





Abstract
Summary: Microorganisms are ubiquitous in nature and constitute intrinsic parts of almost every ecosystem. A culture-independent and powerful way to study microbial communities is metagenomics. In such studies, functional analysis is performed on fragmented genetic material from multiple species in the community. The recent advances in high-throughput sequencing have greatly increased the amount of data in metagenomic projects. At present, there is an urgent need for efficient statistical tools to analyse these data. We have created ShotgunFunctionalizeR, an R-package for functional comparison of metagenomes. The package contains tools for importing, annotating and visualizing metagenomic data produced by shotgun high-throughput sequencing. ShotgunFunctionalizeR contains several statistical procedures for assessing functional differences between samples, both for individual genes and for entire pathways. In addition to standard and previously published methods, we have developed and implemented a novel approach based on a Poisson model. This procedure is highly flexible and thus applicable to a wide range of different experimental designs. We demonstrate the potential of ShotgunFunctionalizeR by performing a regression analysis on metagenomes sampled at multiple depths in the Pacific Ocean.
Availability: http://shotgun.zool.gu.se
Contact: [email protected]; [email protected]
Supplementary information: supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Metagenomics is the analysis of genetic material obtained directly from the environment without isolating and growing the species in a laboratory. Organisms are hence studied in their natural habitats, which is not possible with traditional methods since only a tiny fraction of all microorganisms (∼1%) is cultivable using standard techniques (Hugenholtz et al., 1998). Metagenomics therefore provides means to discover important genetic and functional differences and offers a unique view of the structure and organization of microbial communities (Ritz, 2007). The main obstacle, however, is that data comes in short DNA fragments (reads) without any labels showing to which organism they belong. In addition, unequal species distributions often prevent obtaining a sufficient sequencing depth. Even with high-throughput sequencing techniques, it is often not possible to assemble the genome for any but the most dominant species (Kunin et al., 2008).
A common and powerful approach, which bypasses whole-genome assembly, is to study the abundance of gene families. A gene family is a set of functionally similar genes, including both paralogues from single species and orthologues from different species. Changes in gene family abundance between metagenomes can thus be linked to functional differences based on their corresponding annotation. A similar analysis can also be performed on a higher level using gene family categories, sets of functionally related gene families such as pathways. Analysis of metagenomes using gene families or gene family categories is usually referred to as gene-centric analysis and pathway-centric analysis, respectively (Kunin et al., 2008). Even though these procedures are relatively well-established, there are few generic tools for proper statistical analysis of metagenomes.
2 METHODS
We have created ShotgunFunctionalizeR, an R package for functional comparison of metagenomes. ShotgunFunctionalizeR is a collection of tools for gene- and pathway-centric analysis of metagenomic data produced by shotgun high-throughput sequencing. For gene-centric analysis, standard and previously described methods have been implemented, such as the binomial and hypergeometric tests (Agresti, 2002). In addition, we have developed a novel approach based on a Poisson model. In contrast with previous methods, the Poisson model is highly flexible and thus applicable to a much wider range of experimental designs, including direct comparisons of groups with multiple samples and regression analysis. The implementation is done using a generalized linear model with the Poisson canonical logarithmic link function. The total number of observations (i.e. all reads assigned to gene families) for each metagenome is used as an offset to remove the effects of unequal sample sizes (Fleiss et al., 2003).
ShotgunFunctionalizeR can also perform pathway-centric analysis in a number of different ways. The Gaussian test, currently used by the IMG/M web service (Markowitz et al., 2008), uses a normal approximation to sum the effect of all gene families in a specific gene family category. Another approach is the enrichment test, which assess the overrepresentation of a given gene family category among any ranked list of gene families (Rivals et al., 2007). The novel Poisson model can also be used for pathway-centric analysis, where each gene family in a gene family category has individual estimated baselines to compensate for differences in abundance.
ShotgunFunctionalizeR can analyse metagenomic data annotated as both cluster of orthologous groups (COGs) and Enzyme commission numbers (EC numbers) (Bairoch, 2000; Tatusov et al., 2003). Easy-to-use functions are available for importing, annotating and exporting both these data types. ShotgunFunctionalizeR also contains several types of associated gene family categories, including COG categories, IMG/M COG pathways and KEGG pathways (Kanehisa et al., 2002; Markowitz et al., 2008). All annotations are periodically updated against their corresponding online databases.
The ShotgunFunctionalizeR package also contains tools for data visualization and model validation. One example is the trend plot, which plots regression curves estimated by the Poisson model (see Section 3). Another example is the P-value QQ-plot, which can be used to validate model assumptions. Finally, all tests performed in ShotgunFunctionalizeR can be corrected for multiple testing using various methods such as the Benjamini–Hochberg's false discovery rate (Dudoit and van der Laan, 2008). All mathematical details regarding the procedures implemented in ShotgunFunctionalizeR are available in the Supplementary Material.
Several software tools for functional analysis of metagenomic data have been proposed in the literature. One example is the Metagenome Analyzer (MEGAN), which can be used to visualize differences between metagenomics in a taxonomical context (Mitra et al., 2009). Another example is the web-based tool MG-RAST which provides a pipeline for automatic annotation and functional assignment of metagenomic data (Meyer et al., 2009). In comparison to these methods, ShotgunFunctionalizeR offers a robust and sound statistical framework implemented in R. The novel Poisson model also provides a high degree of flexibility with the ability to analyse a wider range of experimental designs, such as regression analysis.
3 A CASE STUDY: THE OCEAN'S INTERIOR
In this section, we demonstrate ShotgunFunctionalizeR by analysing a public dataset consisting of metagenomes sampled at seven depths in the North Pacific Ocean (DeLong et al., 2006). The raw data were downloaded from Camera (Seshadri et al., 2007) and sequence reads were annotated with COG gene families using RPS-BLAST. Gene-centric regression analysis was performed on the data using the Poisson model. The following commands are sufficient to perform both the regression and displaying the fitted regression curve for the highly significant COG0415 (deoxyribodipyrimidine photolyase).

Deoxyribodipyrmidine photolyase is involved in repair of UV radiation-induced DNA damage. Interestingly, the abundance of this COG is decreasing rapidly after 70 m, which is also close to the maximum reach (∼ 100 m) of UV radiation in the ocean (Fig. 1). Hence, the presence of this gene family is diminishing with depth and indicates a general adaptation to an environment without UV light.
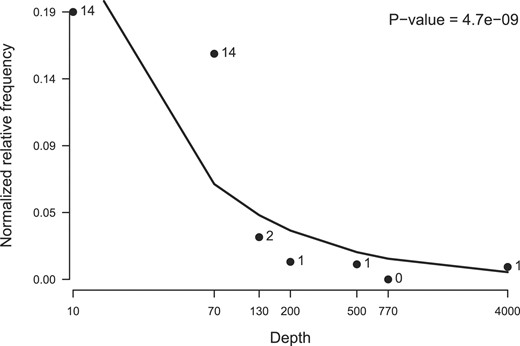
Trend plot for deoxyribodipyrimidine photolyase (COG0415) showing both fitted regression line and the data points. For this gene family, the trend is highly significant and shows a decreasing abundance at increasing depths.
The data from the Ocean's Interior study, annotated as both COGs and EC numbers, are available for further analysis in the ShotgunFunctionalizeR package. Additional examples can be found in the User's Guide which is included as the Supplementary Material.
Funding: This work was funded by the Swedish Research Council Formas and Svenska Sällskapet för Medicinsk Forskning (SSMF).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alex Bateman


{kind=link}