




Abstract
As the abnormalities of long non-coding RNAs (lncRNAs) are closely related to various human diseases, identifying disease-related lncRNAs is important for understanding the pathogenesis of complex diseases. Most of current data-driven methods for disease-related lncRNA candidate prediction are based on diseases and lncRNAs. Those methods, however, fail to consider the deeply embedded node attributes of lncRNA–disease pairs, which contain multiple relations and representations across lncRNAs, diseases and miRNAs. Moreover, the low-dimensional feature distribution at the pairwise level has not been taken into account. We propose a prediction model, VADLP, to extract, encode and adaptively integrate multi-level representations. Firstly, a triple-layer heterogeneous graph is constructed with weighted inter-layer and intra-layer edges to integrate the similarities and correlations among lncRNAs, diseases and miRNAs. We then define three representations including node attributes, pairwise topology and feature distribution. Node attributes are derived from the graph by an embedding strategy to represent the lncRNA–disease associations, which are inferred via their common lncRNAs, diseases and miRNAs. Pairwise topology is formulated by random walk algorithm and encoded by a convolutional autoencoder to represent the hidden topological structural relations between a pair of lncRNA and disease. The new feature distribution is modeled by a variance autoencoder to reveal the underlying lncRNA–disease relationship. Finally, an attentional representation-level integration module is constructed to adaptively fuse the three representations for lncRNA–disease association prediction. The proposed model is tested over a public dataset with a comprehensive list of evaluations. Our model outperforms six state-of-the-art lncRNA–disease prediction models with statistical significance. The ablation study showed the important contributions of three representations. In particular, the improved recall rates under different top |$k$| values demonstrate that our model is powerful in discovering true disease-related lncRNAs in the top-ranked candidates. Case studies of three cancers further proved the capacity of our model to discover potential disease-related lncRNAs.
Introduction
Long non-coding RNAs (lncRNAs), with a length of more than 200 nucleotides, are one type of non-coding RNAs [1–4]. Accumulating evidences demonstrated that the abnormalities of lncRNAs are linked with progression of various diseases [5–9]. Thus, identifying disease-related lncRNAs will contribute to the exploration of the pathogenesis of the diseases and the promotion of disease diagnosis and treatment.
Recently, computerized prediction models have been developed for miRNA–disease association prediction [10, 11], drug–target interaction prediction [12, 13], synergistic drug combination prediction [14, 15] and lncRNA–disease association prediction [16, 17]. The computerized screening can reduce the costs and time consuming in biomedical experiments. Machine learning models have attracted increasing attention in disease-related lncRNA screening because of the capacity in estimating lncRNA–disease associations using diverse data, which provides potential disease-related lncRNA candidates for biologists to discover true lncRNAs with further experiments [18]. According to the diversity of data sources involved in computing, there are two categories of models for disease–lncRNA association prediction. One category focuses on modeling the similarities of lncRNAs and diseases, while the other one utilizes multiple sources of data apart from lncRNAs and diseases such as miRNAs, proteins and genes. The 1st computational model in the field of lncRNA–disease association prediction was proposed by Chen et al. [19]. The model, known as LRLSLDA, is based on Laplacian regularized least squares, which predicts the candidate lncRNA–disease associations from the known lncRNA–disease associations and lncRNA expression profiles. This semi-supervised approach inspires subsequent research on computerized lncRNA–disease association prediction. Ping et al. [20] proposed a flow propagation algorithm based classifier to integrate two kinds of lncRNA similarities and two disease similarities that were calculated by a lncRNA–disease heterogeneous network. Non-negative matrix factorization [21–23] and graph models [24–27] have also been explored to integrate the lncRNA–disease similarities and associations. Sun et al. [28] constructed a graph of lncRNA similarities and exploited random walk with restart (RWR) algorithm for disease-related lncRNA prediction. A further work by Gu et al. [29] proposed a heterogeneous graph to integrate the similarities of diseases, lncRNAs and lncRNA–disease associations to predict the possibility of lncRNA–disease associations. The aforementioned models, however, failed to consider diverse sources of information, which are related with lncRNAs and diseases such as miRNAs and proteins.
The 2nd category of methods considers other sources of data apart from lncRNAs and diseases. For instance, disease-related lncRNAs were predicted based on flow propagation algorithm over a lncRNA–protein–disease network [30]. Lan et al. [31] proposed a support vector machine (SVM) based classifier to integrate two kinds of lncRNA similarities and five disease similarities. Ding et al. [32] estimated the lncRNA–disease association scores by constructing a lncRNA–disease-gene network and resource allocation mechanism. Similarly, a network of lncRNAs, diseases and miRNAs was constructed by Yu et al. [33], where a Naive Bayes classification model was used for association estimation. Fan et al. [34] integrated the expressions of lncRNAs, miRNAs and proteins and used RWR model to derive potential lncRNA–disease associations. There are also a couple of methods based on matrix factorization for multi-source data integration [35–37]. Wang et al. [38] developed a data fusion method based on selective matrix factorization, which collaboratively decomposes multiple interrelated data matrices into low-rank representation matrices for each object type and optimizes their weights. Even though multiple sources of data were considered in the models mentioned above, these models were not able to discover the underlying and complex relationship of lncRNAs and diseases because of the shallow level integration of cross-modality similarities.
With the development of deep learning algorithms, recent models further improved the prediction performance by extracting deep and complementary features. A dual convolutional neural network (CNN) was proposed by Xuan et al. [39] to predict disease-related lncRNAs. A more recent graph CNN model was exploited by [40]. These two deep models, however, failed to integrate the node attributes and feature distributions of lncRNA–disease at pairwise level. Even though deep models demonstrated improved performance in lncRNA–disease association prediction, CNN-based models cannot effectively integrate and learn correlations from non-grid structural heterogenous data such as miRNA, lncRNA and disease. To learn from heterogenous data, recent research in social network and recommender systems shows that attribute information and topological information contribute to the improvement performance in neural networks [41–44].
In this work, we propose a prediction model, VADLP, to learn, encode and adaptively integrate multi-level representations including pairwise topology, node attributes and feature distributions from multi-sourced data. The contributions of our model are included:
A multi-layer heterogenous graph is constructed to benefit the extraction of a newly introduced representation of node attributes for lncRNA–disease association modeling. The graph is composed of weighted inter- and intra-layer edges to embed similarities and correlations across multiple sources of data including lncRNAs, diseases and miRNAs. Pairwise node attributes are derived from the heterogeneous graph by a proposed embedding mechanism based on biological premises that if a pair of lncRNA and disease has associations or similarities with common lncRNAs, miRNAs or diseases, there is high probability that the lncRNA and disease are associated.
We took the initiative to introduce feature distribution, which is modeled by variance autoencoder (VAE), to reveal the underlying relationship and facilitate lncRNA–disease association prediction. The feature distribution is a high-level representation learnt and encoded from original node attributes.
We propose a pairwise topology encoding module to learn the hidden topological structural relations between a pair of lncRNA and disease. The pairwise topology representation is firstly formulated by applying random walk algorithm over the multi-layer heterogeneous graph and then encoded by a convolutional autoencoder (CAE).
An attentional representation-level integration and optimization module is proposed to enable the adaptive fusion of pairwise topology, node attributes and feature distribution. The contributions of three representations and capacity of the proposed model for disease-related lncRNA prediction are demonstrated by a comprehensive list of evaluations including ablation study, comparison with recent published models and case studies of three diseases.
Materials and Methods
The framework of the proposed VADLP model for lncRNA–disease association prediction is given in Figure 1. We firstly construct a triple-layer heterogeneous graph with weighted inter-layer and intra-layer edges to associate the similarities and correlations among lncRNAs, miRNAs and diseases. The representations of node attributes, pairwise topology and feature distribution are learned, respectively. Finally, these three representations are adaptively fused by attentional integration mechanism for further estimating the association possibility of a pair of lncRNA and disease.
Dataset
A public dataset for lncRNA–disease association prediction is obtained from [35], which contains 240 lncRNAs, 495 miRNAs and 405 diseases. The available information includes 2687 lncRNA–disease associations extracted from LncRNADisease database [45], 1002 pairs of lncRNA–miRNA interactions obtained from starBasev2.0 [47] and 13 559 pairs of miRNA–disease association obtained from HMDD [48]. Disease names are obtained from the US National Library of Medicine (MeSH, http://www.ncbi.nlm.nih.gov/mesh).
Triple-layer heterogeneous graph
We construct a weighted graph |$\textrm{G}{ \textrm{=}} \left ( {V,E,\textbf{W}} \right )$| with lncRNA-, miRNA- and disease-based layers, where nodes |$ V = \big \{{V^{lncRNA}}\cup{V^{miRNA}}\cup{V^{disease}}\big \}$| are composed of sets of lncRNA |${V^{lncRNA}}$|, miRNA |${V^{miRNA}}$| and disease |${V^{disease}}$| and an edge |${e_{ij}} \in E$| links a pair of nodes |$v_{i},v_{j} \in V$| undirectedly with weight |$w_{ij}\in \textbf{W}$|. According to the types of nodes connected by the edge, there are intra-layer and inter-layer edges. As shown in Figure 2, we define weight matrix |${\bf W}\, =\, ({\bf A,S})$| with inter-association matrix A and intra-similarity matrix S for inter- and intra-layer edges, respectively. Inter-association matrices denote whether the relations between different types of nodes are available or not. Intra-similarity matrices represent the similarities between the same type of nodes.
|${S_{ij}^{lncRNA}}$| and |$S_{ij}^{miRNA}$| are calculated via related diseases of a pair of lncRNAs or miRNAs by the method proposed by Chen et al. and Wang et al. [51, 52], respectively. Suppose |$i$|-th lncRNA |$v_{i}^{lncRNA}$| is associated with a set of diseases |$\Phi _{i}^{lncRNA} = {\Big \{ {d_{ik}|k=1\ldots N_{\Phi _{i}^{lncRNA}}} \Big \}}$|, lncRNA |$v_{j}^{lncRNA}$| is associated with a set of diseases |$\Phi _{j}^{lncRNA} =\Big \{ {d_{jl}|l=1\ldots N_{\Phi _{j}^{lncRNA}}} \Big \}$|and |$S_{ij}^{lncRNA}$| is obtained by measuring the similarity between |$\Phi _{i}^{lncRNA}$| and |$\Phi _{j}^{lncRNA}$|. Similarly, |$S_{ij}^{miRNA}$| is calculated by the similarity between associated disease sets |$\Phi _{i}^{miRNA}$| and |$\Phi _{j}^{miRNA}$|. Detailed calculations of |${S_{ij}^{lncRNA}}$| and |$S_{ij}^{miRNA}$| are given in Supplementary File SF1. In addition, the calculation results for |${S_{ij}^{lncRNA}}$| and |$S_{ij}^{miRNA}$| are shown in SF2 and SF3, respectively.
Pairwise topology extraction by random walk
To fully utilize the topological structural relations among lncRNA–disease–miRNA embedded in the graph G, we formulate the pairwise topology by random walk algorithm [53] to assist the lncRNA–disease association prediction.
To learn the pairwise topology between |$m$|-th RNA and |$n$|-th disease, we suppose a random walker starts from a node of |$m$|-th lncRNA |$v_{m}^{lncRNA}\in V^{lncRNA}$| and |$n$|-th disease |$v_{n}^{disease}\in V^{disease}$|. The walker iteratively transmits to its neighboring node via the connecting edge with the probability that is proportional to the edge weight. The higher the weight, the easier the transition.
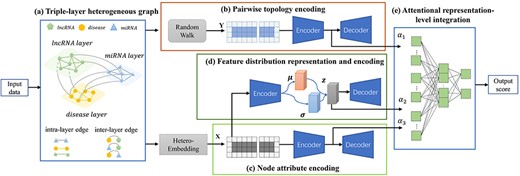
Framework of the proposed VADLP model. Given input data, (A) triple-layer heterogeneous graph is constructed to associate the similarities and correlations across lncRNAs, miRNAs and diseases with inter- and intra-layer weighted edges. Three representations are learned including (B) pairwise topology encoding by random walk and CAE, (C) node attributes by a proposed heterogeneous embedding mechanism and CAE and (D) feature distribution representation and encoding by VAE. The three representations are adaptively fused by (E) attentional representation-level integration for final lncRNA–disease association prediction. Details of each component are given in Fiures 2–4.
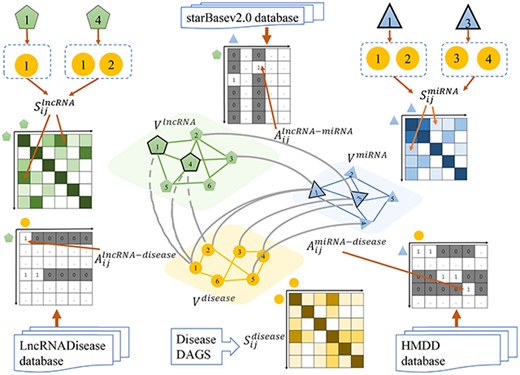
Inter-association and intra-similarity matrices A and S derived from the multi-layer heterogeneous graph of lncRNA, miRNA and disease.
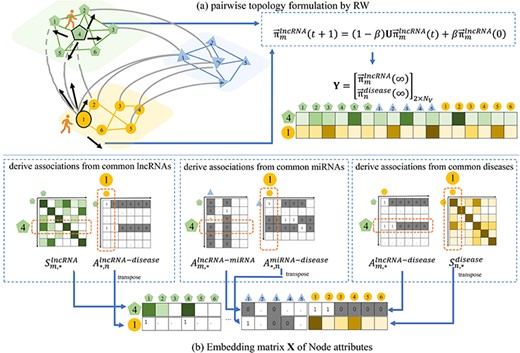
Illustration of extraction process of the proposed pairwise topology and node attributes between a pair of lncRNA and disease. Given 4th lncRNA and 1st disease as an example, (A) the pairwise topology is obtained as the steady-state status of RW over the graph, and (B) the embedding matrix of node attributes are derived from the graph weights via the common lncRNAs, miRNAs and diseases.
Heterogenous node attributes extraction by embedding strategy
We propose an embedding strategy to derive heterogeneous node attribute between a pair of lncRNA and disease from inter-layer associations and intra-layer similarities. The embedding strategy is based on the biological premise that for a pair lncRNA and disease with unknown association, if they have associations or similarities with common lncRNAs, miRNAs or diseases, there are high probabilities that the lncRNA and disease are associated.
Multi-level knowledge encoding
As the matrices of pairwise topology and node attributes obtained from the graph model are sparse high-dimensional matrices, there may be useless and non-presentative information. To learn deep while discriminative node attributes and topology from the original data distribution, CAE is used for encoding and decoding. To derive implicit representative feature distributions to assist prediction, VAE module is used. The overview of the proposed method is shown in Figure 1.
Pairwise topology encoding by CAE
Given matrix Y, a CAE is constructed to encode pairwise topological representation as shown by Figure 4A.
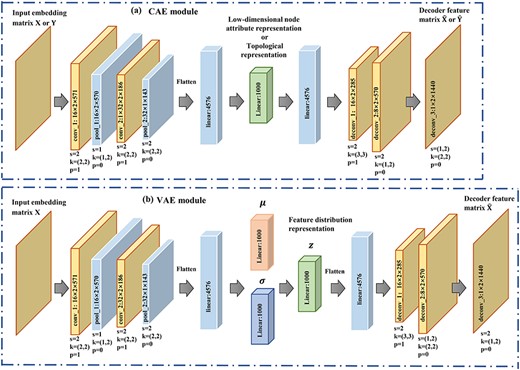
Parameter settings of (A) CAE and (B) VAE. s denotes stride, k denotes kernel and p is the padding.
Node attribute encoding by CAE
Similarly, a CAE module is constructed for the embedding matrix X to encode node attributes of a pair of lncRNA–disease. The settings for encoder and decoder are the same as that for pairwise topology encoding with details given in Figure 4A. We denote the encoded pairwise node attributes of the |$m$|-th lncRNA and |$n$|-th disease after the training process by |$\mathcal{F}_{attribute}$|.
Feature distribution representation and encoding by VAE
Apart from |$\mathcal{F}_{attribute}$| learnt from the original node attributes, we introduce representative feature distribution, which is derived from node attributes by VAE. Our hypothesis is that the feature distribution would serve as complementary information to further improve the prediction performance.
A VAE module is established as shown in Figure 4B. Matrix X is fed into a variational encoder to learn the feature distribution |$z$| and then |$z$| is sent to decoder to obtain the decoded |$\hat{\textbf{X}} $|. The optimization process aims to minimize the error between |$\hat{\textbf{X}}$| and X.
Attentional multi-level representation integration and optimization
To integrate the encoded pairwise topology, node attributes and feature distributions for association prediction, we propose an attentional multi-level representation integration and optimization module. The module can adaptively fuse multi-sourced knowledge by adaptive weights, which are reflected by attentional scores.
Experimental results and discussions
Experimental setup and evaluation metrics
Our method is implemented in PyTorch framework on a Nvidia GeForce GTX 2070Ti graphic card with 64G memory. The detailed parameter settings of CAE and VAE are given in Figures 4A and B. The learning rate was set as 0.0001. Dropout strategy |$\left ({P=0.5}\right )$| is performed to reduce the impact of overfitting. The effects of restarting parameter |$\beta $| in random walk algorithm are investigated and given in Supplemental Table ST1. Given |$\beta \in \left [{0.1,0.9} \right ] $|, the best performance was achieved when |$\beta =0.8$|.
Five-fold cross-validation is performed for performance evaluation of all the models in comparison. There are 2687 known lncRNA–disease associations and |$240*405-2687=94513$| unknown associations. All known lncRNA–disease associations are randomly partitioned into five equal-sized sets, where four of them are used for training and the remaining one is used for testing. In each fold, we randomly select lncRNA–disease samples with unobserved associations whose size is equal to that of samples with known associations for training, and the remaining unobserved lncRNA–disease associations are used for testing. The known lncRNA–disease associations are regarded as positive samples, while the unobserved lncRNA–disease associations are regarded as negative samples. The predicted association scores of testing samples are calculated and ranked. The higher the ranking of the positive examples, the better the prediction performance. In particular, in each round of cross-validation, lncRNA–lncRNA similarity is re-calculated by excluding the positive samples, which are to be used for testing. By such, we guarantee that the intra-lncRNA similarities used for graph construction will not contain information of the testing dataset.
Several evaluation measures include receiver operating characteristic (ROC) curve, true positive rate (TPR) and false positive rate (FPR), precision-recall (PR) curve, area under ROC (AUC), area under PR (AUPR) and recall rates under different top |$k$| values. The average AUC and AUPR in the 5-fold cross-validations are used to evaluate the performance of all the models in comparison.
Considering the candidates in the top part of ranking list are usually selected by the biologists to further validate with wet-lab experiments, it is beneficial to discover the actual lncRNA–disease association. We thus calculate the recall rates when |$k\in $|[30,240]. The higher the top |$k$| recall rate, the more positive samples can be successfully identified by a model in the top |$k$| ranked candidates.
Ablation experiments
We conduct a set of ablation experiments to validate the contributions of the encoded pairwise topology, node attributes and feature distributions. The training strategy is the same as our final model as introduced in Section 3.1. The experimental results are given in Table 1. Without pairwise topology, the prediction performance dropped down by 4.3% and 11.4% in terms of AUC and AUPR when compared with our final model. Without learning feature distribution by VAE, the AUC and AUPR were 1.9% and 5.7% lower than that of our method. Our method achieved 5.3% and 12.1% higher accuracy in terms of AUC and AUPR when compared the model without learning nodes attributes by CAE. The ablation study demonstrated the essential and significant contributions of the three modules. We also conducted ablation experiments to verify the contributions of attention-based fusion at different representation levels. As shown by Table 1, the model improved the AUC and AUPR by 3.2% and 10.6% when compared with our model without multi-level attention, which demonstrated the contribution of attentional fusion mechanism.
Results of ablation experiments on our method
| Pairwise topology | Node attributes | Feature distribution | Multiple-level attention | Average AUC | Average AUPR |
|---|---|---|---|---|---|
| × | ✓ | ✓ | ✓ | 0.913 | 0.335 |
| ✓ | × | ✓ | ✓ | 0.903 | 0.328 |
| ✓ | ✓ | × | ✓ | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | × | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | ✓ | 0.956 | 0.449 |
| Pairwise topology | Node attributes | Feature distribution | Multiple-level attention | Average AUC | Average AUPR |
|---|---|---|---|---|---|
| × | ✓ | ✓ | ✓ | 0.913 | 0.335 |
| ✓ | × | ✓ | ✓ | 0.903 | 0.328 |
| ✓ | ✓ | × | ✓ | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | × | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | ✓ | 0.956 | 0.449 |
The bold values indicate the higher AUC and AUPR.
Results of ablation experiments on our method
| Pairwise topology | Node attributes | Feature distribution | Multiple-level attention | Average AUC | Average AUPR |
|---|---|---|---|---|---|
| × | ✓ | ✓ | ✓ | 0.913 | 0.335 |
| ✓ | × | ✓ | ✓ | 0.903 | 0.328 |
| ✓ | ✓ | × | ✓ | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | × | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | ✓ | 0.956 | 0.449 |
| Pairwise topology | Node attributes | Feature distribution | Multiple-level attention | Average AUC | Average AUPR |
|---|---|---|---|---|---|
| × | ✓ | ✓ | ✓ | 0.913 | 0.335 |
| ✓ | × | ✓ | ✓ | 0.903 | 0.328 |
| ✓ | ✓ | × | ✓ | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | × | 0.937 | 0.392 |
| ✓ | ✓ | ✓ | ✓ | 0.956 | 0.449 |
The bold values indicate the higher AUC and AUPR.
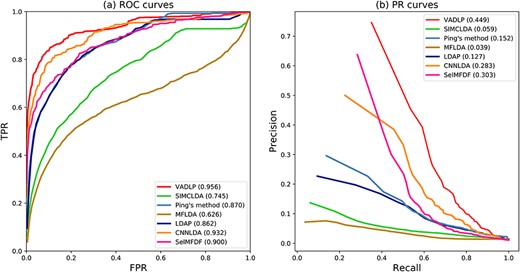
ROC curves and PR curves of all the methods in comparison all the 405 diseases.
Average AUC over all the diseases and AUCs of 10 well-characterized disease*
| Disease name | AUC | ||||||
|---|---|---|---|---|---|---|---|
| VADLP | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF | |
| Breast cancer | 0.963 | 0.886 | 0.872 | 0.742 | 0.830 | 0.517 | 0.906 |
| Stomach cancer | 0.951 | 0.928 | 0.930 | 0.864 | 0.928 | 0.467 | 0.932 |
| Kidney cancer | 0.984 | 0.981 | 0.979 | 0.728 | 0.977 | 0.677 | 0.987 |
| Prostate cancer | 0.961 | 0.932 | 0.826 | 0.874 | 0.710 | 0.553 | 0.948 |
| Gastrointestinal system cancer | 0.953 | 0.926 | 0.896 | 0.784 | 0.867 | 0.582 | 0.933 |
| Liver cancer | 0.978 | 0.953 | 0.910 | 0.799 | 0.898 | 0.634 | 0.943 |
| Hematologic cancer | 0.985 | 0.914 | 0.908 | 0.828 | 0.903 | 0.716 | 0.956 |
| Lung cancer | 0.981 | 0.975 | 0.911 | 0.790 | 0.882 | 0.676 | 0.963 |
| Ovarian cancer | 0.893 | 0.932 | 0.913 | 0.786 | 0.857 | 0.558 | 0.928 |
| Organ system cancer | 0.985 | 0.860 | 0.950 | 0.820 | 0.894 | 0.747 | 0.947 |
| Average AUC of 405 diseases | 0.969 | 0.932 | 0.870 | 0.745 | 0.862 | 0.626 | 0.900 |
| Disease name | AUC | ||||||
|---|---|---|---|---|---|---|---|
| VADLP | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF | |
| Breast cancer | 0.963 | 0.886 | 0.872 | 0.742 | 0.830 | 0.517 | 0.906 |
| Stomach cancer | 0.951 | 0.928 | 0.930 | 0.864 | 0.928 | 0.467 | 0.932 |
| Kidney cancer | 0.984 | 0.981 | 0.979 | 0.728 | 0.977 | 0.677 | 0.987 |
| Prostate cancer | 0.961 | 0.932 | 0.826 | 0.874 | 0.710 | 0.553 | 0.948 |
| Gastrointestinal system cancer | 0.953 | 0.926 | 0.896 | 0.784 | 0.867 | 0.582 | 0.933 |
| Liver cancer | 0.978 | 0.953 | 0.910 | 0.799 | 0.898 | 0.634 | 0.943 |
| Hematologic cancer | 0.985 | 0.914 | 0.908 | 0.828 | 0.903 | 0.716 | 0.956 |
| Lung cancer | 0.981 | 0.975 | 0.911 | 0.790 | 0.882 | 0.676 | 0.963 |
| Ovarian cancer | 0.893 | 0.932 | 0.913 | 0.786 | 0.857 | 0.558 | 0.928 |
| Organ system cancer | 0.985 | 0.860 | 0.950 | 0.820 | 0.894 | 0.747 | 0.947 |
| Average AUC of 405 diseases | 0.969 | 0.932 | 0.870 | 0.745 | 0.862 | 0.626 | 0.900 |
*Ten well-characterized diseases are those which are associated with at least 20 lncRNAs. The bold values indicate the higher AUCs.
Average AUC over all the diseases and AUCs of 10 well-characterized disease*
| Disease name | AUC | ||||||
|---|---|---|---|---|---|---|---|
| VADLP | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF | |
| Breast cancer | 0.963 | 0.886 | 0.872 | 0.742 | 0.830 | 0.517 | 0.906 |
| Stomach cancer | 0.951 | 0.928 | 0.930 | 0.864 | 0.928 | 0.467 | 0.932 |
| Kidney cancer | 0.984 | 0.981 | 0.979 | 0.728 | 0.977 | 0.677 | 0.987 |
| Prostate cancer | 0.961 | 0.932 | 0.826 | 0.874 | 0.710 | 0.553 | 0.948 |
| Gastrointestinal system cancer | 0.953 | 0.926 | 0.896 | 0.784 | 0.867 | 0.582 | 0.933 |
| Liver cancer | 0.978 | 0.953 | 0.910 | 0.799 | 0.898 | 0.634 | 0.943 |
| Hematologic cancer | 0.985 | 0.914 | 0.908 | 0.828 | 0.903 | 0.716 | 0.956 |
| Lung cancer | 0.981 | 0.975 | 0.911 | 0.790 | 0.882 | 0.676 | 0.963 |
| Ovarian cancer | 0.893 | 0.932 | 0.913 | 0.786 | 0.857 | 0.558 | 0.928 |
| Organ system cancer | 0.985 | 0.860 | 0.950 | 0.820 | 0.894 | 0.747 | 0.947 |
| Average AUC of 405 diseases | 0.969 | 0.932 | 0.870 | 0.745 | 0.862 | 0.626 | 0.900 |
| Disease name | AUC | ||||||
|---|---|---|---|---|---|---|---|
| VADLP | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF | |
| Breast cancer | 0.963 | 0.886 | 0.872 | 0.742 | 0.830 | 0.517 | 0.906 |
| Stomach cancer | 0.951 | 0.928 | 0.930 | 0.864 | 0.928 | 0.467 | 0.932 |
| Kidney cancer | 0.984 | 0.981 | 0.979 | 0.728 | 0.977 | 0.677 | 0.987 |
| Prostate cancer | 0.961 | 0.932 | 0.826 | 0.874 | 0.710 | 0.553 | 0.948 |
| Gastrointestinal system cancer | 0.953 | 0.926 | 0.896 | 0.784 | 0.867 | 0.582 | 0.933 |
| Liver cancer | 0.978 | 0.953 | 0.910 | 0.799 | 0.898 | 0.634 | 0.943 |
| Hematologic cancer | 0.985 | 0.914 | 0.908 | 0.828 | 0.903 | 0.716 | 0.956 |
| Lung cancer | 0.981 | 0.975 | 0.911 | 0.790 | 0.882 | 0.676 | 0.963 |
| Ovarian cancer | 0.893 | 0.932 | 0.913 | 0.786 | 0.857 | 0.558 | 0.928 |
| Organ system cancer | 0.985 | 0.860 | 0.950 | 0.820 | 0.894 | 0.747 | 0.947 |
| Average AUC of 405 diseases | 0.969 | 0.932 | 0.870 | 0.745 | 0.862 | 0.626 | 0.900 |
*Ten well-characterized diseases are those which are associated with at least 20 lncRNAs. The bold values indicate the higher AUCs.
Average AUPR over all the diseases and AUPRs of 10 well-characterized disease*
| Disease name | VADLP | CNNLDA | AUPR Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|---|
| Breast cancer | 0.624 | 0.531 | 0.403 | 0.047 | 0.396 | 0.031 | 0.569 |
| Stomach cancer | 0.372 | 0.338 | 0.364 | 0.138 | 0.094 | 0.008 | 0.327 |
| Kidney cancer | 0.238 | 0.709 | 0.663 | 0.030 | 0.462 | 0.034 | 0.722 |
| Prostate cancer | 0.370 | 0.361 | 0.333 | 0.176 | 0.297 | 0.092 | 0.358 |
| Gastrointestinal system cancer | 0.879 | 0.782 | 0.271 | 0.130 | 0.238 | 0.104 | 0.804 |
| Liver cancer | 0.875 | 0.830 | 0.498 | 0.140 | 0.511 | 0.110 | 0.784 |
| Hematologic cancer | 0.461 | 0.425 | 0.403 | 0.216 | 0.370 | 0.121 | 0.435 |
| Lung cancer | 0.628 | 0.526 | 0.437 | 0.131 | 0.363 | 0.171 | 0.581 |
| Ovarian cancer | 0.205 | 0.394 | 0.483 | 0.027 | 0.427 | 0.023 | 0.372 |
| Organ system cancer | 0.837 | 0.781 | 0.765 | 0.411 | 0.628 | 0.338 | 0.757 |
| Average AUC of 405 diseases | 0.481 | 0.283 | 0.152 | 0.059 | 0.127 | 0.039 | 0.303 |
| Disease name | VADLP | CNNLDA | AUPR Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|---|
| Breast cancer | 0.624 | 0.531 | 0.403 | 0.047 | 0.396 | 0.031 | 0.569 |
| Stomach cancer | 0.372 | 0.338 | 0.364 | 0.138 | 0.094 | 0.008 | 0.327 |
| Kidney cancer | 0.238 | 0.709 | 0.663 | 0.030 | 0.462 | 0.034 | 0.722 |
| Prostate cancer | 0.370 | 0.361 | 0.333 | 0.176 | 0.297 | 0.092 | 0.358 |
| Gastrointestinal system cancer | 0.879 | 0.782 | 0.271 | 0.130 | 0.238 | 0.104 | 0.804 |
| Liver cancer | 0.875 | 0.830 | 0.498 | 0.140 | 0.511 | 0.110 | 0.784 |
| Hematologic cancer | 0.461 | 0.425 | 0.403 | 0.216 | 0.370 | 0.121 | 0.435 |
| Lung cancer | 0.628 | 0.526 | 0.437 | 0.131 | 0.363 | 0.171 | 0.581 |
| Ovarian cancer | 0.205 | 0.394 | 0.483 | 0.027 | 0.427 | 0.023 | 0.372 |
| Organ system cancer | 0.837 | 0.781 | 0.765 | 0.411 | 0.628 | 0.338 | 0.757 |
| Average AUC of 405 diseases | 0.481 | 0.283 | 0.152 | 0.059 | 0.127 | 0.039 | 0.303 |
*10 well-characterized diseases are those which are associated with at least 20 lncRNAs.
The bold values indicate the higher AUPRs.
Average AUPR over all the diseases and AUPRs of 10 well-characterized disease*
| Disease name | VADLP | CNNLDA | AUPR Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|---|
| Breast cancer | 0.624 | 0.531 | 0.403 | 0.047 | 0.396 | 0.031 | 0.569 |
| Stomach cancer | 0.372 | 0.338 | 0.364 | 0.138 | 0.094 | 0.008 | 0.327 |
| Kidney cancer | 0.238 | 0.709 | 0.663 | 0.030 | 0.462 | 0.034 | 0.722 |
| Prostate cancer | 0.370 | 0.361 | 0.333 | 0.176 | 0.297 | 0.092 | 0.358 |
| Gastrointestinal system cancer | 0.879 | 0.782 | 0.271 | 0.130 | 0.238 | 0.104 | 0.804 |
| Liver cancer | 0.875 | 0.830 | 0.498 | 0.140 | 0.511 | 0.110 | 0.784 |
| Hematologic cancer | 0.461 | 0.425 | 0.403 | 0.216 | 0.370 | 0.121 | 0.435 |
| Lung cancer | 0.628 | 0.526 | 0.437 | 0.131 | 0.363 | 0.171 | 0.581 |
| Ovarian cancer | 0.205 | 0.394 | 0.483 | 0.027 | 0.427 | 0.023 | 0.372 |
| Organ system cancer | 0.837 | 0.781 | 0.765 | 0.411 | 0.628 | 0.338 | 0.757 |
| Average AUC of 405 diseases | 0.481 | 0.283 | 0.152 | 0.059 | 0.127 | 0.039 | 0.303 |
| Disease name | VADLP | CNNLDA | AUPR Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|---|
| Breast cancer | 0.624 | 0.531 | 0.403 | 0.047 | 0.396 | 0.031 | 0.569 |
| Stomach cancer | 0.372 | 0.338 | 0.364 | 0.138 | 0.094 | 0.008 | 0.327 |
| Kidney cancer | 0.238 | 0.709 | 0.663 | 0.030 | 0.462 | 0.034 | 0.722 |
| Prostate cancer | 0.370 | 0.361 | 0.333 | 0.176 | 0.297 | 0.092 | 0.358 |
| Gastrointestinal system cancer | 0.879 | 0.782 | 0.271 | 0.130 | 0.238 | 0.104 | 0.804 |
| Liver cancer | 0.875 | 0.830 | 0.498 | 0.140 | 0.511 | 0.110 | 0.784 |
| Hematologic cancer | 0.461 | 0.425 | 0.403 | 0.216 | 0.370 | 0.121 | 0.435 |
| Lung cancer | 0.628 | 0.526 | 0.437 | 0.131 | 0.363 | 0.171 | 0.581 |
| Ovarian cancer | 0.205 | 0.394 | 0.483 | 0.027 | 0.427 | 0.023 | 0.372 |
| Organ system cancer | 0.837 | 0.781 | 0.765 | 0.411 | 0.628 | 0.338 | 0.757 |
| Average AUC of 405 diseases | 0.481 | 0.283 | 0.152 | 0.059 | 0.127 | 0.039 | 0.303 |
*10 well-characterized diseases are those which are associated with at least 20 lncRNAs.
The bold values indicate the higher AUPRs.
Comparison of different methods based on AUC and AUPR with the paired Wilcoxon test
| p-value between VADLP and another method | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|
| p-value of AUC | 1.5281e-05 | 2.3362e-05 | 1.0745e-06 | 3.2352e-05 | 3.1702e-07 | 3.26384e-05 |
| p-value of AUPR | 1.1348e-05 | 0.0002 | 3.0745e-07 | 6.3247e-06 | 1.5643e-06 | 5.63271e-05 |
| p-value between VADLP and another method | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|
| p-value of AUC | 1.5281e-05 | 2.3362e-05 | 1.0745e-06 | 3.2352e-05 | 3.1702e-07 | 3.26384e-05 |
| p-value of AUPR | 1.1348e-05 | 0.0002 | 3.0745e-07 | 6.3247e-06 | 1.5643e-06 | 5.63271e-05 |
Comparison of different methods based on AUC and AUPR with the paired Wilcoxon test
| p-value between VADLP and another method | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|
| p-value of AUC | 1.5281e-05 | 2.3362e-05 | 1.0745e-06 | 3.2352e-05 | 3.1702e-07 | 3.26384e-05 |
| p-value of AUPR | 1.1348e-05 | 0.0002 | 3.0745e-07 | 6.3247e-06 | 1.5643e-06 | 5.63271e-05 |
| p-value between VADLP and another method | CNNLDA | Ping’s method | SIMCLDA | LDAP | MFLDA | SelMFDF |
|---|---|---|---|---|---|---|
| p-value of AUC | 1.5281e-05 | 2.3362e-05 | 1.0745e-06 | 3.2352e-05 | 3.1702e-07 | 3.26384e-05 |
| p-value of AUPR | 1.1348e-05 | 0.0002 | 3.0745e-07 | 6.3247e-06 | 1.5643e-06 | 5.63271e-05 |

The recall values at different top |$k$| cutoffs.
The experimental results demonstrated that the contribution of node attributes was the most significant among the three modules. One of the possible reasons is that node attributes embed direct similarities and associations between lncRNAs and diseases and also their neighboring lncRNAs and diseases. Pairwise topology contributed the second most to the results. Compared with node attributes, topological information can be considered as indirect relations between lncRNA and disease, which is hidden in the structure of the heterogeneous graph. Feature distribution encoded from node attributes by VAE is a representation of the underlying relationship between graph nodes, which is latent information. It is an essential component in the prediction model and it can also help with the improvement of prediction performance. In addition, we did investigation of balance controls in imbalanced issues of lncRNA–disease associations, and the statements and experimental results are listed in SF4.
Comparison with other methods
The proposed method is compared with five state-of-the-art methods for lncRNA–disease association prediction including (i) CNNLDA [39], (ii) Ping’s method [20], (iii) LDAP [31], (iv) SIMCLDA [36], (v) MFLDA [35] and (vi) SelMFDF [38]. Our VADLP model and all the methods in comparison were trained and tested using the same dataset in the cross-validation. The best free parameters reported by each method were used in implementation, where |$s = 2\times 2$| was used for CNNLDA, |$\alpha =0.6$| for Ping’s method, |$\alpha _l=0.8$|, |$Gap open=10, Gap extend=10$| for LDAP, |$\alpha _d=0.6$| and |$\lambda =1$| for SIMCLDA, |$\alpha =105$| for MFLDA and |$k=20$| for SelMFDF.
The ROC and PR curves of all the methods in comparison over all the 405 diseases are given in Figure 5. The AUC and AUPR over all the diseases and 10 well-characterized diseases that are associated with at least 20 lncRNAs are given in Tables 2 and 3. As shown by Figure 5A and Table 2, our model achieved the highest average AUC of 0.956, which was 2.4% higher than the 2nd best CNNLDA, 5.6% better than SelMFDF, 8.6% higher than Ping’s method, 9.4% and 21.1% higher than LDAP and SIMCLDA and 33% better than the worst performed MFLDA. In terms of average AUPR over all the diseases, our model achieved the best AUPR of 0.449, which is 16.6%, 14.7%, 29.7%, 39%, 32.2% and 41% higher than that of CNNLDA, SelMFDF, Ping’s method, SIMCLDA, LDAP and MFLDA. For the 10 well-characterized diseases, which are associated with at least 20 lncRNAs, our model achieved the best performance over eight diseases in terms of average AUC and eight diseases with respect to AUPR as shown in Tables 2 and 3. Paired Wilcoxon test results demonstrated that our model statistically significantly (p-value < 0.05) outperformed other methods with respect to both AUC and AUPR as shown in Table 4.
As shown by the results, even though CNNLDA utilizes the neural network, it ignores the feature distribution and attribute information of lncRNA–disease pairwises, so its performance is not as good as our methods. Ping’s method is based on information flow propagation and LDAP is based on SVM, their performance was similar with respect to ROC, PR curve, average AUC and AUPR over all the diseases. One of the possible reasons is that both LDAP and Ping’s method exploited the similarity and association information of lncRNAs and diseases. Without considering intra-lncRNA and disease similarities, SIMCLDA and MFLDA performed much worse than Ping’s method and LDAP. Our method outperformed CNNLDA, Ping’s method and LDAP with the newly introduced pairwise topology, node attributes and deep feature distributions.
The top 15 breast cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | PANDAR | Lnc2Cancer, LncRNADisease | 9 | CCAT2 | Lnc2Cancer, LncRNADisease |
| 2 | XIST | Lnc2Cancer, LncRNADisease | 10 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 3 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease | 11 | MALAT1 | Lnc2Cancer, LncRNADisease |
| 4 | ZFAS1 | Lnc2Cancer, LncRNADisease | 12 | MIR124-2HG | Literature |
| 5 | LINC-ROR | Lnc2Cancer, LncRNADisease | 13 | SOX2-OT | Lnc2Cancer, LncRNADisease |
| 6 | PVT1 | Lnc2Cancer, LncRNADisease | 14 | LINC-PINT | Literature |
| 7 | CCAT1 | Lnc2Cancer, LncRNADisease | 15 | LSINCT5 | Lnc2Cancer |
| 8 | CDKN2B-AS1 | LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | PANDAR | Lnc2Cancer, LncRNADisease | 9 | CCAT2 | Lnc2Cancer, LncRNADisease |
| 2 | XIST | Lnc2Cancer, LncRNADisease | 10 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 3 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease | 11 | MALAT1 | Lnc2Cancer, LncRNADisease |
| 4 | ZFAS1 | Lnc2Cancer, LncRNADisease | 12 | MIR124-2HG | Literature |
| 5 | LINC-ROR | Lnc2Cancer, LncRNADisease | 13 | SOX2-OT | Lnc2Cancer, LncRNADisease |
| 6 | PVT1 | Lnc2Cancer, LncRNADisease | 14 | LINC-PINT | Literature |
| 7 | CCAT1 | Lnc2Cancer, LncRNADisease | 15 | LSINCT5 | Lnc2Cancer |
| 8 | CDKN2B-AS1 | LncRNADisease |
The top 15 breast cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | PANDAR | Lnc2Cancer, LncRNADisease | 9 | CCAT2 | Lnc2Cancer, LncRNADisease |
| 2 | XIST | Lnc2Cancer, LncRNADisease | 10 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 3 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease | 11 | MALAT1 | Lnc2Cancer, LncRNADisease |
| 4 | ZFAS1 | Lnc2Cancer, LncRNADisease | 12 | MIR124-2HG | Literature |
| 5 | LINC-ROR | Lnc2Cancer, LncRNADisease | 13 | SOX2-OT | Lnc2Cancer, LncRNADisease |
| 6 | PVT1 | Lnc2Cancer, LncRNADisease | 14 | LINC-PINT | Literature |
| 7 | CCAT1 | Lnc2Cancer, LncRNADisease | 15 | LSINCT5 | Lnc2Cancer |
| 8 | CDKN2B-AS1 | LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | PANDAR | Lnc2Cancer, LncRNADisease | 9 | CCAT2 | Lnc2Cancer, LncRNADisease |
| 2 | XIST | Lnc2Cancer, LncRNADisease | 10 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 3 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease | 11 | MALAT1 | Lnc2Cancer, LncRNADisease |
| 4 | ZFAS1 | Lnc2Cancer, LncRNADisease | 12 | MIR124-2HG | Literature |
| 5 | LINC-ROR | Lnc2Cancer, LncRNADisease | 13 | SOX2-OT | Lnc2Cancer, LncRNADisease |
| 6 | PVT1 | Lnc2Cancer, LncRNADisease | 14 | LINC-PINT | Literature |
| 7 | CCAT1 | Lnc2Cancer, LncRNADisease | 15 | LSINCT5 | Lnc2Cancer |
| 8 | CDKN2B-AS1 | LncRNADisease |
The top 15 colorectal cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MEG3 | Lnc2Cancer, LncRNADisease | 9 | CASC2 | Lnc2Cancer, LncRNADisease |
| 2 | CCAT2 | Lnc2Cancer, LncRNADisease | 10 | CASC19 | LncRNADisease |
| 3 | GHET1 | Lnc2Cancer, LncRNADisease | 11 | CRNDE | Lnc2Cancer, LncRNADisease |
| 4 | HOTAIRM1 | Lnc2Cancer, LncRNADisease | 12 | TP53COR1 | LncRNADisease |
| 5 | BANCR | Lnc2Cancer, LncRNADisease | 13 | TUG1 | Lnc2Cancer, LncRNADisease |
| 6 | LSINCT5 | Lnc2Cancer | 14 | KCNQ1OT1 | Lnc2Cancer, LncRNADisease |
| 7 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease | 15 | NEAT1 | Lnc2Cancer, LncRNADisease |
| 8 | HOTAIR | Lnc2Cancer, LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MEG3 | Lnc2Cancer, LncRNADisease | 9 | CASC2 | Lnc2Cancer, LncRNADisease |
| 2 | CCAT2 | Lnc2Cancer, LncRNADisease | 10 | CASC19 | LncRNADisease |
| 3 | GHET1 | Lnc2Cancer, LncRNADisease | 11 | CRNDE | Lnc2Cancer, LncRNADisease |
| 4 | HOTAIRM1 | Lnc2Cancer, LncRNADisease | 12 | TP53COR1 | LncRNADisease |
| 5 | BANCR | Lnc2Cancer, LncRNADisease | 13 | TUG1 | Lnc2Cancer, LncRNADisease |
| 6 | LSINCT5 | Lnc2Cancer | 14 | KCNQ1OT1 | Lnc2Cancer, LncRNADisease |
| 7 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease | 15 | NEAT1 | Lnc2Cancer, LncRNADisease |
| 8 | HOTAIR | Lnc2Cancer, LncRNADisease |
The top 15 colorectal cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MEG3 | Lnc2Cancer, LncRNADisease | 9 | CASC2 | Lnc2Cancer, LncRNADisease |
| 2 | CCAT2 | Lnc2Cancer, LncRNADisease | 10 | CASC19 | LncRNADisease |
| 3 | GHET1 | Lnc2Cancer, LncRNADisease | 11 | CRNDE | Lnc2Cancer, LncRNADisease |
| 4 | HOTAIRM1 | Lnc2Cancer, LncRNADisease | 12 | TP53COR1 | LncRNADisease |
| 5 | BANCR | Lnc2Cancer, LncRNADisease | 13 | TUG1 | Lnc2Cancer, LncRNADisease |
| 6 | LSINCT5 | Lnc2Cancer | 14 | KCNQ1OT1 | Lnc2Cancer, LncRNADisease |
| 7 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease | 15 | NEAT1 | Lnc2Cancer, LncRNADisease |
| 8 | HOTAIR | Lnc2Cancer, LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MEG3 | Lnc2Cancer, LncRNADisease | 9 | CASC2 | Lnc2Cancer, LncRNADisease |
| 2 | CCAT2 | Lnc2Cancer, LncRNADisease | 10 | CASC19 | LncRNADisease |
| 3 | GHET1 | Lnc2Cancer, LncRNADisease | 11 | CRNDE | Lnc2Cancer, LncRNADisease |
| 4 | HOTAIRM1 | Lnc2Cancer, LncRNADisease | 12 | TP53COR1 | LncRNADisease |
| 5 | BANCR | Lnc2Cancer, LncRNADisease | 13 | TUG1 | Lnc2Cancer, LncRNADisease |
| 6 | LSINCT5 | Lnc2Cancer | 14 | KCNQ1OT1 | Lnc2Cancer, LncRNADisease |
| 7 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease | 15 | NEAT1 | Lnc2Cancer, LncRNADisease |
| 8 | HOTAIR | Lnc2Cancer, LncRNADisease |
The top 15 hepatocellular cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MALAT1 | Lnc2Cancer, LncRNADisease | 9 | MIR17HG | Literature |
| 2 | SNHG1 | Lnc2Cancer, LncRNADisease | 10 | PANDAR | Lnc2Cancer, LncRNADisease |
| 3 | PCAT1 | Lnc2Cancer, LncRNADisease | 11 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease |
| 4 | IGF2-AS | Lnc2Cancer, LncRNADisease | 12 | DBH-AS1 | Lnc2Cancer, LncRNADisease |
| 5 | MEG3 | Lnc2Cancer, LncRNADisease | 13 | XIST | Lnc2Cancer, LncRNADisease |
| 6 | CDKN2B-AS1 | LncRNADisease | 14 | DANCR | Lnc2Cancer, LncRNADisease |
| 7 | LINC00974 | Lnc2Cancer, LncRNADisease | 15 | H19 | Lnc2Cancer, LncRNADisease |
| 8 | GAS5 | LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MALAT1 | Lnc2Cancer, LncRNADisease | 9 | MIR17HG | Literature |
| 2 | SNHG1 | Lnc2Cancer, LncRNADisease | 10 | PANDAR | Lnc2Cancer, LncRNADisease |
| 3 | PCAT1 | Lnc2Cancer, LncRNADisease | 11 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease |
| 4 | IGF2-AS | Lnc2Cancer, LncRNADisease | 12 | DBH-AS1 | Lnc2Cancer, LncRNADisease |
| 5 | MEG3 | Lnc2Cancer, LncRNADisease | 13 | XIST | Lnc2Cancer, LncRNADisease |
| 6 | CDKN2B-AS1 | LncRNADisease | 14 | DANCR | Lnc2Cancer, LncRNADisease |
| 7 | LINC00974 | Lnc2Cancer, LncRNADisease | 15 | H19 | Lnc2Cancer, LncRNADisease |
| 8 | GAS5 | LncRNADisease |
The top 15 hepatocellular cancer-related lncRNA candidates
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MALAT1 | Lnc2Cancer, LncRNADisease | 9 | MIR17HG | Literature |
| 2 | SNHG1 | Lnc2Cancer, LncRNADisease | 10 | PANDAR | Lnc2Cancer, LncRNADisease |
| 3 | PCAT1 | Lnc2Cancer, LncRNADisease | 11 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease |
| 4 | IGF2-AS | Lnc2Cancer, LncRNADisease | 12 | DBH-AS1 | Lnc2Cancer, LncRNADisease |
| 5 | MEG3 | Lnc2Cancer, LncRNADisease | 13 | XIST | Lnc2Cancer, LncRNADisease |
| 6 | CDKN2B-AS1 | LncRNADisease | 14 | DANCR | Lnc2Cancer, LncRNADisease |
| 7 | LINC00974 | Lnc2Cancer, LncRNADisease | 15 | H19 | Lnc2Cancer, LncRNADisease |
| 8 | GAS5 | LncRNADisease |
| Rank | LncRNA name | Source of verification | Rank | LncRNA name | Source of verification |
|---|---|---|---|---|---|
| 1 | MALAT1 | Lnc2Cancer, LncRNADisease | 9 | MIR17HG | Literature |
| 2 | SNHG1 | Lnc2Cancer, LncRNADisease | 10 | PANDAR | Lnc2Cancer, LncRNADisease |
| 3 | PCAT1 | Lnc2Cancer, LncRNADisease | 11 | AFAP1-AS1 | Lnc2Cancer, LncRNADisease |
| 4 | IGF2-AS | Lnc2Cancer, LncRNADisease | 12 | DBH-AS1 | Lnc2Cancer, LncRNADisease |
| 5 | MEG3 | Lnc2Cancer, LncRNADisease | 13 | XIST | Lnc2Cancer, LncRNADisease |
| 6 | CDKN2B-AS1 | LncRNADisease | 14 | DANCR | Lnc2Cancer, LncRNADisease |
| 7 | LINC00974 | Lnc2Cancer, LncRNADisease | 15 | H19 | Lnc2Cancer, LncRNADisease |
| 8 | GAS5 | LncRNADisease |
According to equation (30), the recall rates under different top |$k$| values are given in Figure 6. Our model consistently outperformed other methods at various |$k$| cutoffs due to the learnt pairwise topology, node attributes and feature distribution. The results demonstrated the capacity of the proposed model in screening real disease-related lncRNAs for the top parts of the prediction results. When |$k$| was 30, the highest recall rate of 85.1% was achieved by our model, and the 2nd best 74.6% was obtained by CNNLDA. 68.9% was obtained by Ping’s method which was slightly higher than the fourth ranked LDAP with a rate of 68.5%. When |$k$| was increased from 60 to 120, our model remained the best performed with recall rates of 91%, 92.4% and 94.4%. The second best was CNNLDA with recall rates of 89.5%, 91.3% and 93.5%. LDAP started to outperform Ping’s method but with a marginal improvement. The corresponding recall rates of LDAP were 81.7%, 88.0% and 93.3% while those of Ping’s method were 81.3%, 87.5% and 92.7%. SIMCLDA was consistently worse than CNNLDA, Ping’s method and LDAP with recall rates of 49.3%, 62.9%, 74.4% and 80.3% when |$k$| was between 30 and 120. The lowest recall values were obtained by MFLDA, which were 42.0%, 53.8%, 60.9% and 65.5%.
Case studies: breast cancer, hepatocellular cancer and colorectal cancer
To further demonstrate the capability of the proposed model in discovering the potential lncRNA–disease associations, we performed case studies over breast, hepatocellular and colorectal cancers. For each cancer, we ranked the lncRNA candidates according to their estimated lncRNA–disease association scores in a descending order.
The top 15 ranked candidates for each cancer are given in Tables 5–7. LncRNADisease, Lnc2Cancer databases and published literatures are used to verify and confirm the predicted lncRNA–disease associations. LncRNADisease provides the information about the lncRNAs and their effects on human diseases, which is obtained from the biological experiments and published literatures [45]. Lnc2Cancer is a manually curated database, which records 4989 lncRNA–disease associations between lncRNAs and human cancers obtained from the biological experiments where dysregulation of lncRNA are further manually confirmed [46].
Firstly, among all the 15 top-ranked breast cancer-related lncRNAs by our model (Table 5), 12 of them are verified by LncRNADisease. This result demonstrated that those lncRNA candidates are indeed related with the disease. Twelve of them are confirmed by Lnc2Cancer, which means that they are upregulated or downregulated in breast cancer tissue. In addition, there are two lncRNA candidates, MIR124-2HG and LINC-PINT, verified by literature. MIR124-2HG is supported by recent study showing that decreased MIR124-2HG expression enhances the proliferation of breast cancer cells targeting BECN1 [59, 60]. As shown by another literature, LINC-PINT is apparently downregulated in breast cancer tissue compared with normal tissue [61, 62].
Secondly, the top 15 hepatocellular cancer-related lncRNAs are given in Table 6. Fourteen of them are confirmed by LncRNADisease, which proved their associations with hepatocellular cancer. Thirteen of the top-ranked candidates are found in Lnc2Cancer, where their expression levels in hepatocellular cancer are significantly different from normal tissue. One candidate is supported by the literature, which has abnormal expression in hypocellular cancer [63, 64].
Lastly, among all the top-ranked lncRNA candidates related to colorectal cancer (Table 7), 14 of them are verified by LncRNADisease and 13 of them are proved by Lnc2Cancer database. The case studies further demonstrated the capability of our model in discovering potential lncRNA–disease associations.
Prediction of novel disease-related lncRNAs
In the end, the proposed model is used for the prediction of lncRNA candidates, which are related with the diseases. The top 50 ranked lncRNA candidates predicted by our model are provided in the ST2 to assist the biologists in discovering true novel disease-related lncRNAs in further wet-lab experiments.
Conclusions
In this paper, we proposed a model to adaptively learn and integrate pairwise topology, node attributes and deep feature distribution encoded from multi-sourced data to predict disease-related lncRNAs. A multi-layer heterogeneous graph was constructed to benefit node attribute embedding and pairwise topology extraction by random walks. A framework based on CAE and VAE was constructed for learning and encoding pairwise topology representation, node attribute representation and feature distribution representation. The attention mechanism was proposed to discriminate the contributions of these three representations and adaptively fuse them. Comparison with five recent lncRNA–disease association prediction models and ablation study demonstrated the improved performance of our model in terms of AUC and AUPR. Notably, our model is more powerful in discovering true lncRNA–disease associations and return them as top-ranked candidates as demonstrated by recall rates under different top |$k$| values. Case studies of three cancers further proved the capacity of our model. Our model can be used as a prioritization tool to screen potential candidates and then discover true lncRNA–disease associations through wet laboratory experiments.
A new multi-layer heterogeneous graph is proposed to benefit the extraction and representation of diverse relations among multiple sources of data, lncRNAs, diseases and miRNAs, for lncRNA–disease association modeling.
We extract three levels of representations between a pair of lncRNA and disease including the novel node attributes to represent the lncRNA–disease associations, which are inferred via their common lncRNAs, diseases and miRNAs, a new high-level feature distribution to reveal the deep and underlying relations across three sources of data and a pairwise topology to represent the learnt hidden topological structural relations.
An attentional representation-level integration and optimization module is proposed to fuse three levels of representations adaptively.
The contributions of each representation and improved performance were demonstrated by ablation study and comparison with six state-of-the-art lncRNA–disease prediction models over a public dataset. The improved recall rates under different top |$k$| values showed that our model was powerful in discovering true disease-related lncRNAs in the top-ranked candidates. Case studies of three cancers further proved the capacity of the proposed model.
Funding
Natural Science Foundation of China (61972135); Natural Science Foundation of Heilongjiang Province (LH2019F049 and LH2019A029); China Postdoctoral Science Foundation (2019M650069); Heilongjiang Postdoctoral Scientific Research Staring Foundation (BHLQ18104); Fundamental Research Foundation of Universities in Heilongjiang Province for Technology Innovation (KJCX201805); Innovation Talents Project of Harbin Science and Technology Bureau (2017RAQXJ094); Fundamental Research Foundation of Universities in Heilongjiang Province for Youth Innovation Team (RCYJTD201805).
Conflict of interest
The Authors declare that there is no conflict of interest.
Nan Sheng is studying for his master’s degree in the School of Computer Science and Technology at Heilongjiang University, Harbin, China. His research interests include complex network analysis and deep learning.
Hui Cui, PhD (The University of Sydney), is a lecturer at Department of Computer Science and Information Technology, La Trobe University, Melbourne, Australia. Her research interests lie in data-driven and computerized models for biomedical and health informatics.
Tiangang Zhang, PhD (the University of Tokyo), is an associate professor of the Department of Mathematical Science, Heilongjiang University, Harbin, China. His current research interests include complex network analysis and computational fluid dynamics.
Ping Xuan, PhD (Harbin Institute of Technology), is a professor at the School of Computer Science and Technology, Heilongjiang University, Harbin, China. Her current research interests include computational biology, complex network analysis and deep learning.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}