





Abstract
Summary: We developed a modular and scalable framework called Eoulsan, based on the Hadoop implementation of the MapReduce algorithm dedicated to high-throughput sequencing data analysis. Eoulsan allows users to easily set up a cloud computing cluster and automate the analysis of several samples at once using various software solutions available. Our tests with Amazon Web Services demonstrated that the computation cost is linear with the number of instances booked as is the running time with the increasing amounts of data.
Availability and implementation: Eoulsan is implemented in Java, supported on Linux systems and distributed under the LGPL License at: http://transcriptome.ens.fr/eoulsan/
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
High-throughput sequencing data analysis requires large computer clusters associated with expensive costs that are only profitable for large genomics centers. Outsourcing data analysis over cloud computing infrastructure is one alternative. Nevertheless, software that use distributed algorithm are not common among bioinformatics developers, and only few solutions have been made available for sequencing data (Langmead et al., 2010; McKenna et al., 2010). Here we present Eoulsan, an open source framework, to facilitate high-throughput sequencing data by the use of distributed computation. This software has been developed in order to automate the analysis of a large number of samples at once, simplify the configuration of the cloud computing infrastructure and work with various already available analysis solutions.
2 SOFTWARE IMPLEMENTATION
We first implemented Eoulsan for differential analysis of transcript expression. The workflow dedicated to this task includes 6 steps (Supplementary Fig. S1). First, the reads generated from the sequencers are subjected to quality control filtering. Second, reads are mapped onto the reference genome. Currently four different mappers are embedded in Eoulsan: BWA (Li and Durbin, 2009), Bowtie (Langmead et al., 2009b), SOAP2 (Li et al., 2009) and GSNAP (Wu and Nacu, 2010). Third, alignments are filtered to keep only single matches on the genome. Fourth, transcript expression calculation is performed using mapped reads associated to an annotation file. Fifth, a normalization step is applied on the different samples in order to correct for biases with either the scaling normalization implemented in the edgeR R package for technical replicates (Robinson et al., 2010) or with the DESeq R package when biological replicates are available (Anders and Huber, 2010). Sixth, detection of differential expression is carried out on the biological entities described in the annotation file by applying a Fisher's Exact Test for technical replicates or, the DESeq R package for biological replicates.
Eoulsan can be run under three modes: standalone, local cluster or cloud computing on Amazon Elastic MapReduce (EMR). The last two execution modes have been implemented using the Hadoop framework, an open source implementation of a MapReduce programming model (Taylor, 2010). In practice, the distributed mode of Eoulsan performs as described in Figure 1.
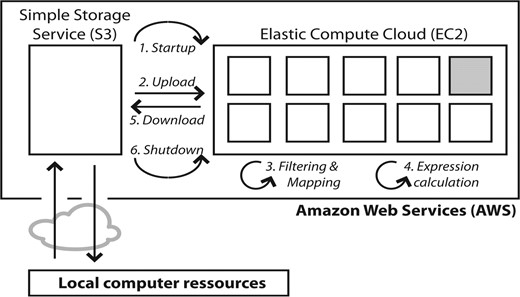
Eoulsan is launched from the user local computer where it uploads to S3 all the data needed to perform the analysis. 1. AWS starts-up the EC2 cluster by booking virtual machines (represented by squares with one master node in gray) and copies the Hadoop distribution using the Amazon EMR system. 2. The data needed for the analysis are transferred from S3 to the EC2 cluster. 3. Eoulsan launches read filtering, mapping and alignment filtering steps. 4. It performs expression estimation. Both these two steps use a MapReduce strategy. 5. Results are downloaded back to S3. 6. AWS shuts-down the EC2 cluster and the user gets back the results from S3 to the local computer where the final statistical analysis is performed (normalization and differential analysis).
3 CLOUD COMPUTING ASSESSMENT
We tested Eoulsan on Amazon Web Services (AWS) with 8 mouse samples from RNA-Seq data (for a total of 188 millions reads) using different read mappers embedded in the workflow. We estimated the time needed to perform the calculation process and the cost charged by AWS on three different Elastic Compute Cloud (EC2) virtual machine types (called instance by AWS): m1.large, m1.xlarge and c1.xlarge (see Supplementary Table S1 for instance selection). The results are presented on Supplementary Figure S2 and in Supplementary Table S2. If we compare the time spent, the fastest result is obtained from the c1.xlarge instance whatever the mapper used. In terms of costs, the computation is always more expensive using m1.xlarge instances with m1.large remaining the most economical choice.
One interest of using cloud computing facilities is to speed up the calculation process by using a large number of computers at once. To evaluate this possibility, we tested how the number of booked instances influences the calculation process (Supplementary Fig. S3 and Supplementary Table S3). We observed that the whole time spent for the calculations strongly drop with low instance number and remained close to linear with more than five instances booked. However, one question remains. How many instances do we need to use for data analysis? This is critical as each hourly booked EC2 instance costs a fixed price. Our tests demonstrate that the cost per hour is linear over the number of instances used (Supplementary Fig. 4). This means that the number of instances can be increased in order to speed up the data analysis process without the risk to fall in a suboptimal configuration.
Finally, we assessed the impact of an increase in raw data on the computation time by running Eoulsan with 16 and 32 samples of 23.5 millions of reads each, respectively, 376 and 752 millions of total reads (Supplementary Fig. S5 and Supplementary Table S4). We saw that the relationship between running time and number of samples is also linear (Supplementary Fig. S6). This demonstrates that Eoulsan is able to handle the increase in raw data coming from future evolutions of Illumina sequencing devices.
4 CONCLUSION
With our framework, we aimed to facilitate high-throughput sequencing analysis on cloud computing services with an automatic, modular and efficient tool. This approach differs from other solutions already available for distributed calculation such as Myrna, CloudBurst or Crossbow (Langmead et al., 2010; Langmead et al., 2009a; Schatz, 2009), where the cloud computing implementation is made around a unique analysis solution. In addition, Eoulsan is also complementary to the customizable Galaxy server solution as it allows for batch analyses and it contains a full automation process able to handle external file locations and distributed file system. More generally, we have first implemented an automated RNA-Seq analysis pipeline but all the tools are already included within Eoulsan for other applications such as SNP calling or ChIP Peak analysis. The Java plug-in system we developed as well as the full documentation we provide, allow for user contribution by the integration of other available solution in Eoulsan.
The distributed calculation process we used is based on Hadoop and it can be installed on numerous cluster server configurations. We made our proof of concept using AWS solution as it includes EMR, an advanced service based on EC2 that easily allows Hadoop distributed computation solutions to be deployed. However, we are working to make our software independent of EMR to work directly on EC2 services. Such an evolution will allow Eoulsan to be run on any other cloud computing solutions such as Open Source initiatives like OpenNebula or OpenStack. This would open future possibilities by creating regional genomic computer infrastructures (like iPlant for example) to be shared among several local high-throughput sequencing users. With a dedicated high-speed network, this can speed up the time transfer process. In addition, this could also favor the standardization of analysis pipelines developed from the bioinformatics community, making high-throughput sequencing technologies really accessible for a wide audience.
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
ACKNOWLEDGEMENTS
We thank J. Le Men and P. Gilardi Hebenstreit for providing RNA-Seq samples and the Pasteur Institute Transcriptomics and Epigenomics platform for the Illumina runs. We express our gratitude to T. Portnoy for helpful discussions and to A. Kandil and J. Bansse for their analysis of the cloud computing market. Finally, we thank P. Surrun for the careful review of the English spelling.
Funding: This work was funded by the network Infrastructures en Biologie Santé et Agronomie (IBiSA) and by an Amazon Web Services research grant.
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Janet Kelso


{kind=link}