results were analysed for probes 1 to 3 only, as probe 4 gave poorly reproducible results.
Gene list establishment
All genes implicated in ataxia by April 2014 were included: AFG3L2, CACNA1A, DNMT1, EEF2, ELOVL5, FGF14, GRID2, IFRD1, ITPR1, KCNC3, KCND3, OPA1, PDYN, POLG, PRKCG, SPTBN2, TGM6, TTBK2, and VAMP1, as mentioned above. Because point mutations in CACNA1A were first recognized to cause episodic ataxia (Ophoff et al., 1996) (EA), then extended to progressive cerebellar ataxia (Yue et al., 1997), and because interictal ataxia may occur in episodic ataxia, three genes involved in episodic ataxia were also included: KCNA1 (EA1; Browne et al., 1994), CACNB4 (EA5; Escayg et al., 2000), and SLC1A3 (EA6; Jen et al., 2005). A few other genes whose mutations are related to other core traits before ataxia were also added: ITM2B, linked to dementia (British-type and Danish-type) with ataxia (Vidal et al., 1999); NOL3, whose mutation in one pedigree caused familial cortical myoclonus, later followed by ataxia (Russell et al., 2012); REEP1 and SPG7, whose variants are associated with prominent hereditary spastic paraplegia, with a frequent cerebellar component (Casari et al., 1998; Zuchner et al., 2006; Klebe et al., 2012). If SPG7 causative variants are classically biallelic in hereditary spastic paraplegia, autosomal dominant transmission has been established in optic neuropathy (Klebe et al., 2012). Relatives of affected individuals that carried heterozygous variants were also shown to harbour frequent cerebellar atrophy, or slight late-onset cerebellar signs (Klebe et al., 2012). STUB1, implicated in autosomal recessive cerebellar ataxia (Shi et al., 2013), was also included to assess a potential implication in SCAs, since we otherwise found a heterozygous variant segregating in one family. Because they had not been described in SCAs by the time we designed the panel, CCDC88C, ELOVL4, TMEM240 and TRPC3 were not included.
The full list of known genes considered for sequencing is described in Supplementary Table 1.
Candidate genes were also included, which brought the number of genes to 65. Among them, CACNA1G was sequenced, which allowed confirmation of its implication in SCA (Coutelier et al., 2015a).
Panel sequencing
Primers to amplify the 1150 amplicons covering the 65 genes selected were designed by Fluidigm Primer D3 Design Assay. DNA was amplified using the Fluidigm Access Array™ system, according to the manufacturer’s protocol (Fluidigm). Briefly, 48 × 48 chips were loaded with, on one side, 50 ng of DNA and a PCR mix containing FastStart™ High Fidelity enzyme (Roche); and, on the other side, pools of up to 12 primer couples diluted in Access Array™ loading reagent. After chip loading, amplification, and chip harvesting, samples were barcoded with an index PCR on 100 × diluted harvested product. Indexed pools were purified with SPRIselect beads (Beckman Coulter), and then pooled equimolarily. Once purified, the final library was sequenced on a MiSeq Illumina Sequencer as 2 × 300 base pair reads using standard protocols (Illumina).
Bioinformatics processing and variant analysis
Fastq sequences were aligned to the human genome v19 reference sequence using BWA (Li and Durbin, 2009), then GATK (McKenna et al., 2010) for local realignment and recalibration, as well as detection of variants. Variants were annotated with Annovar (Wang et al., 2010b), then filtered with the following criteria: (i) quality (PASS GATK filter); (ii) effect on coding sequence (exonic non-synonymous or splice variant); (iii) frequency in public databases (<1% in EVS); (iv) internal database frequency (<5%); and (v) heterozygosity (allelic frequency between 0.3 and 0.7). Manual curation of all variants was performed, with a refinement on public frequency threshold made possible by the release of ExAC database (http://exac.broadinstitute.org; <0.01% for heterozygous variants) and a correlation to the clinical picture. All possibly relevant variants were Sanger sequenced using classical procedures, as well as most of the variants of unknown significance. When available, segregation of the variant was assessed in family members (Table 1 and Supplementary Fig. 3).
Class 1, class 2 and class 3 variants
| Patient ID | Gene | Status | Variant | Pathogenicity predictions | GERP++ | Other | ExAC | Segregation | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| AAD-636-3 | CACNA1A | Class 1 | NM_001127222:c.1745G>A:p.R582Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-414-3 | CACNA1A | Class 1 | NM_001127222:c.2039_2040del:p.680_680del | Frameshift variant | – | – | 0 | NA | Pathogenic |
| AAD-899-13 | CACNA1A | Class 1 | NM_001127222:c.3787G>A:p.E1263K | Known variant | – | – | 0 | TRUE (2 affected) | Pathogenic |
| AAD-555-11 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-870-1 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-202-7 | CACNA1A | Class 1 | NM_001127222:c.4979G>A:p.R1660H | 4/4 | 4.96 | Close to known R1664Q, recurrent in another study | 0 | NA | Likely pathogenic |
| AAD-750-3 | CACNA1A | Class 1 | NM_001127222:c.4996C>T:p.R1666W | Known variant | – | – | 0 | NA | Pathogenic |
| 14GX000698 | CACNA1A | Class 1 | NM_001127222:c.5200G>T:p.E1734X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-82-4 | CACNA1A | Class 1 | NM_001127222:c.5248C>T:p.R1750W | Known variant | – | – | 0 | TRUE (6 affected) | Pathogenic |
| AAD-890-6 | CACNA1A | Class 1 | NM_001127222:c.6028C>T:p.Q2010X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-883-1 | CACNA1A | Class 2 | NM_001127222:c.835C>T:p.R279C (+ NM_001127221:c.5575A>G:p.I1859V (non-canonical transcript)) | 4/4 | 4.24 | Close to known C287Y | 0/0 | NA | Likely pathogenic |
| AAD-603-1 | CACNA1A | Class 2 | NM_001127222:c.880C>T:p.P294S | 4/4 | 5.42 | Close to known G293R | 0 | NA | Likely pathogenic |
| AAD-875-1 | CACNA1A | Class 2 | NM_001127222:c.2026G>A:p.G676R | 4/4 | 4.58 | Close to known E668K | 0 | NA | Likely pathogenic |
| AAD-263-11 | CACNA1A | Class 3 | NM_001127222:c.2056G>A:p.G686S | 2/4 | 3.55 | Close to known E668K | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-747-11 | CACNA1A | Class 3 | NM_001127222:c.4448G>T:p.R1483L | 3/4 | 5.13 | Close to known F1491S | 0 | NA | Likely pathogenic |
| AAD-70-7 | CACNA1A | Class 3 | NM_001127221:c.5578_5579insAT:p.S1860fs (non-canonical transcript) | Frameshift variant | – | Not in canonical transcript | 0 | NA | Pathogenic |
| AAD-785-9 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V; c.858dupT:p.A286fs | A510V + frameshift variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAD-248-12 | SPG7 | Class 1 | NM_003119:c.958G>T:p.E320X; c.1047dupC:p.G349fs | Nonsense + frameshift variants | – | – | 0/0 | NA | Pathogenic/pathogenic |
| AAD-420-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAR-541-13 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant; also detected by WES (Coutelier et al., in preparation) | – | – | 306/0 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-951-11 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-847-18 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-1033-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| SAL-399-1026 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-458-5 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.2249C>T:p.P750L | A510V + known variant | – | – | 306/3/120860 | NA | Pathogenic/pathogenic |
| AAD-886-8 | AFG3L2 | Class 1 | NM_006796:c.1961C>T:p.T654I | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-994-1 | AFG3L2 | Class 1 | NM_006796:c.1996A>G:p.M666V | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-315-17 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-512-11 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-541-19 | AFG3L2 | Class 2 | NM_006796:c.1450G>C:p.A484P | 4/4 | 6.03 | In AAA domain | 1/121360 | NA | Uncertain significance; no criteria for benign |
| AAD-1009-1 | AFG3L2 | Class 2 | NM_006796:c.2062C>G:p.P688A | 4/4 | 5.62 | In proteolytic domain, close to known Y689H | 0 | NA | Likely pathogenic |
| AAD-1005-1 | AFG3L2 | Class 3 | NM_006796:c.1430C>T:p.P477L | 2/4 | 6.03 | In AAA domain | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-722-1 | KCND3 | Class 1 | NM_004980:c.1348C>T:p.L450F | Previously reported patient | – | – | 14/90828 | NA | Likely pathogenic |
| AAD-114-9 | KCND3 | Class 1 | NM_004980:c.1897C>T:p.P633S | Previously reported patient | – | – | 3/121388 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-374-1 | KCND3 | Class 2 | NM_004980:c.1086T>G:p.I362M | 4/4 | −8. but conserved amino acid across species | Close to known T352P and M373I | 0 | TRUE (3 affected) | Likely pathogenic |
| AAD-484-1 | KCND3 | Class 2 | NM_004980:c.1094T>C:p.M365T | 4/4 | 5.59 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-350-9 | KCNA1 | Class 1 | NM_000217:c.677C>T:p.T226M | Known variant | – | – | 0 | NA | Pathogenic |
| ENE-13-18 | KCNA1 | Class 2 | NM_000217:c.508G>T:p.A170S | 4/4 | 4.91 | Heterozygous A510V in SPG7 | 0 | TRUE (two affected) | Likely pathogenic |
| AAD-982-3 | KCNC3 | Class 1 | NM_004977:c.1268G>A:p.R423H | Known variant | – | – | 0 | TRUE (two affected) | Pathogenic |
| AAD-449-3 | KCNC3 | Class 3 | NM_004977:c.1420G>A:p.A474T | 3/4 | 3.36 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-479-5 | SPTBN2 | Class 2 | NM_006946:c.1294C>A:p.R432S | 4/4 | 3.7 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-483-1 | SPTBN2 | Class 3 | NM_006946:c.1261G>A:p.E421K | 3/4 | 4.62 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-285-3 | ELOVL5 | Class 1 | NM_001242828:c.214C>G:p.L72V | Previously reported patient | – | – | 0 | NA | Likely pathogenic |
| AAD-958-1 | FGF14 | Class 2 | NM_004115:c.351G>T:p.Q117H | 4/4 | 5.87 | – | 0 | NA | Uncertain significance; no criteria for benign |
| 14GX000693 | STUB1 | Class 3 | NM_005861:c.433A>C:p.K145Q;c.502C>T:p.L168F | 3/4/4/4 | 4.59/4.59 | Trans, close to W147C and L165F/concordant phenotype, mild cognitive impairment | 82/116864/0 | NA | Pathogenic/pathogenic |
| AAD-915-6 | TTBK2 | Class 1 | NM_173500:c.1306_1307del:p.D436fs | Previously reported patient | – | – | 0 | NA | Pathogenic |
| Patient ID | Gene | Status | Variant | Pathogenicity predictions | GERP++ | Other | ExAC | Segregation | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| AAD-636-3 | CACNA1A | Class 1 | NM_001127222:c.1745G>A:p.R582Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-414-3 | CACNA1A | Class 1 | NM_001127222:c.2039_2040del:p.680_680del | Frameshift variant | – | – | 0 | NA | Pathogenic |
| AAD-899-13 | CACNA1A | Class 1 | NM_001127222:c.3787G>A:p.E1263K | Known variant | – | – | 0 | TRUE (2 affected) | Pathogenic |
| AAD-555-11 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-870-1 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-202-7 | CACNA1A | Class 1 | NM_001127222:c.4979G>A:p.R1660H | 4/4 | 4.96 | Close to known R1664Q, recurrent in another study | 0 | NA | Likely pathogenic |
| AAD-750-3 | CACNA1A | Class 1 | NM_001127222:c.4996C>T:p.R1666W | Known variant | – | – | 0 | NA | Pathogenic |
| 14GX000698 | CACNA1A | Class 1 | NM_001127222:c.5200G>T:p.E1734X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-82-4 | CACNA1A | Class 1 | NM_001127222:c.5248C>T:p.R1750W | Known variant | – | – | 0 | TRUE (6 affected) | Pathogenic |
| AAD-890-6 | CACNA1A | Class 1 | NM_001127222:c.6028C>T:p.Q2010X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-883-1 | CACNA1A | Class 2 | NM_001127222:c.835C>T:p.R279C (+ NM_001127221:c.5575A>G:p.I1859V (non-canonical transcript)) | 4/4 | 4.24 | Close to known C287Y | 0/0 | NA | Likely pathogenic |
| AAD-603-1 | CACNA1A | Class 2 | NM_001127222:c.880C>T:p.P294S | 4/4 | 5.42 | Close to known G293R | 0 | NA | Likely pathogenic |
| AAD-875-1 | CACNA1A | Class 2 | NM_001127222:c.2026G>A:p.G676R | 4/4 | 4.58 | Close to known E668K | 0 | NA | Likely pathogenic |
| AAD-263-11 | CACNA1A | Class 3 | NM_001127222:c.2056G>A:p.G686S | 2/4 | 3.55 | Close to known E668K | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-747-11 | CACNA1A | Class 3 | NM_001127222:c.4448G>T:p.R1483L | 3/4 | 5.13 | Close to known F1491S | 0 | NA | Likely pathogenic |
| AAD-70-7 | CACNA1A | Class 3 | NM_001127221:c.5578_5579insAT:p.S1860fs (non-canonical transcript) | Frameshift variant | – | Not in canonical transcript | 0 | NA | Pathogenic |
| AAD-785-9 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V; c.858dupT:p.A286fs | A510V + frameshift variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAD-248-12 | SPG7 | Class 1 | NM_003119:c.958G>T:p.E320X; c.1047dupC:p.G349fs | Nonsense + frameshift variants | – | – | 0/0 | NA | Pathogenic/pathogenic |
| AAD-420-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAR-541-13 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant; also detected by WES (Coutelier et al., in preparation) | – | – | 306/0 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-951-11 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-847-18 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-1033-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| SAL-399-1026 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-458-5 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.2249C>T:p.P750L | A510V + known variant | – | – | 306/3/120860 | NA | Pathogenic/pathogenic |
| AAD-886-8 | AFG3L2 | Class 1 | NM_006796:c.1961C>T:p.T654I | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-994-1 | AFG3L2 | Class 1 | NM_006796:c.1996A>G:p.M666V | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-315-17 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-512-11 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-541-19 | AFG3L2 | Class 2 | NM_006796:c.1450G>C:p.A484P | 4/4 | 6.03 | In AAA domain | 1/121360 | NA | Uncertain significance; no criteria for benign |
| AAD-1009-1 | AFG3L2 | Class 2 | NM_006796:c.2062C>G:p.P688A | 4/4 | 5.62 | In proteolytic domain, close to known Y689H | 0 | NA | Likely pathogenic |
| AAD-1005-1 | AFG3L2 | Class 3 | NM_006796:c.1430C>T:p.P477L | 2/4 | 6.03 | In AAA domain | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-722-1 | KCND3 | Class 1 | NM_004980:c.1348C>T:p.L450F | Previously reported patient | – | – | 14/90828 | NA | Likely pathogenic |
| AAD-114-9 | KCND3 | Class 1 | NM_004980:c.1897C>T:p.P633S | Previously reported patient | – | – | 3/121388 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-374-1 | KCND3 | Class 2 | NM_004980:c.1086T>G:p.I362M | 4/4 | −8. but conserved amino acid across species | Close to known T352P and M373I | 0 | TRUE (3 affected) | Likely pathogenic |
| AAD-484-1 | KCND3 | Class 2 | NM_004980:c.1094T>C:p.M365T | 4/4 | 5.59 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-350-9 | KCNA1 | Class 1 | NM_000217:c.677C>T:p.T226M | Known variant | – | – | 0 | NA | Pathogenic |
| ENE-13-18 | KCNA1 | Class 2 | NM_000217:c.508G>T:p.A170S | 4/4 | 4.91 | Heterozygous A510V in SPG7 | 0 | TRUE (two affected) | Likely pathogenic |
| AAD-982-3 | KCNC3 | Class 1 | NM_004977:c.1268G>A:p.R423H | Known variant | – | – | 0 | TRUE (two affected) | Pathogenic |
| AAD-449-3 | KCNC3 | Class 3 | NM_004977:c.1420G>A:p.A474T | 3/4 | 3.36 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-479-5 | SPTBN2 | Class 2 | NM_006946:c.1294C>A:p.R432S | 4/4 | 3.7 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-483-1 | SPTBN2 | Class 3 | NM_006946:c.1261G>A:p.E421K | 3/4 | 4.62 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-285-3 | ELOVL5 | Class 1 | NM_001242828:c.214C>G:p.L72V | Previously reported patient | – | – | 0 | NA | Likely pathogenic |
| AAD-958-1 | FGF14 | Class 2 | NM_004115:c.351G>T:p.Q117H | 4/4 | 5.87 | – | 0 | NA | Uncertain significance; no criteria for benign |
| 14GX000693 | STUB1 | Class 3 | NM_005861:c.433A>C:p.K145Q;c.502C>T:p.L168F | 3/4/4/4 | 4.59/4.59 | Trans, close to W147C and L165F/concordant phenotype, mild cognitive impairment | 82/116864/0 | NA | Pathogenic/pathogenic |
| AAD-915-6 | TTBK2 | Class 1 | NM_173500:c.1306_1307del:p.D436fs | Previously reported patient | – | – | 0 | NA | Pathogenic |
List of variants of Classes 1, 2 and 3 (as described in main text) found by sequencing 412 index patients with dominantly inherited cerebellar ataxia. Pathogenicity predictions were assessed by Annovar annotation with SIFT, PolyPhen 2 HDIV, LRT and MutationTaster. WES = whole exome sequencing.
Class 1, class 2 and class 3 variants
| Patient ID | Gene | Status | Variant | Pathogenicity predictions | GERP++ | Other | ExAC | Segregation | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| AAD-636-3 | CACNA1A | Class 1 | NM_001127222:c.1745G>A:p.R582Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-414-3 | CACNA1A | Class 1 | NM_001127222:c.2039_2040del:p.680_680del | Frameshift variant | – | – | 0 | NA | Pathogenic |
| AAD-899-13 | CACNA1A | Class 1 | NM_001127222:c.3787G>A:p.E1263K | Known variant | – | – | 0 | TRUE (2 affected) | Pathogenic |
| AAD-555-11 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-870-1 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-202-7 | CACNA1A | Class 1 | NM_001127222:c.4979G>A:p.R1660H | 4/4 | 4.96 | Close to known R1664Q, recurrent in another study | 0 | NA | Likely pathogenic |
| AAD-750-3 | CACNA1A | Class 1 | NM_001127222:c.4996C>T:p.R1666W | Known variant | – | – | 0 | NA | Pathogenic |
| 14GX000698 | CACNA1A | Class 1 | NM_001127222:c.5200G>T:p.E1734X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-82-4 | CACNA1A | Class 1 | NM_001127222:c.5248C>T:p.R1750W | Known variant | – | – | 0 | TRUE (6 affected) | Pathogenic |
| AAD-890-6 | CACNA1A | Class 1 | NM_001127222:c.6028C>T:p.Q2010X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-883-1 | CACNA1A | Class 2 | NM_001127222:c.835C>T:p.R279C (+ NM_001127221:c.5575A>G:p.I1859V (non-canonical transcript)) | 4/4 | 4.24 | Close to known C287Y | 0/0 | NA | Likely pathogenic |
| AAD-603-1 | CACNA1A | Class 2 | NM_001127222:c.880C>T:p.P294S | 4/4 | 5.42 | Close to known G293R | 0 | NA | Likely pathogenic |
| AAD-875-1 | CACNA1A | Class 2 | NM_001127222:c.2026G>A:p.G676R | 4/4 | 4.58 | Close to known E668K | 0 | NA | Likely pathogenic |
| AAD-263-11 | CACNA1A | Class 3 | NM_001127222:c.2056G>A:p.G686S | 2/4 | 3.55 | Close to known E668K | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-747-11 | CACNA1A | Class 3 | NM_001127222:c.4448G>T:p.R1483L | 3/4 | 5.13 | Close to known F1491S | 0 | NA | Likely pathogenic |
| AAD-70-7 | CACNA1A | Class 3 | NM_001127221:c.5578_5579insAT:p.S1860fs (non-canonical transcript) | Frameshift variant | – | Not in canonical transcript | 0 | NA | Pathogenic |
| AAD-785-9 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V; c.858dupT:p.A286fs | A510V + frameshift variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAD-248-12 | SPG7 | Class 1 | NM_003119:c.958G>T:p.E320X; c.1047dupC:p.G349fs | Nonsense + frameshift variants | – | – | 0/0 | NA | Pathogenic/pathogenic |
| AAD-420-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAR-541-13 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant; also detected by WES (Coutelier et al., in preparation) | – | – | 306/0 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-951-11 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-847-18 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-1033-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| SAL-399-1026 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-458-5 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.2249C>T:p.P750L | A510V + known variant | – | – | 306/3/120860 | NA | Pathogenic/pathogenic |
| AAD-886-8 | AFG3L2 | Class 1 | NM_006796:c.1961C>T:p.T654I | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-994-1 | AFG3L2 | Class 1 | NM_006796:c.1996A>G:p.M666V | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-315-17 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-512-11 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-541-19 | AFG3L2 | Class 2 | NM_006796:c.1450G>C:p.A484P | 4/4 | 6.03 | In AAA domain | 1/121360 | NA | Uncertain significance; no criteria for benign |
| AAD-1009-1 | AFG3L2 | Class 2 | NM_006796:c.2062C>G:p.P688A | 4/4 | 5.62 | In proteolytic domain, close to known Y689H | 0 | NA | Likely pathogenic |
| AAD-1005-1 | AFG3L2 | Class 3 | NM_006796:c.1430C>T:p.P477L | 2/4 | 6.03 | In AAA domain | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-722-1 | KCND3 | Class 1 | NM_004980:c.1348C>T:p.L450F | Previously reported patient | – | – | 14/90828 | NA | Likely pathogenic |
| AAD-114-9 | KCND3 | Class 1 | NM_004980:c.1897C>T:p.P633S | Previously reported patient | – | – | 3/121388 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-374-1 | KCND3 | Class 2 | NM_004980:c.1086T>G:p.I362M | 4/4 | −8. but conserved amino acid across species | Close to known T352P and M373I | 0 | TRUE (3 affected) | Likely pathogenic |
| AAD-484-1 | KCND3 | Class 2 | NM_004980:c.1094T>C:p.M365T | 4/4 | 5.59 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-350-9 | KCNA1 | Class 1 | NM_000217:c.677C>T:p.T226M | Known variant | – | – | 0 | NA | Pathogenic |
| ENE-13-18 | KCNA1 | Class 2 | NM_000217:c.508G>T:p.A170S | 4/4 | 4.91 | Heterozygous A510V in SPG7 | 0 | TRUE (two affected) | Likely pathogenic |
| AAD-982-3 | KCNC3 | Class 1 | NM_004977:c.1268G>A:p.R423H | Known variant | – | – | 0 | TRUE (two affected) | Pathogenic |
| AAD-449-3 | KCNC3 | Class 3 | NM_004977:c.1420G>A:p.A474T | 3/4 | 3.36 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-479-5 | SPTBN2 | Class 2 | NM_006946:c.1294C>A:p.R432S | 4/4 | 3.7 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-483-1 | SPTBN2 | Class 3 | NM_006946:c.1261G>A:p.E421K | 3/4 | 4.62 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-285-3 | ELOVL5 | Class 1 | NM_001242828:c.214C>G:p.L72V | Previously reported patient | – | – | 0 | NA | Likely pathogenic |
| AAD-958-1 | FGF14 | Class 2 | NM_004115:c.351G>T:p.Q117H | 4/4 | 5.87 | – | 0 | NA | Uncertain significance; no criteria for benign |
| 14GX000693 | STUB1 | Class 3 | NM_005861:c.433A>C:p.K145Q;c.502C>T:p.L168F | 3/4/4/4 | 4.59/4.59 | Trans, close to W147C and L165F/concordant phenotype, mild cognitive impairment | 82/116864/0 | NA | Pathogenic/pathogenic |
| AAD-915-6 | TTBK2 | Class 1 | NM_173500:c.1306_1307del:p.D436fs | Previously reported patient | – | – | 0 | NA | Pathogenic |
| Patient ID | Gene | Status | Variant | Pathogenicity predictions | GERP++ | Other | ExAC | Segregation | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| AAD-636-3 | CACNA1A | Class 1 | NM_001127222:c.1745G>A:p.R582Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-414-3 | CACNA1A | Class 1 | NM_001127222:c.2039_2040del:p.680_680del | Frameshift variant | – | – | 0 | NA | Pathogenic |
| AAD-899-13 | CACNA1A | Class 1 | NM_001127222:c.3787G>A:p.E1263K | Known variant | – | – | 0 | TRUE (2 affected) | Pathogenic |
| AAD-555-11 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-870-1 | CACNA1A | Class 1 | NM_001127222:c.4034G>A:p.R1345Q | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-202-7 | CACNA1A | Class 1 | NM_001127222:c.4979G>A:p.R1660H | 4/4 | 4.96 | Close to known R1664Q, recurrent in another study | 0 | NA | Likely pathogenic |
| AAD-750-3 | CACNA1A | Class 1 | NM_001127222:c.4996C>T:p.R1666W | Known variant | – | – | 0 | NA | Pathogenic |
| 14GX000698 | CACNA1A | Class 1 | NM_001127222:c.5200G>T:p.E1734X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-82-4 | CACNA1A | Class 1 | NM_001127222:c.5248C>T:p.R1750W | Known variant | – | – | 0 | TRUE (6 affected) | Pathogenic |
| AAD-890-6 | CACNA1A | Class 1 | NM_001127222:c.6028C>T:p.Q2010X | Nonsense variant | – | – | 0 | NA | Pathogenic |
| AAD-883-1 | CACNA1A | Class 2 | NM_001127222:c.835C>T:p.R279C (+ NM_001127221:c.5575A>G:p.I1859V (non-canonical transcript)) | 4/4 | 4.24 | Close to known C287Y | 0/0 | NA | Likely pathogenic |
| AAD-603-1 | CACNA1A | Class 2 | NM_001127222:c.880C>T:p.P294S | 4/4 | 5.42 | Close to known G293R | 0 | NA | Likely pathogenic |
| AAD-875-1 | CACNA1A | Class 2 | NM_001127222:c.2026G>A:p.G676R | 4/4 | 4.58 | Close to known E668K | 0 | NA | Likely pathogenic |
| AAD-263-11 | CACNA1A | Class 3 | NM_001127222:c.2056G>A:p.G686S | 2/4 | 3.55 | Close to known E668K | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-747-11 | CACNA1A | Class 3 | NM_001127222:c.4448G>T:p.R1483L | 3/4 | 5.13 | Close to known F1491S | 0 | NA | Likely pathogenic |
| AAD-70-7 | CACNA1A | Class 3 | NM_001127221:c.5578_5579insAT:p.S1860fs (non-canonical transcript) | Frameshift variant | – | Not in canonical transcript | 0 | NA | Pathogenic |
| AAD-785-9 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V; c.858dupT:p.A286fs | A510V + frameshift variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAD-248-12 | SPG7 | Class 1 | NM_003119:c.958G>T:p.E320X; c.1047dupC:p.G349fs | Nonsense + frameshift variants | – | – | 0/0 | NA | Pathogenic/pathogenic |
| AAD-420-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant | – | – | 306/0 | NA | Pathogenic/pathogenic |
| AAR-541-13 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.1519C>T:p.Q507X | A510V + nonsense variant; also detected by WES (Coutelier et al., in preparation) | – | – | 306/0 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-951-11 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-847-18 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | TRUE (3 affected) | Pathogenic/pathogenic |
| AAD-1033-1 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| SAL-399-1026 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V x2 | Homozygous A510V | – | – | 306/306 | NA | Pathogenic/pathogenic |
| AAD-458-5 | SPG7 | Class 1 | NM_003119:c.1529C>T:p.A510V;c.2249C>T:p.P750L | A510V + known variant | – | – | 306/3/120860 | NA | Pathogenic/pathogenic |
| AAD-886-8 | AFG3L2 | Class 1 | NM_006796:c.1961C>T:p.T654I | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-994-1 | AFG3L2 | Class 1 | NM_006796:c.1996A>G:p.M666V | Known variant | – | – | 0 | NA | Pathogenic |
| AAD-315-17 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-512-11 | AFG3L2 | Class 1 | NM_006796:c.2059A>G:p.K687E | Recurrent variant, 3/4 | 5.62 | In proteolytic domain | 0 | NA | Likely pathogenic |
| AAD-541-19 | AFG3L2 | Class 2 | NM_006796:c.1450G>C:p.A484P | 4/4 | 6.03 | In AAA domain | 1/121360 | NA | Uncertain significance; no criteria for benign |
| AAD-1009-1 | AFG3L2 | Class 2 | NM_006796:c.2062C>G:p.P688A | 4/4 | 5.62 | In proteolytic domain, close to known Y689H | 0 | NA | Likely pathogenic |
| AAD-1005-1 | AFG3L2 | Class 3 | NM_006796:c.1430C>T:p.P477L | 2/4 | 6.03 | In AAA domain | 0 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-722-1 | KCND3 | Class 1 | NM_004980:c.1348C>T:p.L450F | Previously reported patient | – | – | 14/90828 | NA | Likely pathogenic |
| AAD-114-9 | KCND3 | Class 1 | NM_004980:c.1897C>T:p.P633S | Previously reported patient | – | – | 3/121388 | NA | Uncertain significance; no strict criteria for benign, divergent in silico predictions |
| AAD-374-1 | KCND3 | Class 2 | NM_004980:c.1086T>G:p.I362M | 4/4 | −8. but conserved amino acid across species | Close to known T352P and M373I | 0 | TRUE (3 affected) | Likely pathogenic |
| AAD-484-1 | KCND3 | Class 2 | NM_004980:c.1094T>C:p.M365T | 4/4 | 5.59 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-350-9 | KCNA1 | Class 1 | NM_000217:c.677C>T:p.T226M | Known variant | – | – | 0 | NA | Pathogenic |
| ENE-13-18 | KCNA1 | Class 2 | NM_000217:c.508G>T:p.A170S | 4/4 | 4.91 | Heterozygous A510V in SPG7 | 0 | TRUE (two affected) | Likely pathogenic |
| AAD-982-3 | KCNC3 | Class 1 | NM_004977:c.1268G>A:p.R423H | Known variant | – | – | 0 | TRUE (two affected) | Pathogenic |
| AAD-449-3 | KCNC3 | Class 3 | NM_004977:c.1420G>A:p.A474T | 3/4 | 3.36 | – | 0 | NA | Uncertain significance; no criteria for benign |
| AAD-479-5 | SPTBN2 | Class 2 | NM_006946:c.1294C>A:p.R432S | 4/4 | 3.7 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-483-1 | SPTBN2 | Class 3 | NM_006946:c.1261G>A:p.E421K | 3/4 | 4.62 | In second spectrin repeat | 0 | NA | Likely pathogenic |
| AAD-285-3 | ELOVL5 | Class 1 | NM_001242828:c.214C>G:p.L72V | Previously reported patient | – | – | 0 | NA | Likely pathogenic |
| AAD-958-1 | FGF14 | Class 2 | NM_004115:c.351G>T:p.Q117H | 4/4 | 5.87 | – | 0 | NA | Uncertain significance; no criteria for benign |
| 14GX000693 | STUB1 | Class 3 | NM_005861:c.433A>C:p.K145Q;c.502C>T:p.L168F | 3/4/4/4 | 4.59/4.59 | Trans, close to W147C and L165F/concordant phenotype, mild cognitive impairment | 82/116864/0 | NA | Pathogenic/pathogenic |
| AAD-915-6 | TTBK2 | Class 1 | NM_173500:c.1306_1307del:p.D436fs | Previously reported patient | – | – | 0 | NA | Pathogenic |
List of variants of Classes 1, 2 and 3 (as described in main text) found by sequencing 412 index patients with dominantly inherited cerebellar ataxia. Pathogenicity predictions were assessed by Annovar annotation with SIFT, PolyPhen 2 HDIV, LRT and MutationTaster. WES = whole exome sequencing.
Statistical analysis of clinical features of patients
Clinical features were compared between (i) patients with a molecular diagnosis after screening and patients without diagnosis; (ii) patients carrying a conventional mutation in a channel gene (CACNA1A, CACNA1G, KCNA1, KCNC3, KCND3), a conventional mutation in the SPG7 pathway (heterozygous in AFG3L2, biallelic in SPG7), and patients with a CAG repeat expansion in ATXN1, ATXN2 or ATXN3; and (iii) patients carrying a conventional mutation in CACNA1A and patients carrying an expansion in CACNA1A (SCA6). For all comparisons, relatives of index cases in which segregation of the variant had been confirmed were included. For comparisons (ii) and (iii), patients identified in a parallel study using whole exome sequencing in an independent autosomal recessive ataxia cohort (Coutelier et al., submitted), were added. The full list of included patients, along with their mutations, is available in Supplementary Table 4. Statistical analyses were performed with the IBM SPSS Statistics version 21 software. Mean ages at onset, at examination and disease durations were compared with a parametric ANOVA. Clinical features frequencies were compared using non-parametric Fisher exact test.
Results
Panel sequencing allows satisfying coverage of about 90% of amplicons
Mean coverage per patient was 450×, with an average 90% of bases covered more than 30×. We found the difference between the mean and the median coverage to be a better indicator of sequencing quality for each individual than the mean coverage alone. Indeed, for five subjects, even though mean coverage was high (472× to 633µ), median coverage per base was much lower: some amplicons had been covered up to >100 000× while most of them were not covered at all (Supplementary Fig. 1). We found this indicator to generally correlate with percentage of bases above 30×. The three most extreme patients had median coverage of 1×, 4×, and 14×; and percentage of bases above 30× of 19.4%, 46.6%, and 46.7%, respectively. Because of the poor quality of the results, all three were excluded from further analysis.
Coverage was satisfactory in ∼90% of the 27 studied SCA gene amplicons (n = 344/382), i.e. average percentage of 10× coverage was above 90%. All amplicons were, however, not equally covered; Supplementary Fig. 2 shows the proportion of amplicons (y) with more than (x)% of coverage over both thresholds of 10 and 30×. A median of 301 amplicons (79%) was perfectly covered (100% at 30× coverage), while 24 (6%) were completely missed (0% at 30×) (Supplementary Fig. 2). Ninety per cent of amplified regions were covered above 10× for 18 genes (18/27, 67%); between 80 and 90% for an additional six (Supplementary Table 2).
Among all 65 genes, from 286 to 1428 (mean 357) SNPs, and from 51 to 80 (mean 67) indels were detected per patient; in known SCA genes, these numbers decreased to 107–652 (mean 152) SNPs and 12–33 (mean 25) indels.
Diagnostic rate reaches 7.2% of the naïve SCA cohort
After analysis, as described above, 0 to 4 variants were sequenced per patient, with a median number of 0 and an average of 0.51 in known genes; 87% of them were true positives. As clinical presentations are rarely specific in dominant forms of cerebellar ataxia, we established a classification of variant pathogenicity based on stringent genetic criteria only. The variants fitting the characteristics used for filtering were distinguished into (Table 1 and Supplementary Table 5) (i) Class 1 variants: pathogenic variants (known or recurrent or null variant, or confirmed segregation in family; (ii) Class 2 variants: probably pathogenic variants (four concordant pathogenicity predictions by SIFT, PolyPhen HDIV, LRT and MutationTaster and GERP++ >3); (iii) Class 3 variants: possibly causative variants (convincing genetic arguments but one of the above is missed); (iv) variants of unknown significance. All Class 1, 2, and 3 variants have been submitted to ClinVar. We chose not to strictly use American College of Medical Genetics and Genomics (ACMG) criteria for pathogenicity (Richards et al., 2015), since we believe they are not perfectly suited for rare diseases. Variants sometimes fail to be classified as pathogenic due to the lack of fulfilled criteria linked to those disease group specificities, but not because they show evidence towards benign classification. Cohorts are indeed too small to reach statistical significance of variant frequency (criterion PS4). Furthermore, in cerebellar ataxias, de novo mutations (criteria PS2 and PM6) are not as classically expected as they are, for example, in autism spectrum disorders. Finally, the marked clinical and genetic heterogeneity of this disease group renders criterion PS1 (previous report of the variant) and PP4 (strict clinicogenetic concordance) difficult to achieve, while we believe that intra-study recurrence and clinical presumption should be taken into account. However, we assessed ACMG criteria for each Class 1, 2, and 3 variant, and they are summarized in Table 1 and detailed in Supplementary Table 3.
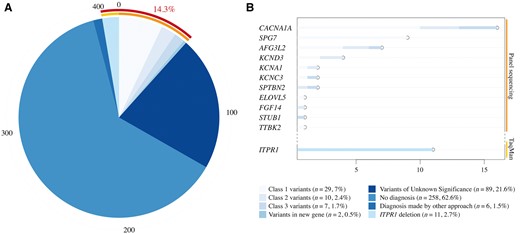
Sequencing conclusions and gene mutations frequencies across a large cohort of 412 patients with dominant cerebellar ataxias. (A) Distribution of pathogenic classes across our cohort. Relevant genetic variants were identified in 14.3% of patients. (B) Distribution of mutations in implicated genes across pathogenicity Classes 1, 2 and 3.
The TaqMan® PCR assay aiming at ITPR1 allowed consistent detection of probe deletions in 11 patients (Supplementary Table 6), four of whom had been previously reported by other techniques (Marelli et al., 2011b), validating the approach. The combined approach of panel sequencing and TaqMan® PCR assay allowed the identification of relevant genetic variants in 14.3% of patients (59 patients). Since polyglutamine expansions had previously been excluded in the cohort and are estimated to account for about half the patients with SCA, those patients would constitute 7.2% of the original cohort.
Possible risk factor variants are detected
Of interest were four specific variants, one in SPG7 and three in POLG, which were recurrently found in our cohort. Their frequency in public databases is too high to consider them pathogenic alone; however, they were more frequent than expected in the SCA cohort. The SPG7 p.A510V variant is found in 306/121348 chromosomes (0.25%) in the ExAC database; whereas in our study it was found in 20/824 chromosomes (2.43%). Even when considering only heterozygous patients, it represented 8/808 (0.99%). These results are statistically significant, with a Fisher’s exact test P-value of 1.837 × 10−13 if patients with biallelic variants are considered, and 0.00129 if they are excluded.
Three POLG variants were also recurrent: 576/121362 (0.47%) chromosomes harbour the p.G517V variant in ExAC; 10/824 in our study (1.21%). The p.G268A variant is present in 422/121102 chromosomes (0.35%) in ExAC, and 7/824 (0.85%) in our cohort. Finally, 195/120460 ExAC chromosomes harbour the p.L392V change (0.16%), while it is the case for 4/824 chromosomes (0.49%) we sequenced. We calculated the significance of these results using a Fisher’s test, at a 5% threshold of significance. All variants were shown to be statistically more frequent in our cohort (Table 2). No Dunn-Sidak correction was applied, since data are independent (in our cohort at least, no patient carries two of these four variants).
Potential risk factors identified
| ExAC | ADCA cohort | EVS – European Americans | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Wild- type count | Mutant count | Frequency | Wild- type count | Mutant count | Frequency | P | Wild- type count | Mutant count | Frequency | P |
| SPG7 | p.A510V | 121042 | 306 | 0.0025 | 804 | 20 | 0.0243 | 1.84 × 10−13 | 8561 | 39 | 0.004555543 | 5.66 × 10−8 |
| POLG | p.G517V | 120786 | 576 | 0.0047 | 814 | 10 | 0.0121 | 0.007156 | 8526 | 72 | 0.008444757 | 0.241075 |
| POLG | p.G268A | 120680 | 422 | 0.0035 | 817 | 7 | 0.0085 | 0.02796 | 8551 | 47 | 0.005496433 | 0.326859 |
| POLG | p.L392V | 120265 | 195 | 0.0016 | 820 | 4 | 0.0049 | 0.04772 | 8584 | 14 | 0.001630941 | 0.066154 |
| ExAC | ADCA cohort | EVS – European Americans | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Wild- type count | Mutant count | Frequency | Wild- type count | Mutant count | Frequency | P | Wild- type count | Mutant count | Frequency | P |
| SPG7 | p.A510V | 121042 | 306 | 0.0025 | 804 | 20 | 0.0243 | 1.84 × 10−13 | 8561 | 39 | 0.004555543 | 5.66 × 10−8 |
| POLG | p.G517V | 120786 | 576 | 0.0047 | 814 | 10 | 0.0121 | 0.007156 | 8526 | 72 | 0.008444757 | 0.241075 |
| POLG | p.G268A | 120680 | 422 | 0.0035 | 817 | 7 | 0.0085 | 0.02796 | 8551 | 47 | 0.005496433 | 0.326859 |
| POLG | p.L392V | 120265 | 195 | 0.0016 | 820 | 4 | 0.0049 | 0.04772 | 8584 | 14 | 0.001630941 | 0.066154 |
Four recurrent rare variants were identified in our cohort, with higher frequencies than observed in the ExAC (http://exac.broadinstitute.org/) population. They were all significantly more present in our SCA cohort (bold font), as established with a Fischer test, at the threshold of 5%. Compared to the frequencies observed in the European American population of the Exome Variant Server (EVS, http://evs.gs.washington.edu) database, the difference is only statistically significant for the SPG7 p.A510V variant.
Potential risk factors identified
| ExAC | ADCA cohort | EVS – European Americans | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Wild- type count | Mutant count | Frequency | Wild- type count | Mutant count | Frequency | P | Wild- type count | Mutant count | Frequency | P |
| SPG7 | p.A510V | 121042 | 306 | 0.0025 | 804 | 20 | 0.0243 | 1.84 × 10−13 | 8561 | 39 | 0.004555543 | 5.66 × 10−8 |
| POLG | p.G517V | 120786 | 576 | 0.0047 | 814 | 10 | 0.0121 | 0.007156 | 8526 | 72 | 0.008444757 | 0.241075 |
| POLG | p.G268A | 120680 | 422 | 0.0035 | 817 | 7 | 0.0085 | 0.02796 | 8551 | 47 | 0.005496433 | 0.326859 |
| POLG | p.L392V | 120265 | 195 | 0.0016 | 820 | 4 | 0.0049 | 0.04772 | 8584 | 14 | 0.001630941 | 0.066154 |
| ExAC | ADCA cohort | EVS – European Americans | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Wild- type count | Mutant count | Frequency | Wild- type count | Mutant count | Frequency | P | Wild- type count | Mutant count | Frequency | P |
| SPG7 | p.A510V | 121042 | 306 | 0.0025 | 804 | 20 | 0.0243 | 1.84 × 10−13 | 8561 | 39 | 0.004555543 | 5.66 × 10−8 |
| POLG | p.G517V | 120786 | 576 | 0.0047 | 814 | 10 | 0.0121 | 0.007156 | 8526 | 72 | 0.008444757 | 0.241075 |
| POLG | p.G268A | 120680 | 422 | 0.0035 | 817 | 7 | 0.0085 | 0.02796 | 8551 | 47 | 0.005496433 | 0.326859 |
| POLG | p.L392V | 120265 | 195 | 0.0016 | 820 | 4 | 0.0049 | 0.04772 | 8584 | 14 | 0.001630941 | 0.066154 |
Four recurrent rare variants were identified in our cohort, with higher frequencies than observed in the ExAC (http://exac.broadinstitute.org/) population. They were all significantly more present in our SCA cohort (bold font), as established with a Fischer test, at the threshold of 5%. Compared to the frequencies observed in the European American population of the Exome Variant Server (EVS, http://evs.gs.washington.edu) database, the difference is only statistically significant for the SPG7 p.A510V variant.
All four variants have slightly higher frequencies in the European American population of Exome Variant Server (Table 2). While considering those, the SPG7 p.A510V is the only statistically significantly associated with ataxic presentation. However, the tendency for the others to be more frequent in our cohort remains true.
Of interest, we could identify the POLG p.G517V variant in a 5-plex family. Among the five patients, only three carried the variant, which establishes that it is not causative per se.
Diagnosed patients have earlier presentations than undiagnosed patients
Patients carrying Class 1 (known, recurrent, null or segregating) and Class 2 (unanimously predicted pathogenic and conserved) mutations were compared to patients without molecular diagnosis, either only harbouring variants of unknown significance, or no mutation at all in screened genes (Table 3). Age at onset was significantly earlier in diagnosed cases (32.6 versus 45.7), as was age at examination (48.5 versus 57.5), while disease duration was longer (16.8 versus 12). Severity index (disease stage divided by disease duration) was significantly lower in diagnosed patients (0.38 versus 0.58), which means that the disease progression was slower.
Clinical comparison between patients with dominant inherited cerebellar ataxias with or without mutations of diagnostic value after screening
| Diagnostic mutation | Non-diagnostic mutation/VUS | P | |
|---|---|---|---|
| n = 410 | 63 | 347 | |
| Females/males | 22/41 | 174/173 | 0.03 |
| Age at onset in years (range) | 32.6 ± 20 (0–78) | 45.7 ± 17 (0–69) | 0.001 |
| Age at examination in years (range) | 48.5 ± 17 (3–79) | 57.5 ± 13 (13–88) | 0.001 |
| Disease duration in years (range) | 16.8 ± 14 (1–60) | 12.0 ± 11 (1–70) | 0.005 |
| Functional stage (0–7) | 2.8 ± 1.2 | 3.1 ± 1.3 | 0.09 |
| Severity index (functional stage/ duration) | 0.38 ± 0.53 | 0.58 ± 0.69 | 0.04 |
| Cerebellar dysarthria | 71% | 69% | 0.5 |
| Extensor plantar reflex | 29% | 29% | 1 |
| Increased reflexes | 13% | 15% | 0.08 |
| Abolished reflexes | 8% | 13% | |
| Distal wasting | 5% | 11% | 0.23 |
| Decreased vibration sense at ankles | 39% | 46% | 0.31 |
| Dystonia | 5% | 5% | 1 |
| Parkinsonian syndrome | 7% | 11% | 0.48 |
| Ophthalmoplegia | 7% | 9% | 0.71 |
| Dysphagia | 20% | 29% | 0.24 |
| Urinary dysfunction | 26% | 31% | 0.53 |
| Late cognitive impairment | 14% | 20% | 0.34 |
| Early intellectual deficiency | 14% | 5% | 0.018 |
| Diagnostic mutation | Non-diagnostic mutation/VUS | P | |
|---|---|---|---|
| n = 410 | 63 | 347 | |
| Females/males | 22/41 | 174/173 | 0.03 |
| Age at onset in years (range) | 32.6 ± 20 (0–78) | 45.7 ± 17 (0–69) | 0.001 |
| Age at examination in years (range) | 48.5 ± 17 (3–79) | 57.5 ± 13 (13–88) | 0.001 |
| Disease duration in years (range) | 16.8 ± 14 (1–60) | 12.0 ± 11 (1–70) | 0.005 |
| Functional stage (0–7) | 2.8 ± 1.2 | 3.1 ± 1.3 | 0.09 |
| Severity index (functional stage/ duration) | 0.38 ± 0.53 | 0.58 ± 0.69 | 0.04 |
| Cerebellar dysarthria | 71% | 69% | 0.5 |
| Extensor plantar reflex | 29% | 29% | 1 |
| Increased reflexes | 13% | 15% | 0.08 |
| Abolished reflexes | 8% | 13% | |
| Distal wasting | 5% | 11% | 0.23 |
| Decreased vibration sense at ankles | 39% | 46% | 0.31 |
| Dystonia | 5% | 5% | 1 |
| Parkinsonian syndrome | 7% | 11% | 0.48 |
| Ophthalmoplegia | 7% | 9% | 0.71 |
| Dysphagia | 20% | 29% | 0.24 |
| Urinary dysfunction | 26% | 31% | 0.53 |
| Late cognitive impairment | 14% | 20% | 0.34 |
| Early intellectual deficiency | 14% | 5% | 0.018 |
Clinical comparison of natural evolution of the disease and main phenotypic traits in 63 patients with a diagnostic mutation (46 index cases, including 28 Class 1 variants, two causative CACNA1G variants, 10 Class 2 variants, and six ITPR1 deletions; and 17 affected relatives) versus 347 index patients without molecular diagnosis [89 variant of unknown significance (VUS) and 258 without any variant after panel analysis].
Clinical comparison between patients with dominant inherited cerebellar ataxias with or without mutations of diagnostic value after screening
| Diagnostic mutation | Non-diagnostic mutation/VUS | P | |
|---|---|---|---|
| n = 410 | 63 | 347 | |
| Females/males | 22/41 | 174/173 | 0.03 |
| Age at onset in years (range) | 32.6 ± 20 (0–78) | 45.7 ± 17 (0–69) | 0.001 |
| Age at examination in years (range) | 48.5 ± 17 (3–79) | 57.5 ± 13 (13–88) | 0.001 |
| Disease duration in years (range) | 16.8 ± 14 (1–60) | 12.0 ± 11 (1–70) | 0.005 |
| Functional stage (0–7) | 2.8 ± 1.2 | 3.1 ± 1.3 | 0.09 |
| Severity index (functional stage/ duration) | 0.38 ± 0.53 | 0.58 ± 0.69 | 0.04 |
| Cerebellar dysarthria | 71% | 69% | 0.5 |
| Extensor plantar reflex | 29% | 29% | 1 |
| Increased reflexes | 13% | 15% | 0.08 |
| Abolished reflexes | 8% | 13% | |
| Distal wasting | 5% | 11% | 0.23 |
| Decreased vibration sense at ankles | 39% | 46% | 0.31 |
| Dystonia | 5% | 5% | 1 |
| Parkinsonian syndrome | 7% | 11% | 0.48 |
| Ophthalmoplegia | 7% | 9% | 0.71 |
| Dysphagia | 20% | 29% | 0.24 |
| Urinary dysfunction | 26% | 31% | 0.53 |
| Late cognitive impairment | 14% | 20% | 0.34 |
| Early intellectual deficiency | 14% | 5% | 0.018 |
| Diagnostic mutation | Non-diagnostic mutation/VUS | P | |
|---|---|---|---|
| n = 410 | 63 | 347 | |
| Females/males | 22/41 | 174/173 | 0.03 |
| Age at onset in years (range) | 32.6 ± 20 (0–78) | 45.7 ± 17 (0–69) | 0.001 |
| Age at examination in years (range) | 48.5 ± 17 (3–79) | 57.5 ± 13 (13–88) | 0.001 |
| Disease duration in years (range) | 16.8 ± 14 (1–60) | 12.0 ± 11 (1–70) | 0.005 |
| Functional stage (0–7) | 2.8 ± 1.2 | 3.1 ± 1.3 | 0.09 |
| Severity index (functional stage/ duration) | 0.38 ± 0.53 | 0.58 ± 0.69 | 0.04 |
| Cerebellar dysarthria | 71% | 69% | 0.5 |
| Extensor plantar reflex | 29% | 29% | 1 |
| Increased reflexes | 13% | 15% | 0.08 |
| Abolished reflexes | 8% | 13% | |
| Distal wasting | 5% | 11% | 0.23 |
| Decreased vibration sense at ankles | 39% | 46% | 0.31 |
| Dystonia | 5% | 5% | 1 |
| Parkinsonian syndrome | 7% | 11% | 0.48 |
| Ophthalmoplegia | 7% | 9% | 0.71 |
| Dysphagia | 20% | 29% | 0.24 |
| Urinary dysfunction | 26% | 31% | 0.53 |
| Late cognitive impairment | 14% | 20% | 0.34 |
| Early intellectual deficiency | 14% | 5% | 0.018 |
Clinical comparison of natural evolution of the disease and main phenotypic traits in 63 patients with a diagnostic mutation (46 index cases, including 28 Class 1 variants, two causative CACNA1G variants, 10 Class 2 variants, and six ITPR1 deletions; and 17 affected relatives) versus 347 index patients without molecular diagnosis [89 variant of unknown significance (VUS) and 258 without any variant after panel analysis].
Conventional channelopathies have earlier onsets and more slowly progressive courses
Clinical comparison between patients with cerebellar ataxias due to distinct mutational subgroups
| SCAs polyQa | Channelopathiesb | SPG7-relatedc | P | |
|---|---|---|---|---|
| n = 184 (index cases: n = 162) | 109 (106) | 37 (25) | 38 (31) | |
| Females/males | 53/56 | 14/23 | 11/27 | 0.088 |
| Age at onset in years (range) | 40.9 ± 12.1 (12–78) | 24.6 ± 22 (0–78) | 37.8 ± 12.5 (10–70) | 0.001 |
| Age at examination in years (range) | 50.3 ± 12.9 (22–79) | 44.7 ± 19.3 (3–79) | 51.5 ± 10.1 (31–78) | 0.073 |
| Disease duration in years (range) | 9.3 ± 6.8 (1–30) | 20.5 ± 15.7 (1–60) | 13.7 ± 11.4 (2–48) | 0.001 |
| Functional stage (0–7) | 3.3 ± 1.5 | 3.1 ± 1.4 | 3.2 ± 1.1 | 0.602 |
| Severity index (functional stage/duration) | 0.55 ± 0.4 | 0.39 ± 0.66 | 0.43 ± 0.36 | 0.149 |
| Cerebellar dysarthria | 79% | 70% | 69% | 0.4 |
| Extensor plantar reflex | 42% | 27% | 54% | 0.08 |
| Increased reflexes | 20% | 13% | 32% | 0.001 |
| Abolished reflexes | 31% | 3% | 3% | |
| Distal wasting | 4% | 6% | 16% | 0.034 |
| Decreased vibration sense at ankles | 55% | 38% | 49% | 0.21 |
| Dystonia | 13% | 6% | 8% | 0.52 |
| Parkinsonian syndrome | 2% | 3% | 8% | 0.21 |
| Ophthalmoplegia | 21% | 3% | 25% | 0.03 |
| Dysphagia | 42% | 16% | 29% | 0.017 |
| Urinary dysfunction | 32% | 18% | 47% | 0.03 |
| Late cognitive impairment | 10% | 11% | 14% | 0.79 |
| Early intellectual deficiency | 0% | 23% | 0% | 0.005 |
| CACNA1A polyglutamine expansions | CACNA1A point mutations | P | ||
| n = 45 (index cases n = 27) | 23 (11) | 22 (16) | ||
| Females/males | 11/12 | 10/12 | 1 | |
| Age at onset in years (range) | 47.3 ± 11.4 (24–67) | 25.2 ± 21.6 (0–65) | <0.001 | |
| Age at examination in years (range) | 59.0 ± 14.3 (24–80) | 43.9 ± 19.7 (3–70) | 0.005 | |
| Disease duration in years (range) | 10.9 ± 8.3 (0–31) | 18.7 ± 16.2 (1–60) | 0.054 | |
| Functional stage (0–7) | 3.8 ± 1.9 | 3.1 ± 1.5 | 0.24 | |
| Severity index (functional stage/duration) | 0.44 ± 0.27 | 0.39 ± 0.71 | 0.79 | |
| Cerebellar dysarthria | 78% | 55% | 0.22 | |
| Extensor plantar reflex | 23% | 23% | 0.18 | |
| Increased reflexes | 35% | 9% | 0.01 | |
| Abolished reflexes | 0% | 14% | ||
| Distal wasting | 9% | 4.5% | 0.49 | |
| Extrapyramidal syndrome | 13% | 9% | 0.1 | |
| Decreased vibration sense at ankles | 52% | 32% | 0.34 | |
| Ophthalmoplegia | 0% | 4.5% | 0.46 | |
| Cognitive impairment | 9% | 14% | 0.87 | |
| Intellectual deficiency | 4.30% | 24% | 0.09 |
| SCAs polyQa | Channelopathiesb | SPG7-relatedc | P | |
|---|---|---|---|---|
| n = 184 (index cases: n = 162) | 109 (106) | 37 (25) | 38 (31) | |
| Females/males | 53/56 | 14/23 | 11/27 | 0.088 |
| Age at onset in years (range) | 40.9 ± 12.1 (12–78) | 24.6 ± 22 (0–78) | 37.8 ± 12.5 (10–70) | 0.001 |
| Age at examination in years (range) | 50.3 ± 12.9 (22–79) | 44.7 ± 19.3 (3–79) | 51.5 ± 10.1 (31–78) | 0.073 |
| Disease duration in years (range) | 9.3 ± 6.8 (1–30) | 20.5 ± 15.7 (1–60) | 13.7 ± 11.4 (2–48) | 0.001 |
| Functional stage (0–7) | 3.3 ± 1.5 | 3.1 ± 1.4 | 3.2 ± 1.1 | 0.602 |
| Severity index (functional stage/duration) | 0.55 ± 0.4 | 0.39 ± 0.66 | 0.43 ± 0.36 | 0.149 |
| Cerebellar dysarthria | 79% | 70% | 69% | 0.4 |
| Extensor plantar reflex | 42% | 27% | 54% | 0.08 |
| Increased reflexes | 20% | 13% | 32% | 0.001 |
| Abolished reflexes | 31% | 3% | 3% | |
| Distal wasting | 4% | 6% | 16% | 0.034 |
| Decreased vibration sense at ankles | 55% | 38% | 49% | 0.21 |
| Dystonia | 13% | 6% | 8% | 0.52 |
| Parkinsonian syndrome | 2% | 3% | 8% | 0.21 |
| Ophthalmoplegia | 21% | 3% | 25% | 0.03 |
| Dysphagia | 42% | 16% | 29% | 0.017 |
| Urinary dysfunction | 32% | 18% | 47% | 0.03 |
| Late cognitive impairment | 10% | 11% | 14% | 0.79 |
| Early intellectual deficiency | 0% | 23% | 0% | 0.005 |
| CACNA1A polyglutamine expansions | CACNA1A point mutations | P | ||
| n = 45 (index cases n = 27) | 23 (11) | 22 (16) | ||
| Females/males | 11/12 | 10/12 | 1 | |
| Age at onset in years (range) | 47.3 ± 11.4 (24–67) | 25.2 ± 21.6 (0–65) | <0.001 | |
| Age at examination in years (range) | 59.0 ± 14.3 (24–80) | 43.9 ± 19.7 (3–70) | 0.005 | |
| Disease duration in years (range) | 10.9 ± 8.3 (0–31) | 18.7 ± 16.2 (1–60) | 0.054 | |
| Functional stage (0–7) | 3.8 ± 1.9 | 3.1 ± 1.5 | 0.24 | |
| Severity index (functional stage/duration) | 0.44 ± 0.27 | 0.39 ± 0.71 | 0.79 | |
| Cerebellar dysarthria | 78% | 55% | 0.22 | |
| Extensor plantar reflex | 23% | 23% | 0.18 | |
| Increased reflexes | 35% | 9% | 0.01 | |
| Abolished reflexes | 0% | 14% | ||
| Distal wasting | 9% | 4.5% | 0.49 | |
| Extrapyramidal syndrome | 13% | 9% | 0.1 | |
| Decreased vibration sense at ankles | 52% | 32% | 0.34 | |
| Ophthalmoplegia | 0% | 4.5% | 0.46 | |
| Cognitive impairment | 9% | 14% | 0.87 | |
| Intellectual deficiency | 4.30% | 24% | 0.09 |
Top: Clinical comparison between patients with cerebellar ataxias due to polyglutamine expansions (SCAs polyQ), mutations in channels genes (channelopathies) and mutations in SPG7 linked genes (SPG7-related). Bottom: Clinical comparison between patients with cerebellar ataxias due to mutations in CACNA1A, either polyglutamine expansions or point mutations. Values with a significant P-value of <5% are highlighted in bold.
aSCA1 = 28; SCA2 = 24; SCA3 = 57.
bCACNA1A = 22; CACNA1G = 4; KCNA1 = 3; KCNC3 = 2; KCND3 = 6.
cBiallelic SPG7 = 30; AFG3L2 = 8.
Clinical comparison between patients with cerebellar ataxias due to distinct mutational subgroups
| SCAs polyQa | Channelopathiesb | SPG7-relatedc | P | |
|---|---|---|---|---|
| n = 184 (index cases: n = 162) | 109 (106) | 37 (25) | 38 (31) | |
| Females/males | 53/56 | 14/23 | 11/27 | 0.088 |
| Age at onset in years (range) | 40.9 ± 12.1 (12–78) | 24.6 ± 22 (0–78) | 37.8 ± 12.5 (10–70) | 0.001 |
| Age at examination in years (range) | 50.3 ± 12.9 (22–79) | 44.7 ± 19.3 (3–79) | 51.5 ± 10.1 (31–78) | 0.073 |
| Disease duration in years (range) | 9.3 ± 6.8 (1–30) | 20.5 ± 15.7 (1–60) | 13.7 ± 11.4 (2–48) | 0.001 |
| Functional stage (0–7) | 3.3 ± 1.5 | 3.1 ± 1.4 | 3.2 ± 1.1 | 0.602 |
| Severity index (functional stage/duration) | 0.55 ± 0.4 | 0.39 ± 0.66 | 0.43 ± 0.36 | 0.149 |
| Cerebellar dysarthria | 79% | 70% | 69% | 0.4 |
| Extensor plantar reflex | 42% | 27% | 54% | 0.08 |
| Increased reflexes | 20% | 13% | 32% | 0.001 |
| Abolished reflexes | 31% | 3% | 3% | |
| Distal wasting | 4% | 6% | 16% | 0.034 |
| Decreased vibration sense at ankles | 55% | 38% | 49% | 0.21 |
| Dystonia | 13% | 6% | 8% | 0.52 |
| Parkinsonian syndrome | 2% | 3% | 8% | 0.21 |
| Ophthalmoplegia | 21% | 3% | 25% | 0.03 |
| Dysphagia | 42% | 16% | 29% | 0.017 |
| Urinary dysfunction | 32% | 18% | 47% | 0.03 |
| Late cognitive impairment | 10% | 11% | 14% | 0.79 |
| Early intellectual deficiency | 0% | 23% | 0% | 0.005 |
| CACNA1A polyglutamine expansions | CACNA1A point mutations | P | ||
| n = 45 (index cases n = 27) | 23 (11) | 22 (16) | ||
| Females/males | 11/12 | 10/12 | 1 | |
| Age at onset in years (range) | 47.3 ± 11.4 (24–67) | 25.2 ± 21.6 (0–65) | <0.001 | |
| Age at examination in years (range) | 59.0 ± 14.3 (24–80) | 43.9 ± 19.7 (3–70) | 0.005 | |
| Disease duration in years (range) | 10.9 ± 8.3 (0–31) | 18.7 ± 16.2 (1–60) | 0.054 | |
| Functional stage (0–7) | 3.8 ± 1.9 | 3.1 ± 1.5 | 0.24 | |
| Severity index (functional stage/duration) | 0.44 ± 0.27 | 0.39 ± 0.71 | 0.79 | |
| Cerebellar dysarthria | 78% | 55% | 0.22 | |
| Extensor plantar reflex | 23% | 23% | 0.18 | |
| Increased reflexes | 35% | 9% | 0.01 | |
| Abolished reflexes | 0% | 14% | ||
| Distal wasting | 9% | 4.5% | 0.49 | |
| Extrapyramidal syndrome | 13% | 9% | 0.1 | |
| Decreased vibration sense at ankles | 52% | 32% | 0.34 | |
| Ophthalmoplegia | 0% | 4.5% | 0.46 | |
| Cognitive impairment | 9% | 14% | 0.87 | |
| Intellectual deficiency | 4.30% | 24% | 0.09 |
| SCAs polyQa | Channelopathiesb | SPG7-relatedc | P | |
|---|---|---|---|---|
| n = 184 (index cases: n = 162) | 109 (106) | 37 (25) | 38 (31) | |
| Females/males | 53/56 | 14/23 | 11/27 | 0.088 |
| Age at onset in years (range) | 40.9 ± 12.1 (12–78) | 24.6 ± 22 (0–78) | 37.8 ± 12.5 (10–70) | 0.001 |
| Age at examination in years (range) | 50.3 ± 12.9 (22–79) | 44.7 ± 19.3 (3–79) | 51.5 ± 10.1 (31–78) | 0.073 |
| Disease duration in years (range) | 9.3 ± 6.8 (1–30) | 20.5 ± 15.7 (1–60) | 13.7 ± 11.4 (2–48) | 0.001 |
| Functional stage (0–7) | 3.3 ± 1.5 | 3.1 ± 1.4 | 3.2 ± 1.1 | 0.602 |
| Severity index (functional stage/duration) | 0.55 ± 0.4 | 0.39 ± 0.66 | 0.43 ± 0.36 | 0.149 |
| Cerebellar dysarthria | 79% | 70% | 69% | 0.4 |
| Extensor plantar reflex | 42% | 27% | 54% | 0.08 |
| Increased reflexes | 20% | 13% | 32% | 0.001 |
| Abolished reflexes | 31% | 3% | 3% | |
| Distal wasting | 4% | 6% | 16% | 0.034 |
| Decreased vibration sense at ankles | 55% | 38% | 49% | 0.21 |
| Dystonia | 13% | 6% | 8% | 0.52 |
| Parkinsonian syndrome | 2% | 3% | 8% | 0.21 |
| Ophthalmoplegia | 21% | 3% | 25% | 0.03 |
| Dysphagia | 42% | 16% | 29% | 0.017 |
| Urinary dysfunction | 32% | 18% | 47% | 0.03 |
| Late cognitive impairment | 10% | 11% | 14% | 0.79 |
| Early intellectual deficiency | 0% | 23% | 0% | 0.005 |
| CACNA1A polyglutamine expansions | CACNA1A point mutations | P | ||
| n = 45 (index cases n = 27) | 23 (11) | 22 (16) | ||
| Females/males | 11/12 | 10/12 | 1 | |
| Age at onset in years (range) | 47.3 ± 11.4 (24–67) | 25.2 ± 21.6 (0–65) | <0.001 | |
| Age at examination in years (range) | 59.0 ± 14.3 (24–80) | 43.9 ± 19.7 (3–70) | 0.005 | |
| Disease duration in years (range) | 10.9 ± 8.3 (0–31) | 18.7 ± 16.2 (1–60) | 0.054 | |
| Functional stage (0–7) | 3.8 ± 1.9 | 3.1 ± 1.5 | 0.24 | |
| Severity index (functional stage/duration) | 0.44 ± 0.27 | 0.39 ± 0.71 | 0.79 | |
| Cerebellar dysarthria | 78% | 55% | 0.22 | |
| Extensor plantar reflex | 23% | 23% | 0.18 | |
| Increased reflexes | 35% | 9% | 0.01 | |
| Abolished reflexes | 0% | 14% | ||
| Distal wasting | 9% | 4.5% | 0.49 | |
| Extrapyramidal syndrome | 13% | 9% | 0.1 | |
| Decreased vibration sense at ankles | 52% | 32% | 0.34 | |
| Ophthalmoplegia | 0% | 4.5% | 0.46 | |
| Cognitive impairment | 9% | 14% | 0.87 | |
| Intellectual deficiency | 4.30% | 24% | 0.09 |
Top: Clinical comparison between patients with cerebellar ataxias due to polyglutamine expansions (SCAs polyQ), mutations in channels genes (channelopathies) and mutations in SPG7 linked genes (SPG7-related). Bottom: Clinical comparison between patients with cerebellar ataxias due to mutations in CACNA1A, either polyglutamine expansions or point mutations. Values with a significant P-value of <5% are highlighted in bold.
aSCA1 = 28; SCA2 = 24; SCA3 = 57.
bCACNA1A = 22; CACNA1G = 4; KCNA1 = 3; KCNC3 = 2; KCND3 = 6.
cBiallelic SPG7 = 30; AFG3L2 = 8.
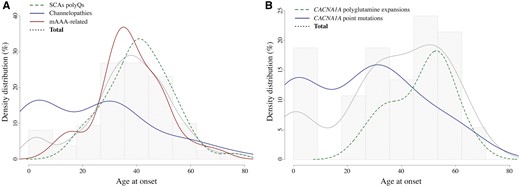
Age at onset density distribution for dominant cerebellar ataxias. (A) Age at onset density distribution for dominant cerebellar ataxias due to mutations in channel genes (CACNA1A, CACNA1G, KCNC3, KCND3, KCNA1) (solid blue), mAAA-related ataxias (biallelic SPG7, monoallelic AFG3L2) (solid red), or SCA1, 2 and 3 (dotted green). (B) Age at onset density distribution for dominant cerebellar ataxias due to CACNA1A polyglutamine expansions (dotted green) versus point mutations (solid blue). Light dotted dark lines and grey background histograms show the age at onset distribution for all considered patients as a whole.
![Brain MRI of patients carrying mutations in CACNA1A (point mutations and expansion), SPG7 and AFG3L2, with similar severity indexes. (A) T2-weighted sagittal and axial brain MRI showing marked cerebellar atrophy of Patient AAD-870, CACNA1A mutation p.R1345Q; age at onset: 28; age at examination: 52; severity index 0.13. (B) T1-weighted sagittal and T2 FLAIR axial brain MRI showing cerebellar and brainstem atrophy of Patient AAD-980, SCA6 [(CAG)n = 27]; age at onset: 45; age at examination: 47; severity index 0.5. (C) T1-weighted sagittal and axial brain MRI showing mild cerebellar atrophy of Patient AAD-1033, SPG7 homozygous mutation p.A510V; age at onset: 57; age at examination: 70; severity index 0.15. (D) T1-weighted sagittal and axial brain MRI showing slight vermian atrophy of Patient AAD-1009; AFG3L2 mutation p.P688A; age at onset: 47; age at examination: 57; severity index 0.1. Brainstem atrophy (arrow) is only noticed in the SCA6 patient, which is in line with the observed phenotypic differences. Cerebellar atrophy is more prominent in CACNA1A-related ataxias, and in particular in point mutation cases, than in mAAA-linked cases.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/140/6/10.1093_brain_awx081/2/m_awx081f3.jpeg?Expires=1750304561&Signature=3VTiLZvsHdzruMsHIf0SSR9d3iciqeHwUuVNUsgTgmlTFUPUOJnunL~-Mf5bBct14dXQw6fluTS8rUZ8R18G5CFiW1yZfXER7AKZFxqeU~N8mP1Zfrtm7vQEdWIZmhFChmKBJJVhMY4CbOZy14Aj5gyhshYRVvj4i0465ACSbH5yJHtwATMQb~mtS0dMQnbiQbIcTk7NX3ODMWreK7a2YuS7Pho1RB6f0PlnkkXBDh7SjWXtvmJKiwRAA2lGHzu~-65wHIZCCE0GpL-6jasVkFQLxEGaj9K8Ir2H7zPkRz5lLTpYlyKR~MQ4TFvM0Xmk6Ww53wvGJdAdkWAZqtKqCg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Brain MRI of patients carrying mutations in CACNA1A (point mutations and expansion), SPG7 and AFG3L2, with similar severity indexes. (A) T2-weighted sagittal and axial brain MRI showing marked cerebellar atrophy of Patient AAD-870, CACNA1A mutation p.R1345Q; age at onset: 28; age at examination: 52; severity index 0.13. (B) T1-weighted sagittal and T2 FLAIR axial brain MRI showing cerebellar and brainstem atrophy of Patient AAD-980, SCA6 [(CAG)n = 27]; age at onset: 45; age at examination: 47; severity index 0.5. (C) T1-weighted sagittal and axial brain MRI showing mild cerebellar atrophy of Patient AAD-1033, SPG7 homozygous mutation p.A510V; age at onset: 57; age at examination: 70; severity index 0.15. (D) T1-weighted sagittal and axial brain MRI showing slight vermian atrophy of Patient AAD-1009; AFG3L2 mutation p.P688A; age at onset: 47; age at examination: 57; severity index 0.1. Brainstem atrophy (arrow) is only noticed in the SCA6 patient, which is in line with the observed phenotypic differences. Cerebellar atrophy is more prominent in CACNA1A-related ataxias, and in particular in point mutation cases, than in mAAA-linked cases.
Specific assessment of patients with CACNA1A expansions versus conventional mutations yielded similar observations. We compared 23 patients with CACNA1A expansions (SCA6) to 22 patients with CACNA1A point mutations identified in this study or in another independent whole exome sequencing study of sporadic and recessive cases (Coutelier et al., submitted for publication). Clinical characteristics are summarized in Table 4. Individuals with conventional mutations had lower average age at onset (25.2 versus 47.3 years) and longer disease duration (18.7 versus 10.9). The severity index was higher for SCA6 patients (0.44 versus 0.39), indicating more rapid progression. The limited size of the reported groups might explain the lack of statistical power of the analysis, but this confirms the tendency observed between ataxias linked to channel gene point mutations and SCA1, 2 and 3. Pyramidal involvement is more frequent in SCA6 cases with expansions, whereas ophthalmoplegia is limited to patients with conventional mutations.
Clinical features of patients with SPG7 mutations
Nine patients carried Class 1 SPG7 mutations; four were homozygous for the p.A510V variant, four carried the p.A510V in compound heterozygosity with either a null variant (n = 3, two nonsense and one frameshift) or a missense mutation (n = 1), and one carried both a nonsense and a frameshift mutation. Average age at onset was 38 years, with a tendency for homozygous p.A510V carriers to have a later onset (42.5 years) than heterozygous carriers (33 years), even though the patient with two null mutations had an onset at 40 years. Seven of nine patients had brisk deep tendon reflexes, 6/9 had an extensor plantar reflex, and 3/9 were clearly spastic. On MRI, 7/9 patients presented cerebellar atrophy. Among the nine probands, five had a clear dominant history, with cerebellar signs reported in the mother (deceased or not examined, without DNA available for sequencing). Two pedigrees were multiplex one-generation families, with 3/10 and 3/6 affected probands, respectively; motor impairment in the grandfather and alcoholism in the father were reported in one; and dementia in the father in the ninth patient.
Discussion
In summary, we aimed to explore the nosology of dominantly inherited cerebellar ataxias linked to conventional mutations through assessment of their relative frequency in a large cohort, and genotype–phenotype correlations in the most numerous groups. We used an amplicon-based panel sequencing technique for 65 genes, including 27 known genes, on 412 index patients. We obtained good coverage of most amplicons (90% of the sequence of 90% of the amplicons was at least covered at 10×), with an acceptable rate of false positive variants detected (<15%). We combined the panel study with a TaqMan® PCR assay to detect deletions in ITPR1.
We yielded a very probable or definite diagnostic in 50 patients (12.1%), a possible diagnostic in seven, and confirmed the implication of a new gene in two, for a total of 14.3% (n = 59). Considering that about half the patients with dominantly inherited cerebellar ataxia bear (CAG)n repeat expansions, which had been previously excluded in this population, the diagnostic percentage amongst general population falls to 7.2%. This is rather low compared to other studies using panel sequencing or exome sequencing on undiagnosed cerebellar ataxia cohorts. Indeed, diagnostic rate in previous studies, on smaller cohorts though, ranged between 18 and 46% (Nemeth et al., 2013; Fogel et al., 2014; Gomez and Das, 2014; Sawyer et al., 2014; Pyle et al., 2015). However, most of these studies aimed at patients with undiagnosed ataxias, regardless of the transmission mode or age at onset. Among adult patients, diagnostic rate was usually somehow lower, with the identification of the genetic background in 8.3% of 24 undiagnosed adult-onset progressive patients (capture sequencing; Nemeth et al., 2013), 21% of 76 adult and sporadic cases (whole exome sequencing; Fogel et al., 2014), and 33% of 12 sporadic patients (Keogh et al., 2015). Moreover, many variants in the latter studies were described in genes whose mutations are transmitted on a recessive mode, which for the most part, were not included in our study.
This is low compared to diagnostic yield in other Mendelian diseases, which usually reaches about a third on average (Gomez and Das, 2014; Novarino et al., 2014), and which could have several explanations. First, the most frequent mutations in SCAs remain polyglutamine expansions. Conventional mutations were estimated to account for less than 10% of SCA cases (Durr, 2010). This, however, remained to be formally demonstrated, as, in many studies, only polyglutamine expansions were consistently looked for. We were thus able to confirm that estimate of ∼10%, when reporting to the total number of cases with ataxia, including polyglutamine ataxias. Furthermore, we also report a relatively high number of variants of unknown significance (21.6%). This is not very surprising in cerebellar ataxias either, for which the phenotype is rarely pathognomonic, since most heterozygous rare variants within a known gene with some damaging effect predicted by in silico tools may often be considered. To further confirm pathogenicity of those variants, a convergence of evidences should be used, as described in the ACMG guidelines (Richards et al., 2015). In the absence of biomarkers, and since functional studies cannot be performed for each variant, it is difficult to obtain higher confidence in pathogenicity. A major criterion, however, is the recurrence of (extremely) rare variants in patients with similar phenotypes, hence the importance of publicly sharing data. Finally, our cohort comes from a highly specialized university hospital, including a national reference centre for rare diseases, and some of the patients have already been variably screened for several genes and partly cleaned out through more than 20 years of genetic screening efforts. The study is thus biased in that the cohort is, for the most part, not naïve. This is confirmed by the identification of already known variants in some of the patients, which were found elsewhere during the study process. All in all, this low identification rate raises the attention of other genes, mutation types, and pathogenic mechanisms, which remain to be discovered.
Despite these facts, we could still identify the genetic origin of the ataxic phenotype in ∼15% of the patients (∼7.2% considering all ataxic patients before expansion screening). More importantly, among these patients, most carried variants in two genes, previously little recognized in progressive ataxia: CACNA1A and SPG7. Of note, this panel study also allowed us to describe patients with mutations in new SCA genes such as CACNA1G, which has been described elsewhere (Coutelier et al., 2015a). Deletions in ITPR1 are also relatively frequent, and should be looked for (Marelli et al., 2011b).
CACNA1A point mutations were first recognized in episodic ataxia type 2 (EA2, loss-of-function variants) and familial hemiplegic migraine type 1 (gain-of-function variants) (Ophoff et al., 1996). Small size polyglutamine expansions were then implicated in SCA6 (Zhuchenko et al., 1997). They were shortly after described in EA2 (Jodice et al., 1997), while point mutations were conversely implicated in SCA6 (Yue et al., 1997). However, diagnostic work-up in progressive ataxia often fails to identify point mutations in CACNA1A, rather focusing on searching for polyglutamine expansions. We establish here that these are frequent causes of undiagnosed ataxia. Interestingly, no correlation seems to exist between specific variants and the phenotypic presentation, whether hemiplegic migraine, progressive, or episodic ataxia.
However, clinical presentations associated with point mutations differ from those linked to polyglutamine expansions, with an earlier age at onset for a similar functional disability score after a longer evolution. This observation was made while exclusively regarding mutations in CACNA1A, or considering wider SCA gene groups. The associated phenotype also differs. Channelopathies show a pure cerebellar syndrome, frequently combined with intellectual deficiency. This is in line with more general observations comparing the clinical presentation of patients with mutations in channel genes (CACNA1A, CACNA1G, KCND3, KCNC3), and patients with classical polyglutamine ataxias (SCA1, SCA2, SCA3). Those observations are of importance in the clinically heterogeneous group of cerebellar ataxias, where genetic counselling might prove difficult even amongst families. While phenotypic variability still lacks convincing genetic explanation, this is a first step in the delineation of firm genotype–phenotype correlation tendencies.
Though point mutations in CACNA1A are thus frequent in progressive ataxia, this does not seem to be the case for mutations in other episodic ataxia genes. Two causative or possibly causative variants were found in KCNA1, but the clinical picture of the patients included episodic ataxia. No variant was detected in SLC1A3. For CACNB4, the only variant we detected, p.R473H, is carried by a patient with a causative CACNA1A mutation. It is, however, conserved (GERP++ 5.26) and predicted to be pathogenic by all four software programs used, and might be a modifying factor, although the rarity of the observation makes it impossible to demonstrate. While we did not identify episodic symptoms in patients carrying KCND3 or KCNC3 variants, blurring of phenotypes seems to occur bidirectionally, and testing of SCA genes, especially channel genes, might prove to be successful in diagnosing episodic ataxias.
SPG7-linked phenotype has largely been remodelled since its first implication in autosomal recessive spastic paraplegia (Casari et al., 1998), then optic neuropathy with a clear dominant pattern in one family (Klebe et al., 2012). In the latter study, cerebellar ataxia co-occurred in 39% of patients (with cerebellar atrophy at MRI in almost all cases), and in some related heterozygous carriers. Biallelic mutations in SPG7 were recently recognized as a major cause of undiagnosed ataxia (Pfeffer et al., 2015). We confirm these results in our cohort, with nine patients identified as undoubtedly SPG7-mutated, carrying previously reported or loss-of-function biallelic mutations. All but one eventually developed pyramidal signs; 3/8 (37.5%) were clearly spastic. SPG7 biallelic variants hence account for ∼2% of our patients’ cohort (9/412 = 2.2%). This is not as high as reported in Pfeffer’s study (15/72 = 20.8%). Our cohort, however, predominantly includes patients with autosomal dominant transmission. This also sheds light on the fact that mutations in recessive cerebellar ataxia genes might be implicated in some of them, but missed because of the use of a panel strategy, which was noted in other studies (Fogel et al., 2014; Keogh et al., 2015). However, among those nine patients, at least five had a clear history of dominant transmission of cerebellar signs. This raises, again, the question of dominant inheritance of SPG7 mutations, possibly with later-onset cerebellar signs in heterozygous carriers, as previously described in two individual patients carrying a heterozygous inframe deletion (Klebe et al., 2012). Unavailability of both DNA and patients for thorough examination keeps us from reaching more definite conclusions in our families.
Those examples of phenotype remodelling, linked to mutations in a given gene, raise the question of other overlapping genes, which we would not have included in our panel, and whose mutations may also give rise to cerebellar ataxia, even though not yet recognized. This is a main disadvantage of our choice of sequencing technique, and the next step in evaluating those patients would be to use whole exome or genome sequencing. However, amplicon-based panel sequencing has proven fast, still efficient, and markedly cost-effective.
Some variants of interest identified in our study may be risk factors in cerebellar ataxia; they are rare (<1%), but too frequent to be considered pathogenic alone.
The pathogenicity of SPG7 p.A510V variant has long been discussed; it is now recognized to be causative while homozygous, or in compound heterozygosity with a second pathogenic heterozygous variant (Elleuch et al., 2006; Roxburgh et al., 2013; Sanchez-Ferrero et al., 2013). Among the nine patients we identified with SPG7 mutations, respectively, four were homozygous for that change, and four were compound heterozygous, with a loss-of-function or previously reported variant. The overall frequency of p.A510V variant in our cohort was significantly higher than in ExAC chromosomes, which was still the case when biallelic SPG7 patients were excluded. It might thus be a hypomorphic allele conferring a risk for the disease, even though not causative on its own. This is similar to what has been described for the p.S44L variant in SPG4; even though not pathogenic alone in most if not all cases, and quite frequent in general population, these variants are pathogenic at the homozygous state, and accentuate or induce a phenotype in combination with another pathogenic variant (Chinnery et al., 2004).
The pathogenicity of POLG variant p.G517V has also been debated. It was first described in several pedigrees with dominant transmission (Horvath et al., 2006; Burusnukul and de los Reyes, 2009; Hopkins et al., 2010). Due to its occurrence in patients presenting variable ages of onset and clinical pictures, the mutant protein polymerase activity was assessed and shown to be similar to wild-type (Kasiviswanathan and Copeland, 2011). We demonstrated, in a 5-plex family that the variant does not segregate, being present in three patients only. This would argue against its being causative in SCA. POLG p.G268A variant is also reported in patients with diverse presentations (Graziewicz et al., 2006). Mutant protein was shown to induce an altered growth phenotype in yeast, in a recessive pattern (Baruffini et al., 2006). To the best of our knowledge, no functional study has been performed on variant p.L392V. Insufficient evidence has been gathered to consider POLG heterozygous variants causative in ataxia.
In some SCA genes, no variant of interest was identified (SLC1A3, PDYN, ITM2B); in several others, all (rare) identified changes were classified as variants of unknown significance (EEF2, IFRD1, NOL3). This sheds light on the fact that these genes are, at best, rarely implicated in ataxia, if at all. The case of IFRD1 is enlightening. Only one variant has been described so far (Brkanac et al., 2009). Its rarity and lack of conservation made its implication in dominant forms of cerebellar ataxia doubtful; the fact that we could not identify any other pathogenic variant in such a large cohort reinforces the presumption that the variant identified by Brkanac and colleagues (2009) may not be causative.
In conclusion, we sequenced a large cohort of index cases with dominantly inherited cerebellar ataxia, with an amplicon-based panel including 27 known genes. We reached a satisfactory coverage on most regions, though the obtained results will not allow direct implementation of the technique for diagnostic purposes. Combining the panel approach with a TaqMan® PCR assay aiming ITPR1, we identified (possibly) causative variations in up to 14.3% of cases, which is low compared to other diseases, but expected in SCAs. We confirmed the prominent implication of CACNA1A and SPG7 point mutations in the disease, and underlined genotype–phenotype correlations in CACNA1A-linked progressive ataxias, with an earlier and purer phenotype for point mutations compared to polyglutamine expansions. This observation is more generally corroborated in ataxia-channelopathies.
Acknowledgements
We are deeply indebted to all patients for their participations and their patience. Many thanks to the DNA and Cell bank of the Institut du Cerveau et de la Moelle Epinière for technical assistance. We are grateful for referrals of patients for research purposes by M. Vidailhet, M. Tchikviladzé, R. Debs, A. Tataru, M. Maillet-Vioud, P. Calvas, P. Damier, C. Lubetzki, A. Echaniz, M. Gouttard, E. Lagrange, R. Padovani, M. Vercelletto, J. M. Blard, C. De Barace, and all the clinicians of the SPATAX network.
Funding
M.C. was the recipient of a fellowship from the Fond National de la Recherche Scientifique (aspirant Fonds National de la Recherche Scientifique). C.T. received a fellowship from the French Ministry for Research through the Doctoral School 472-EPHE. This work received financial support from the French National Agency for Research (ANR; to G.S.), the French Ministry of Health PHRC 2009 program (AOM 09178 to A.D.), the European Union (Omics call: NeurOmics; to A.B. and A.D.), the Verum Foundation (to A.B. and G.S.), the Fondation Roger de Spoelberch (to A.B.), the French patient association ‘Connaitre les Syndromes Cérébelleux’ (to G.S.) and the program Investissements d’Avenir (ANR-10-IAIHU-06; to the ICM).
Supplementary material
Supplementary material is available at Brain online.
Appendix 1
SPATAX network collaborators
Alexandra Durr; Giovanni Stevanin; Soraya Belarbi; Abdelmadjid Hamri; Meriem Tazir; Sylvia Boesch; Massimo Pandolfo; Urielle Ullmann; Laura Jardim; Velina Guergueltcheva; Ivalo Tournev; Bing-Wen Soong; Olga Lucia Pedraza Linarès; Jørgen E. Nielsen; Kirsten Svenstrup; Maha Zaki; Jean-Philippe Azulay; Guillaume Banneau; Odile Boesfplug-Tanguy; Alexis Brice; Andrea Burgo; Cécile Cazeneuve; Perrine Charles; Frédéric Darios; Christel Depienne; Charles Duyckaerts; Bertrand Fontaine; Sylvie Forlani; Cyril Goizet; Didier Hannequin; Jamilé Hazan; Michel Koenig; Pierre Labauge; Cecilia Marelli; Karine N’guyen; Diana Rodriguez; Annie Sittler; Christophe Verny; Peter Bauer; Lüdger Schöls; Rebecca Schüle; Georgios Koutsis; Alexander Lossos; Antonella Antenora; Maria Teresa Bassi; Manuela Basso; Enrico Bertini; Alfredo Brusco; Carlo Casali; Giorgio Casari; Chiara Criscuolo; Alessandro Filla; Maria Lieto; Laura Orsi; Filippo M. Santorelli; Enza Maria Valente; Marinela Vavla; Giovanni Vazza; André Megarbane; Ali Benomar; Richard Roxburgh; Anne Kjersti Erichsen; Chantal Tallaksen; Isabel Alonso; Paula Coutinho; José Léal Loureiro; Jorge Sequeiros; Mustapha Salih; Vladimir S Kostic; Idoia Rouco Axpe; Samir Roumani; Berry Kremer; Willeke Van Roon-Mom; Amir Boukhris; Chokri Mhiri; Arzu Karabay; Suran Nethisinghe; Cahir O’kane; Megan Oliva; Evan Reid; Thomas Warner; Nicholas Wood.
Abbreviation
References
Author notes
See Appendix 1 and Supplementary material for SPATAX network collaborators.


{kind=link}
{kind=link}