





Abstract
Summary: The preparation of an appropriate sequence dataset is the starting point of all genomic analyses. We present retrieve-ensembl-seq, an application that considerably eases the retrieval of sequences from the Ensembl database, via our user-friendly web site or web services. The user provides Ensembl identifiers or gene names, and the program returns corresponding upstream, downstream, intronic, exonic, UTR or whole gene sequences. retrieve-ensembl-seq also offers a multiple organism mode to retrieve sequences from homologous genes at any taxonomical level. And we introduce various original filters such as the masking of coding fragments and the avoidance of sequence redundancy for genes with multiple transcripts. retrieve-ensembl-seq is included in the software suite regulatory sequence analysis tools (RSAT), allowing instant submission of retrieved sequences to further analysis tools.
Availability: retrieve-ensembl-seq is integrated in the RSAT suite: http://rsat.ulb.ac.be/rsat. Web site: http://rsat.ulb.ac.be/rsat/retrieve-ensembl-seq_form.cgi. Web services: http://rsat.ulb.ac.be/rsat/web_services/RSATWS.wsdl. Stand-alone distribution: freely available under an academic licence to download from the RSAT web site. The complete manual, a convenient tutorial and demos are available from the RSAT website. Additional help can be found on the RSAT public forum.
Contact: [email protected]
1 TOOL FUNCTIONALITY
Any genomic study starts with the preparation of a sequence dataset to analyse. The Ensembl database (Hubbard et al., 2009) is a key provider of vertebrates and other eukaryotes genomic data, with 50 species supported in version 53. Moreover, it is currently expanding to a wider taxonomic range with bacteria, fungi, plants and protists. To retrieve personalized datasets, Ensembl offers access via BioMart (Kasprzyk et al., 2004) and application programming interfaces (APIs). However, BioMart does not fully exploits the richness of annotations provided by Ensembl (e.g. introns) and offers very limited support for multi-species sequence retrieval (maximum five species per query). The use of APIs requires programming skills which not all interested users possess.
We present here retrieve-ensembl-seq, a user-friendly program that allows to retrieve a large variety of sequence types (upstream, downstream, introns, exons, UTRs and whole genes) from the Ensembl database. At each query, retrieve-ensembl-seq establishes a direct connection to Ensembl via its Core and Compara APIs, thereby ensuring a permanent access to up-to-date sequences. Useful and original options are introduced here. These include masking of coding fragments (converted to N characters), and the possibility to get only non-redundant sequences for genes with multiple transcripts. Furthermore, when retrieving upstream or downstream sequences, one can prevent overlapping with a neighbouring gene or an open reading frame (ORF).
Comparative genomics takes a growing importance for the detection of conserved features across related genomes. Yet, fetching sequences remains a tedious almost organism-per-organism task. retrieve-ensembl-seq proposes an original multiple organism mode to automatically retrieve homologous sequences at any taxonomical level. The integration of retrieve-ensembl-seq in the regulatory sequence analysis tools (RSAT; Thomas-Chollier et al., 2008; van Helden et al., 2000) ensures its seamless interconnection with pattern discovery and matching programs, plus convenient utilities. To suit all types of users, the program is accessible via web site, SOAP web services and command line. The next sections detail the parameters of retrieve-ensembl-seq.
2 SINGLE ORGANISM MODE
2.1 Organism
All Ensembl-supported organisms are available. The web site always queries the latest Ensembl version. Previous versions can be accessed via web services or command line.
2.2 Query (Gene, transcript or protein ID)
The query can be one or several Ensembl IDs or gene names. Identifiers from other databases are also accepted as long as they are valid external references in Ensembl. If a transcript or protein ID is provided, the program first recovers the corresponding gene ID before sequence retrieval. It is thus presently not possible to query for one given transcript or protein when there are alternative ones.
2.3 Sequence type
The sequence type parameter determines the kind of sequence to retrieve (Fig. 1A). It can be of type upstream/downstream, gene (whole gene sequence, from 5′-most transcription start site (TSS) to 3′-most transcription termination site (TTS), intron (all intron sequences), first intron, exon (all exon sequences), non-coding exon (only non-coding parts of exons sequences), UTRs (both UTR sequences), 5′ UTR or 3′ UTR.
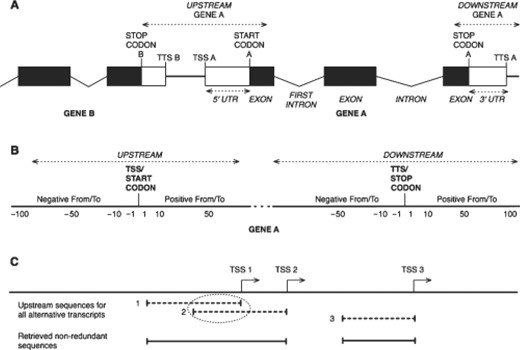
Schematic representation of genomic regions that can be retrieved with the program retrieve-ensembl-seq. (A) Sequence types (shown in italics). Boxes represent exons, broken lines represent introns and straight lines represent intergenic regions. Black boxes represent coding exons, white boxes represent non-coding exons. (B) Coordinate system used for upstream and downstream sequence types. Negative coordinates refer to positions upstream the reference feature, positive ones refer to positions downstream the reference feature. (C) Treatment of redundancy for genes with alternative transcripts. The dotted oval highlights mutually overlapping fragments in upstream sequences of transcripts 1 and 2. These will be merged in the retrieved sequences. Upstream sequence for transcript 3 will be fully retrieved.
With the upstream/downstream type (Fig. 1B), it is necessary to specify the feature taken as reference and the positions relative to this feature (‘From’ and ‘To’ values). The ‘Relative to feature’ option value can be CDS (coding sequence; start or stop codon, depending on the upstream or downstream sequence position choice), mRNA (TSS for upstream sequences, TTS for downstream sequences) or gene (5′-most TSS for upstream sequences, 3′-most TTS for downstream sequences). ‘From’ and ‘To’ values can be negative or positive. A negative value refers to a coordinate upstream the feature chosen as reference, whereas a positive value refers to a coordinate downstream the feature chosen as reference. If a negative value is set for the ‘From’ parameter and a positive value is set for the ‘To’ parameter, the retrieved sequence will span over the reference feature.
2.4 Additional options
With the option ‘Prevent overlap with neighbouring’ ORF or gene, the sequence is truncated at the neighbouring ORF or gene limit.
Two masking options are available: ‘repeats’ (if annotated) and ‘coding sequences’.
It is possible to avoid redundant sequences due to alternative transcripts (Fig. 1C). When the query gene has more than one transcript, the retrieved sequences for this gene may have overlapping regions. This option removes, for each individual query, any redundancy in the retrieved sequence set, by merging mutually overlapping fragments.
The organism name in the sequence fasta header can be set to scientific, common or none (no organism name in the fasta header).
The type of output can be defined as display (results printed on result page), server (link to results printed on result page) or email (link to results sent by email).
3 MULTIPLE ORGANISM MODE
In multiple organism mode, retrieve-ensembl-seq automatically collects not only the sequences of the query organism, but also the sequences from its homologs in all other organisms. More refined results can be obtained by filtering the homologs on their homology type and/or on their taxonomic level (taxon).
The homology type filter supports all Ensembl-defined homologies: all homologs (no filter), orthologs (all orthologs), ortholog_one2one, ortholog_one2many, ortholog_many2many, apparent_ortholog_one2one, paralogs (all paralogs), within_species_paralog, between_species_paralog.
The taxon filter enables to restrict the retrieval of homologous sequences to a given taxonomic level (example: Mammalia).
All options described for the single organism mode apply here as well.
4 EXAMPLES AND HELP
To help first time users, two demos are available from the web site. DEMO_1 shows an example of single organism mode use and DEMO_2 a multiple organism one.
There is also a tutorial on the web site to guide beginners through the various options. Finally, there is a manual describing every parameter. For quick reference, each parameter of the web site form is linked to its description in the manual. A flowchart on the RSAT web site tutorials page shows typical analysis paths.
Funding: This work was supported by the BioSapiens Network of Excellence funded under the sixth Framework program of the European Communities (LSHG-CT-2003-503265) and the Vrije Universiteit Brussel (Geconcerteerde Onderzoeksactie 29). The BiGRe Laboratory is part of the Belgian Program on Interuniversity Attraction Poles, initiated by the Belgian Federal Science Policy Office, project P6/25 (BioMaGNet).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Dmitrij Frishman


{kind=link}