




Abstract
Human leukocyte antigen class I (HLA-I) molecules are encoded by major histocompatibility complex (MHC) class I loci in humans. The binding and interaction between HLA-I molecules and intracellular peptides derived from a variety of proteolytic mechanisms play a crucial role in subsequent T-cell recognition of target cells and the specificity of the immune response. In this context, tools that predict the likelihood for a peptide to bind to specific HLA class I allotypes are important for selecting the most promising antigenic targets for immunotherapy. In this article, we comprehensively review a variety of currently available tools for predicting the binding of peptides to a selection of HLA-I allomorphs. Specifically, we compare their calculation methods for the prediction score, employed algorithms, evaluation strategies and software functionalities. In addition, we have evaluated the prediction performance of the reviewed tools based on an independent validation data set, containing 21 101 experimentally verified ligands across 19 HLA-I allotypes. The benchmarking results show that MixMHCpred 2.0.1 achieves the best performance for predicting peptides binding to most of the HLA-I allomorphs studied, while NetMHCpan 4.0 and NetMHCcons 1.1 outperform the other machine learning-based and consensus-based tools, respectively. Importantly, it should be noted that a peptide predicted with a higher binding score for a specific HLA allotype does not necessarily imply it will be immunogenic. That said, peptide-binding predictors are still very useful in that they can help to significantly reduce the large number of epitope candidates that need to be experimentally verified. Several other factors, including susceptibility to proteasome cleavage, peptide transport into the endoplasmic reticulum and T-cell receptor repertoire, also contribute to the immunogenicity of peptide antigens, and some of them can be considered by some predictors. Therefore, integrating features derived from these additional factors together with HLA-binding properties by using machine-learning algorithms may increase the prediction accuracy of immunogenic peptides. As such, we anticipate that this review and benchmarking survey will assist researchers in selecting appropriate prediction tools that best suit their purposes and provide useful guidelines for the development of improved antigen predictors in the future.
Introduction
The binding of peptides to specific human leukocyte antigen (HLA) allomorphs and the subsequent recognition of peptide-HLA complexes (pHLAs) by T cells establish the antigenicity of the peptide, and this process forms the basis of immune surveillance [1]. HLA molecules can be divided into two main categories, namely HLA class I (HLA-I) and HLA class II (HLA-II). The HLA-I molecules are encoded by three I loci (HLA-A, -B and -C), and the encoded proteins are expressed on the surface of all nucleated cells. In contrast, HLA-II molecules encoded by HLA class II loci (HLA-DR, -DQ and -DP) can only be expressed in professional antigen-presenting cells (APC) such as dendritic cells, mononuclear phagocytes and B cells [1]. HLA-I molecules mainly bind short peptides of 8–12 amino acids in length, typically derived from proteasome-mediated degradation of intracellular proteins. These pHLAs are then presented on the cell surface for recognition by CD8+ T cells. HLA-II molecules tend to bind longer peptides (12–20 amino acids in length) liberated from extracellular proteins within the endosomal compartments. These pHLAs are presented on the surface of professional APC for recognition by CD4+ T cells [2]. The interactions between HLA molecules and peptides and subsequent recognition of these complexes by T cells control the magnitude and effectiveness of the immune response. Thus, a major goal in vaccinology and immunotherapy resides in the accurate prediction of peptide-HLA binding and the ability of these complexes to induce a desired immune response [3]. Understanding which peptides are selected for display in the context of an individual’s HLA type can aid the design of vaccines designed to induce protective or therapeutic immunity towards various pathogens [4, 5]. Equally, several studies have found that neo-antigens generated as a result of non-synonymous mutations in cancer cells play a significant role in the dynamics of the anti-tumour immune response [6–9]. Indeed, vaccines based on such neo-antigens have been shown to benefit clinical outcomes [10, 11]. Typically, identifying neo-antigens first requires characterization of the non-synonymous mutations from primary tumours using next-generation sequencing (NGS) platforms such as RNAseq. In the second step, peptide sequences that contain mutations are further assessed by predicting their individual probability to bind to patient HLA allomorphs [12]. By filtering out potential allomorph-specific HLA ligands, the number of candidates can be substantially decreased, thereby accelerating the final step of experimentally verifying neo-epitopes [13, 14]. These and other considerations have increased the interest in predicting peptide binding to HLA molecules over the past few years.
Over the last few decades, the availability of experimentally verified HLA ligand sequences has increased, with sequences often deposited in public peptide ligand databases. Up until recently, most of these data have been generated using in vitro binding assays [15, 16], but the use of mass spectrometry (MS)-based identification of purified HLA-binding peptides has now come into the forefront [17–19]. The Immune Epitope Database (IEDB) is the largest public resource for HLA ligands and T-cell epitopes [20]. It contains detailed information on curated peptides collected from published journal articles with appended metadata including the experimental modality used for data acquisition. Due to the increasing availability of high-quality HLA allele-specific data sets, a number of new computational tools have been developed for predicting peptide binding to HLA molecules. However, the main focus has been on the prediction of HLA-I ligands, since the more complex nature (longer and more heterogeneous peptide sequences) of HLA-II ligands makes their prediction more difficult [21]. Here we classify these publically available tools into three major categories based on the methodologies they used, namely (i) methods based on sequence-scoring functions, including SYFPEITHI [22], RANKPEP [23], PickPocket 1.1 [24], stabilized matrix method (SMM)—peptide:major histocompatibility complex (MHC)-binding energy covariance (SMMPMBEC) [25], PSSMHCpan 1.0 [26] and MixMHCpred 2.0.1 [18, 27]; (ii) methods based on machine learning algorithms, including NetMHC 4.0 [28], NetMHCstabpan 1.0 [29], NetMHCPan 4.0 [30], MHCflurry 1.2.0 [31], MHCnuggets 2.0 [32], ConvMHC [33] and HLA-CNN [34]; and (iii) methods based on the integration of different peptide-binding predictors, including NetMHCcons 1.1 [35] and IEDB-analysis resource-consensus (IEDB-AR-Consensus) [25]. It should be noted that structure-based methods can also contribute to HLA-I peptide-binding prediction. These methods have achieved accurate binding prediction performance by modelling the docking between the HLA protein and peptide ligands [36–51]. However, structure-based approaches are not suitable for all allotypes and rely on homologous structures. Therefore, the present review has not included the structure-based peptide-binding prediction methods.
Several attempts have been made to provide benchmark tests of prediction tools; however, each study had certain limitations: either they did not include a performance evaluation of all reviewed tools, or several state-of-the-art prediction tools were not considered and benchmarked or did not have a detailed algorithm description for each of the reviewed tools [52–56]. For example, a recent review comprehensively discussed currently available peptide-binding prediction tools, but it lacked a uniform validation approach to allow performance comparison of the different predictors [2]. To overcome these issues, here we provide a comprehensive performance benchmarking and assessment of currently available, state-of-the-art tools for predicting peptide binding to HLA-I molecules. In total, 15 prediction tools have been systematically benchmarked in terms of their underlying algorithms, feature selection, performance evaluation strategy and webserver and/or software functionality. Most importantly, we also performed an independent test to evaluate the performance of these tools by using a newly generated peptide data set containing HLA molecule ligands across 19 HLA-I allomorphs. Following our review, we give some suggestions for the design and development of future prediction tools. Lastly, we hope our review will assist and inspire scientists with interest in this field to facilitate their efforts in developing improved tools for the prediction of T-cell epitopes.
A list of currently available tools for HLA-I peptide-binding prediction assessed in this review
| Category | Toola | Year | Software availabilityb | Webserver availabilityb | Max data upload | Step-by-step instructionb,c | Parameters setting functionb,c | Email of resultb,c | Features | Algorithmc | Training–test splitc | Performance evaluation strategyc | Last updated date |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scoring function based | SYFPEITHI | 1999 | No | Yes | Single sequence | No | No | No | PSSM | NM | N.A. | August 2012 | |
| RANKPEP | 2002 | No | Yes | N.M. | Yes | Yes | No | PSSM | NM | Independent test | January 2019 | ||
| PickPocket 1.1 | 2009 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | PSSM | NM | LOO, 5-fold CV | January 2017 | ||
| SMMPMBEC | 2009 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | PSSM | NM | 5-fold CV | January 2014 | ||
| PSSMHCpan 1.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | 10-fold CV, independent test | February 2017 | ||
| MixMHCpred 2.0.1 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | Independent test | August 2018 | ||
| Machine learning based | NetMHC 4.0 | 2014 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based features | NN | 4:1 | 5-fold CV | October 2017 |
| NetMHCstabpan 1.0 | 2016 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Physicochemical features | NN | 4:1 | 5-fold CV, independent test | September 2018 | |
| NetMHCPan 4.0 | 2017 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based & binary features | NN | 4:1 | 5-fold CV, independent test | January 2018 | |
| MHCnuggets 2.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & physicochemical features | NN | NM | 3-fold CV | August 2018 | |
| ConvMHC | 2017 | No | Yes | N.M. | No | No | No | Sequence-based & physicochemical features | NN | 75:1 | LOO, 5-fold CV, independent test | July 2018 | |
| HLA-CNN | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based features | NN | 7:3 | 5-fold CV, independent test | August 2017 | |
| MHCflurry 1.2.0 | 2018 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & binary features | NN | 9:1 | Independent test | January 2019 | |
| Consensus | IEDB-AR-Consensus | 2012 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | Con | NM | Independent data set | May 2012 | |
| NetMHCcons 1.1 | 2012 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Con | NM | Independent data set | January 2017 |
| Category | Toola | Year | Software availabilityb | Webserver availabilityb | Max data upload | Step-by-step instructionb,c | Parameters setting functionb,c | Email of resultb,c | Features | Algorithmc | Training–test splitc | Performance evaluation strategyc | Last updated date |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scoring function based | SYFPEITHI | 1999 | No | Yes | Single sequence | No | No | No | PSSM | NM | N.A. | August 2012 | |
| RANKPEP | 2002 | No | Yes | N.M. | Yes | Yes | No | PSSM | NM | Independent test | January 2019 | ||
| PickPocket 1.1 | 2009 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | PSSM | NM | LOO, 5-fold CV | January 2017 | ||
| SMMPMBEC | 2009 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | PSSM | NM | 5-fold CV | January 2014 | ||
| PSSMHCpan 1.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | 10-fold CV, independent test | February 2017 | ||
| MixMHCpred 2.0.1 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | Independent test | August 2018 | ||
| Machine learning based | NetMHC 4.0 | 2014 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based features | NN | 4:1 | 5-fold CV | October 2017 |
| NetMHCstabpan 1.0 | 2016 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Physicochemical features | NN | 4:1 | 5-fold CV, independent test | September 2018 | |
| NetMHCPan 4.0 | 2017 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based & binary features | NN | 4:1 | 5-fold CV, independent test | January 2018 | |
| MHCnuggets 2.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & physicochemical features | NN | NM | 3-fold CV | August 2018 | |
| ConvMHC | 2017 | No | Yes | N.M. | No | No | No | Sequence-based & physicochemical features | NN | 75:1 | LOO, 5-fold CV, independent test | July 2018 | |
| HLA-CNN | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based features | NN | 7:3 | 5-fold CV, independent test | August 2017 | |
| MHCflurry 1.2.0 | 2018 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & binary features | NN | 9:1 | Independent test | January 2019 | |
| Consensus | IEDB-AR-Consensus | 2012 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | Con | NM | Independent data set | May 2012 | |
| NetMHCcons 1.1 | 2012 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Con | NM | Independent data set | January 2017 |
aThe URL addresses for accessing the listed tools are as follows: SYFPEITHI, http://www.syfpeithi.de/index.html/; RANKPEP, http://imed.med.ucm.es/Tools/rankpep.html/; PickPocket 1.1, http://www.cbs.dtu.dk/services/PickPocket/; SMMPMBEC, https://github.com/ykimbiology/smmpmbec/; PSSMHCpan 1.0, https://github.com/BGI2016/PSSMHCpan/; MixMHCpred 2.0.1, https://github.com/GfellerLab/MixMHCpred/; NetMHC4.0, http://www.cbs.dtu.dk/services/NetMHC/; NetMHCstabpan 1.0, http://www.cbs.dtu.dk/services/NetMHCstabpan/; NetMHCPan-4.0, http://www.cbs.dtu.dk/services/NetMHCpan/; MHCnuggets 2.0, https://github.com/KarchinLab/mhcnuggets-2.0/; ConvMHC, http://jumong.kaist.ac.kr:8080/convmhc/; HLA-CNN, https://github.com/uci-cbcl/HLA-bind/; MHCflurry 1.2.0, https://github.com/openvax/mhcflurry/; IEDB-AR-Consensus, http://tools.iedb.org/mhci/; NetMHCcons-1.1, http://www.cbs.dtu.dk/services/NetMHCcons/.
bYes: the publication has developed the function according to the column; No: the publication has not developed the function according to the column.
cAbbreviations: N.M., not mentioned; N.A., not applicable; PSSM, position-specific scoring matrix; NN, Neural network; Con, Consensus-based method; CV, cross-validation; LOO, leave-one-out.
A list of currently available tools for HLA-I peptide-binding prediction assessed in this review
| Category | Toola | Year | Software availabilityb | Webserver availabilityb | Max data upload | Step-by-step instructionb,c | Parameters setting functionb,c | Email of resultb,c | Features | Algorithmc | Training–test splitc | Performance evaluation strategyc | Last updated date |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scoring function based | SYFPEITHI | 1999 | No | Yes | Single sequence | No | No | No | PSSM | NM | N.A. | August 2012 | |
| RANKPEP | 2002 | No | Yes | N.M. | Yes | Yes | No | PSSM | NM | Independent test | January 2019 | ||
| PickPocket 1.1 | 2009 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | PSSM | NM | LOO, 5-fold CV | January 2017 | ||
| SMMPMBEC | 2009 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | PSSM | NM | 5-fold CV | January 2014 | ||
| PSSMHCpan 1.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | 10-fold CV, independent test | February 2017 | ||
| MixMHCpred 2.0.1 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | Independent test | August 2018 | ||
| Machine learning based | NetMHC 4.0 | 2014 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based features | NN | 4:1 | 5-fold CV | October 2017 |
| NetMHCstabpan 1.0 | 2016 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Physicochemical features | NN | 4:1 | 5-fold CV, independent test | September 2018 | |
| NetMHCPan 4.0 | 2017 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based & binary features | NN | 4:1 | 5-fold CV, independent test | January 2018 | |
| MHCnuggets 2.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & physicochemical features | NN | NM | 3-fold CV | August 2018 | |
| ConvMHC | 2017 | No | Yes | N.M. | No | No | No | Sequence-based & physicochemical features | NN | 75:1 | LOO, 5-fold CV, independent test | July 2018 | |
| HLA-CNN | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based features | NN | 7:3 | 5-fold CV, independent test | August 2017 | |
| MHCflurry 1.2.0 | 2018 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & binary features | NN | 9:1 | Independent test | January 2019 | |
| Consensus | IEDB-AR-Consensus | 2012 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | Con | NM | Independent data set | May 2012 | |
| NetMHCcons 1.1 | 2012 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Con | NM | Independent data set | January 2017 |
| Category | Toola | Year | Software availabilityb | Webserver availabilityb | Max data upload | Step-by-step instructionb,c | Parameters setting functionb,c | Email of resultb,c | Features | Algorithmc | Training–test splitc | Performance evaluation strategyc | Last updated date |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scoring function based | SYFPEITHI | 1999 | No | Yes | Single sequence | No | No | No | PSSM | NM | N.A. | August 2012 | |
| RANKPEP | 2002 | No | Yes | N.M. | Yes | Yes | No | PSSM | NM | Independent test | January 2019 | ||
| PickPocket 1.1 | 2009 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | PSSM | NM | LOO, 5-fold CV | January 2017 | ||
| SMMPMBEC | 2009 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | PSSM | NM | 5-fold CV | January 2014 | ||
| PSSMHCpan 1.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | 10-fold CV, independent test | February 2017 | ||
| MixMHCpred 2.0.1 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | PSSM | NM | Independent test | August 2018 | ||
| Machine learning based | NetMHC 4.0 | 2014 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based features | NN | 4:1 | 5-fold CV | October 2017 |
| NetMHCstabpan 1.0 | 2016 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Physicochemical features | NN | 4:1 | 5-fold CV, independent test | September 2018 | |
| NetMHCPan 4.0 | 2017 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Sequence-based & binary features | NN | 4:1 | 5-fold CV, independent test | January 2018 | |
| MHCnuggets 2.0 | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & physicochemical features | NN | NM | 3-fold CV | August 2018 | |
| ConvMHC | 2017 | No | Yes | N.M. | No | No | No | Sequence-based & physicochemical features | NN | 75:1 | LOO, 5-fold CV, independent test | July 2018 | |
| HLA-CNN | 2017 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based features | NN | 7:3 | 5-fold CV, independent test | August 2017 | |
| MHCflurry 1.2.0 | 2018 | Yes | No | N.A. | N.A. | N.A. | N.A. | Sequence-based & binary features | NN | 9:1 | Independent test | January 2019 | |
| Consensus | IEDB-AR-Consensus | 2012 | Yes | Yes | ≤200 sequences or ≤10 MB | Yes | No | Yes | Con | NM | Independent data set | May 2012 | |
| NetMHCcons 1.1 | 2012 | Yes | Yes | ≤5000 sequences and ≤20 000 amino acids | Yes | Yes | Yes | Con | NM | Independent data set | January 2017 |
aThe URL addresses for accessing the listed tools are as follows: SYFPEITHI, http://www.syfpeithi.de/index.html/; RANKPEP, http://imed.med.ucm.es/Tools/rankpep.html/; PickPocket 1.1, http://www.cbs.dtu.dk/services/PickPocket/; SMMPMBEC, https://github.com/ykimbiology/smmpmbec/; PSSMHCpan 1.0, https://github.com/BGI2016/PSSMHCpan/; MixMHCpred 2.0.1, https://github.com/GfellerLab/MixMHCpred/; NetMHC4.0, http://www.cbs.dtu.dk/services/NetMHC/; NetMHCstabpan 1.0, http://www.cbs.dtu.dk/services/NetMHCstabpan/; NetMHCPan-4.0, http://www.cbs.dtu.dk/services/NetMHCpan/; MHCnuggets 2.0, https://github.com/KarchinLab/mhcnuggets-2.0/; ConvMHC, http://jumong.kaist.ac.kr:8080/convmhc/; HLA-CNN, https://github.com/uci-cbcl/HLA-bind/; MHCflurry 1.2.0, https://github.com/openvax/mhcflurry/; IEDB-AR-Consensus, http://tools.iedb.org/mhci/; NetMHCcons-1.1, http://www.cbs.dtu.dk/services/NetMHCcons/.
bYes: the publication has developed the function according to the column; No: the publication has not developed the function according to the column.
cAbbreviations: N.M., not mentioned; N.A., not applicable; PSSM, position-specific scoring matrix; NN, Neural network; Con, Consensus-based method; CV, cross-validation; LOO, leave-one-out.
Materials and methods
Construction of the positive validation data set
In order to evaluate the performance of currently available peptide-binding prediction tools and provide an overall comparison between them, we extracted the annotations of peptides including peptide sequences, source proteins where peptides were derived from, binding experimental results and the type of HLA molecules that the peptides bound to, from the latest versions of several widely used public databases including IEDB [57], SYFPEITHI [22], MHCBN [58] and EPIMHC [59]. Of note, SYFPEITHI, MHCBN and EPIMHC only store binary data (i.e. positive or negative) to distinguish whether a peptide has been experimentally verified to be a binder or not, while for some peptides in the IEDB database, quantitative measurements (e.g. binding affinity) have been recorded in addition to the binary result. To construct the positive data set, we collected all positive peptides from all the above four public databases, regardless of any quantitative information provided in IEDB. Next, we removed the sequence redundancy by adopting the following procedures: (i) only selecting confirmed allotype-specific peptides, (ii) removing duplicate peptides if they were associated with the same HLA allotype according to the databases, (iii) removing any peptides contained within the training data sets of the reviewed prediction tools and (iv) removing the peptides that had unnatural amino acids [26]. In addition, since most peptides presented by the HLA-I complexes are of 9–11 amino acids long [60], we only retained those peptides with such lengths in the constructed validation data set. Overall, we obtained an independent validation data set with a total of 21 101 non-redundant peptide ligands across 19 HLA allotypes. A statistical summary of the final constructed validation data set is provided in Table S1.
Construction of the negative validation data set
In order to generate a balanced data set with an equal number of non-binding peptides to those positive peptides associated with a certain HLA allotype, we used non-binding regions of the source proteins of the peptides in the positive data set. This strategy has been commonly used for previous performance benchmarking studies [28, 30, 61, 62]. Specifically, we first randomly selected the source proteins from the positive peptide data set. Then we generated a set of peptide sequences by splitting the sequences of the source proteins into 9, 10 or 11 amino acids-long peptides. Those peptides were further pooled according to their lengths and randomly selected to form the negative data set after filtering for those already contained in our independent positive data set or in the training data sets of the reviewed prediction tools. The random selection of negative peptides was performed in a way such that the numbers of negative peptides were identical to the numbers of positive peptides for each length (9, 10 and 11 mers) for each HLA-I allotype. It should be noted that this strategy might falsely classify peptides as non-binders since their binding potential was not formally assessed. However, as binding to these HLA allotypes are very specific and there would only exist a few allotype-specific HLA binders originating from one source protein, the proportion of misclassified peptides is likely very small [62, 63].
Existing peptide-binding prediction tools
Table 1 provides a summary of currently available tools for HLA-peptide-binding prediction, which are grouped into three major categories in terms of their availability, employed peptide features (for machine learning-based predictors), algorithms and performance evaluation strategies. Figure 1 provides an overall workflow for the three tool categories as an illustration of their underlying general methodologies for constructing peptide-binding predictors. Most tools constructed the prediction models in a five-step manner [64], which involves data collection and pre-processing as the 1st step, feature encoding and selection as the 2nd step, followed by model construction and optimization, performance evaluation and webserver/stand-alone software construction [65–69].
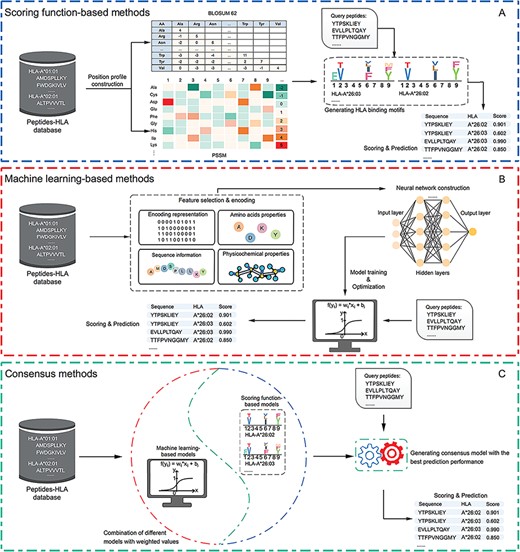
Graphical illustrations of (A) scoring function-based methods; (B) machine learning-based methods and (C) consensus-based methods. For each type of methods, the key steps are summarized and visualized. Scoring function-based methods predict peptide binding using a scoring function to generate the motifs of specific HLA alleles. Machine learning-based methods perform the prediction using well-trained models based on the training data sets. Consensus-based methods can predict peptide binding by integrating different peptide-binding prediction models.
Computational methods developed based on statistical scoring functions score the candidate peptide sequences by calculating certain features such as the sequence similarity and amino acid frequencies. Other statistical scores depend on the position-specific amino acid profiles of peptides. For instance, the position-specific scoring matrix (PSSM) and BLOSUM 62 matrix [70] are two widely used scoring matrices for specifying the evolutionary information of amino acids at different positions of a peptide sequence [71]. After the PSSM matrices are generated, the score of a peptide can be calculated by multiplying the frequencies of the corresponding amino acids at each position.
Machine learning-based methods classify a peptide as a binder or non-binder by generating a score using the training model based on the extracted representative features. Construction of a machine learning-based model for predicting a peptide’s binding probability generally involves four major steps: (i) construction of training data sets where the binding between the HLA allotype and peptide ligands have been experimentally verified, (ii) feature encoding based on the peptide and/or HLA allotype sequences, (iii) selection of a best-performing machine-learning algorithm and training of the corresponding machine-learning model and (iv) optimization of the model and its performance evaluation. As shown in Table 1, the neural network (NN) is the dominant machine-learning algorithm used by currently available peptide-binding prediction tools. Therefore, we use NN as an example to illustrate the workflow of how to construct machine learning-based tools in Figure 1.
As the 3rd major category of methods, consensus-based methods integrate several peptide-binding predictors in a weighted manner and generate the final prediction score based on the results of all individual predictors. The rationale of these methods is that combining the results of several individual predictors might help improve the performance of the final prediction compared to that of individual predictors [72]. Such consensus-based tools can be implemented based on a combination of similar binding prediction models, as is the case for NetMHCcons 1.1 [35], which will be discussed later.
Scoring function-based tools
The major difference between different scoring function-based tools is the statistical approach that they used to calculate the binding score for a candidate peptide sequence. From this perspective, SYFPEITHI [22] calculates the prediction score of a peptide sequence by adding the corresponding value of each amino acid at each position. For a given HLA allotype, amino acids that frequently occur at anchor positions are given the value of 10. The less frequent amino acids are assigned with lower values. The final score of a sequence is the sum of values at each position. As for the result, for a given HLA-I molecule and peptide length, SYFPEITHI calculates the 10 highest-scoring peptides among all possible amino acids with the same length of a given sequence.
RANKPEP [23] predicts the MHC class I-binding peptides using profile motifs by calculating the PSSM of ligands bound to a given HLA allotype. Briefly, the ligands of each HLA allotype are parsed by the length in five sets of 8, 9, 10, 11 and 12+ mers. The PSSM of each length set, as generated by using the PROFILEWEIGHT protocol [73], defines the sequence-weighted frequency of each amino acid observed at each position of the peptide. These values are then normalized to the corresponding expected background frequency of that amino acid in the proteome. The prediction score is calculated by aligning the PSSM with the peptides and adding up the scores that match the residue type and position. The scoring starts at the beginning of each sequence, and the PSSM is slid over the sequence one residue at a time until the end of the sequence. A binding threshold is set to a value. Thus, peptides with a score equal or above the binding threshold are predicted as binders.
PSSMHCpan 1.0 [26] is a recently published scoring function-based tool that also uses the PSSM features to predict peptide binding to HLA-I molecules. It can predict both characterized HLA-I allotypes (with binders in the training data set) and uncharacterized HLA-I allotypes (with no binders in the training data set). The PSSM of each characterized HLA-I allotype is defined as a matrix of M rows (M = 20, number of amino acid types) and N columns (N = 8–25, peptide sequence length). Each element |${\mathrm{p}}_{\mathrm{a},\mathrm{i}}$| in the matrix is calculated as |${\mathrm{p}}_{\mathrm{a},\mathrm{i}}=\log \frac{{\mathrm{F}}_{\mathrm{a},\mathrm{i}}+\omega}{{\mathrm{BG}}_{\mathrm{a}}}$|, where |${\mathrm{F}}_{\mathrm{a},\mathrm{i}}$| is the frequency of amino acid |$a$| at position |$i$| in a peptide from the training data set; |${\mathrm{BG}}_a$| is the background frequency of amino acid |$a$| in the proteome; and |$\omega$| is a random value ranging from 0 to 1 [77]. Then, the tool defined the binding score of a given peptide by summing the corresponding values of |${\mathrm{p}}_{\mathrm{a},\mathrm{i}}$| of the amino acid at each position in the PSSM of the corresponding characterized HLA allotype as follows: |$\mathrm{binding}\ \mathrm{score}=\frac{\sum_{\mathrm{i}=1}^{\mathrm{N}}{\mathrm{p}}_{\mathrm{a},\mathrm{i}}}{\mathrm{N}}$|, where N is the length of the peptide. Finally, PSSMHCpan 1.0 converts the binding score of each peptide to an |$\mathrm{IC}50$| value and uses this value as the prediction result for each peptide.
Machine learning-based tools
The limitation of scoring function-based approaches is that their methods for calculating the prediction scores are relatively simplistic, since they only handle linear features such as sequence similarity and pattern. In the last decade, machine learning-based algorithms have been increasingly used for constructing models to predict peptide binding to HLA allotypes. Such algorithms are capable of identifying non-linear patterns underlying the peptide-binding data [79], which cannot be easily captured by scoring function-based methods. Among various machine learning-based tools, the most commonly used algorithm is NN.
As shown in Table 1, most machine learning-based tools reviewed here utilized NN to construct the prediction models. Generally, the NNs in HLA-peptide-binding prediction models have a layered feed-forward architecture. Briefly, for a typical multi-layer feed-forward NN, the layers are composed of the input, hidden and output layers. Each layer can contain neurons or units to represent the signal. Different units of the layer can be connected to other units of the neighbouring layer through weights and biases. The signal of a unit |${x}_i$|can be transformed and used as the input of its connecting unit |${\mathrm{y}}_{\mathrm{j}}$|through the function |${\mathrm{y}}_{\mathrm{j}}={\mathrm{w}}_{\mathrm{i}\mathrm{j}}{\mathrm{x}}_{\mathrm{i}}+{\theta}_{\mathrm{j}}$|, where |${w}_{ij}$| is the weight value with respect to the units |${x}_i$| and |${y}_j$|, and |${\theta}_j$| is the bias of unit |${y}_j$|. Then, the input outcome of unit |${\mathrm{y}}_{\mathrm{j}}$| is transformed by an activation function like the sigmoid function |$\mathrm{f}({\mathrm{y}}_{\mathrm{j}})=\frac{1}{1+{\mathrm{e}}^{-{\mathrm{y}}_{\mathrm{j}}}}$|. Then, |$\mathrm{f}({\mathrm{y}}_{\mathrm{j}})$| is the output of unit |${\mathrm{y}}_{\mathrm{j}}$| and can be used by the next layer. For optimization of the parameters, back propagation (BP) is a widely used algorithm to optimize the |${\mathrm{w}}_{\mathrm{ij}}$| and |${\theta}_{\mathrm{j}}$|. The theory of the BP algorithm is based on the error-correction-learning rule. Briefly, the NN parameters are optimized by modifying the weights and biases via using error function according to the difference between the network output and the actual label [80].
In this section, we will discuss in detail the current strategies of constructing the architecture of NN for peptide-binding prediction.
NetMHC 4.0 [28] uses an ensemble method to generate the NN and assigns the binding core of a given peptide based on the majority vote of the networks in the ensemble. Briefly, the top 10 networks with the highest test set Pearson correlation coefficient within the 50 networks for each training/test set configuration were selected as the final network ensemble. It uses a BP algorithm to update the weights between units [81]. In particular, it uses both BLOSUM62 and sparse encoding schemes to encode the peptide sequences into nine amino acid-binding cores. For peptides longer or shorter than nine amino acids, deletion or insertion methods are applied to reconcile or extend the original sequence to a core of nine amino acids [28]. In addition, other complementary sequence-based features such as the number of deletions/insertions, the length and compositions of the terminal regions flanking the predicted binding core were also incorporated to enable the algorithm to learn the complex binding patterns from the peptide-HLA-I molecule pairs.
NetMHCstabpan 1.0 [29] is a prediction tool that predicts the stability of peptide-HLA-I complexes based on an NN-based algorithm. The stability of the peptide-HLA-I complex is defined as the half-life of the pHLA complex, which is determined by a scintillation proximity-based peptide-HLA-I dissociation assay [82]. Then, the half-life values are transformed to a value ranging between 0 and 1 as follows: |$\mathrm{s}={2}^{-\mathrm{t}0/\mathrm{th}}$|, where |$s$| is the transformed value, |$\mathrm{th}$| is the measured pHLA complex half-life and |$t0$| is a threshold value that is fitted to obtain a suitable distribution of the data for training purposes, which was set as 1 after optimization of the prediction performance. The tool uses BLOSUM50 or the sparse encoding scheme to encode peptide sequences.
NetMHCpan 4.0 [30] is also an NN-based tool similar to NetMHC 4.0. The major differences between the two tools include two aspects: first, NetMHCpan 4.0 was trained using peptides generated from both binding affinity assays and naturally presented peptide ligands identified by MS; second, the amino acids from the HLA heavy chain that contact the peptide ligand are extracted. These sequences are called pseudo-sequences and enable the tool to predict binding to HLA allotypes with little available binding data. The assumption is that similar HLA allotypes will bind similar peptides. In NetMHCpan 4.0, the similarity between two HLA molecules is defined as the pseudo-distance |$\mathrm{d}=1-\frac{\mathrm{s}(\mathrm{A},\mathrm{B})}{\sqrt{\mathrm{s}(\mathrm{A},\mathrm{A})\times \mathrm{s}(\mathrm{B},\mathrm{B})}}$|, where |$s(A,B)$| is the BLOSUM50 similarity score between pseudo-sequences A and B. The algorithm takes the peptides and the chosen HLA allotype in terms of a pseudo-sequence as inputs. All peptides are represented as 9-mer binding cores by using the same method described in NetMHC 4.0 [28].
MHCnuggets 2.0 [32] is a prediction tool developed based on deep learning (DL) methods. It contains two DL models: (i) long short-term memory networks [83] and (ii) gated recurrent units [84]. The architecture of both networks is a fully connected single layer of 64 hidden units. The network is regularized with a dropout and recurrent dropout probability is 0.2, which means during each time of optimization, the NN algorithm will randomly choose to ignore 20% of units to avoid overfitting. The input sequence is encoded as a 21-dimensional vector using a sparse encoding scheme. Both models were trained using the Adam optimizer [85] with a learning rate of 0.001. Instead of using pseudo-sequences as in NetMHCpan 4.0, MHCnuggets 2.0 designed a transfer-learning protocol through an empirical, bottom-up approach to regenerate weights between two similar HLA allotypes. This protocol has shown to improve the prediction performance for most alleles predicted by the tool.
ConvMHC [33] is a machine learning-based tool, which uses a deep convolutional NN (DCNN) for pan-specific peptide-MHC class I-binding prediction. The algorithm generates a ‘pixel’-like matrix that encodes the residues of a peptide as the height (H) and the sequence of the binding area of a corresponding HLA molecule as the width (W). In this image-like array (ILA) data, each contact area between a peptide and an HLA molecule is defined as a ‘pixel’. In addition, a C channel vector that contains the value of physicochemical properties of the amino acid interaction pair in the ‘pixel’ is also included in this ILA data. ConvMHC selects nine physicochemical properties and converts them into scores assigned to corresponding amino acids. The size of the C channel is 18 as each pair of ‘pixels’ has 2 amino acids. ConvMHC uses 34 HLA-I contact residues provided by NetMHCpan [86] and is trained on 9-mer peptides. Thus, the size of ILA is 34 (W) |$\times$| 9 (H) |$\times$| 18 (channels). The algorithm of ConvMHC is based on the DCNN architecture described in [87] and uses the dropout [88] as the regularization method. The network uses ReLU [89] as an activation function to transfer the non-linear output of each layer and the Adam optimizer with a learning rate of 0.001 for optimization.
Consensus methods
The idea of consensus methods is that prediction performance can be further improved by integrating the outputs from several individual tools based on a weighted scheme. Several benchmarking studies have shown that an improved prediction performance can be achieved by consensus methods that average the prediction scores from multiple individual predictors [24, 54, 92]. IEDB-AR-Consensus [25] is such a method, whose results are based on prediction outcomes from three sources: (i) NetMHC 4.0; (ii) SMM [75] and (iii) CombLib [93]. IEDB-AR-Consensus is recommended by the IEDB peptide-binding prediction platform [25]. The platform includes several prediction tools, for which the query peptide and the HLA allotype exist in training data sets of the consensus method.
Webserver/software functionality
All peptide-HLA-binding prediction tools reviewed here are either accessible via an online web server and/or are available for download as local stand-alone software. This enables researchers to conduct the prediction in an easy and productive manner. In this section, the general functionalities of the currently available tools are discussed.
For prediction tools with accessible web servers, users normally need to submit the peptide sequence(s) of interest and specify the HLA allotype for which binding is to be predicted. RANKPEP, Pickpocket 1.1, SMMPMBEC, NetMHC 4.0, NetMHCpan 4.0, ConvMHC, IEDB-AR-Consensus, NetMHCcons 1.1 and NetMHCstabpan 1.0 allow users to upload a file with multiple sequences in the FASTA format or submit sequences in the FASTA/PEPTIDE format directly. However, SYFPEITHI can only accept a single sequence at each run of prediction. Note that for all tools that can predict multiple sequences at a time, the maximum number of sequences is still limited (i.e. allowing either |$\le$|200 sequences or |$\le$|10 MB of the uploaded file for SMMPMBEC and IEDB-AR-Consensus, |$\le$|5000 sequences and |$\le$|20 000 amino acids of each sequence for other tools that have mentioned the data size). In addition to being implemented as a web server, Pickpocket 1.1, SMMPMBEC, NetMHC 4.0, NetMHC 4.0, IEDB-AR-Consensus, NetMHCcons 1.1 and NetMHCstabpan 1.0 have also been made available as stand-alone software for download from their websites.
For a tool that has been implemented as an online web server, a well-designed user-friendly interface can undoubtedly enhance the efficiency during operation and save unnecessary time otherwise consumed in familiarizing with the tool. To this point, detailed instructions of step-by-step operations have been provided for all tools with webserver, with an exception of SYFPEITHI and ConvMHC. In addition, interpretable outputs can also be found in well-marked places at the web servers of Pickpocket 1.1, SMMPMBEC, NetMHC 4.0, NetMHCpan 4.0, IEDB-AR-Consensus, NetMHCcons 1.1 and NetMHCstabpan 1.0, which has further improved the interpretation of the generated results in terms of the detailed explanation of outcomes. Moreover, tools like RANKPEP, Pickpocket 1.1, NetMHC 4.0, NetMHCpan 4.0, NetMHCcons 1.1 and NetMHCstabpan 1.0 also allow for specific parameters, including the prediction thresholds and weights, to be user-defined or to be set to default values. Also, except for RANKPEP, optional email notifications of accomplished predictions with job IDs can be selected in those tools mentioned above as well as in SMMPEBMEC and IEDB-AR-Consensus, which facilitates a future revisit of results. Moreover, users can also choose to download the results directly from these tools for a further in-depth analysis off-line.
Performance evaluation strategy
Performance evaluation strategies including k-fold cross-validation (CV) and independent tests are widely used for performance evaluation of peptide-binding prediction tools and for further optimization. For k-fold CV, the data set of the tool is divided into k partitions. The validation procedure will be performed k times. Each time one of the k partitions is selected as a validation data set, while the remaining (i.e. k|$-1$|) partitions are used to train the model. The prediction performance of the trained model then would be evaluated using the validation data set. The final performance is calculated as the average performance over those k individual performances. The leave-one-out (LOO) CV is called when k equals the total number of data entries. This method is much more thorough compared with k-fold CV. The data set is first divided into N parts, where N is the number of entries in the entire data set. The training process is carried out N times, and each time one entry will be validated based on the model that has been trained on the remaining (|$N\hbox{--} 1$|) entries. The overall performance is the average over all N training processes. Normally, the CV method is used to test the internal performance and to avoid the overfitting of the model. Independent test is another popular strategy used for performance evaluation of the tools. Here, the independent test data, which is non-overlapping with the training data of the tools, is collected and becomes an independent test data set. This independent test data set is used as a uniform validation data source to test the performance of different tools. Therefore, compared to the CV method, the performance of different tools evaluated on the same independent data test are comparatively more objective, indicative of the tools’ generalization ability and can be compared mutually.
As shown in Table 1, except for SYFPEITHI for which there was no information about the evaluation methods used, all remaining tools reviewed here were evaluated by performing CV and/or independent tests. Tools such as PSSMHCpan 1.0, NetMHCpan 4.0, ConvMHC, HLA-CNN and NetMHCstabpan 1.0 used both methods for performance evaluation. Tools that were evaluated using CV only included Pickpocket 1.1, SMMPMBEC, NetMHC 4.0 and MHCnuggets 2.0, while RANKPEP, MixMHCpred 2.0.1, MHCflurry 1.2.0, IEDB-AR-Consensus and NetMHCcons 1.1 were evaluated using the independent test only in their original studies. Here, to allow a fair comparison, the performance benchmarking of all these tools is conducted using a curated up-to-date independent test data set aforementioned.
In addition, in this study, the receiver operating characteristic (ROC) curves were also used to visualize the performance of different methods, with the area under the curve (AUC) calculated to quantify their performance.
Results and discussion
Conservation analysis of sequence motifs of the HLA-I ligands
The binding motif of HLA-I allotypes reflects sequence characteristics of peptide ligands that facilitate binding to the antigen-binding cleft of specific HLA-I allotypes. HLA-I allotype-specific scoring matrices can be established based on these conserved peptide-binding motifs. We analysed the positional preferences of amino acids for allotype-specific peptide ligands across our curated validation data set. For each HLA allotype we generated consensus-binding motifs using the pLogo program [98], which can be visualized in Figure 2 and Figure S1. Irrespective of the ligand length, peptides derived from the same HLA-I allotype show the consensus-binding motifs. For instance, the peptides binding to HLA-A*02:04 and HLA-A*24:06 prefer Leu and Tyr at position 2 (‘P2’), as well as Leu/Val or Phe at their C-termini (Figure 2D–F; Figure S2A, B, C). In addition, it can be seen that preferential amino acid patterns exist in the peptides binding to closely related alleles, for example for HLA-B*27:x (x = 01, 07, 08 and 09), where Arg is often required at P2 (Figure 2G–I; Figure S2G–O). However, a closer look revealed that HLA-B*27:01 also preferred to have Arg at the P1 position, while other allotypes did not have this requirement. Moreover, the C-termini of ligands binding to HLA-B*27:01 were relatively diverse compared to HLA-B*27:x (x = 07, 08, 09) that had a strong preference for Leu and Phe at this position.

Position and residue specificity of four HLA-I alleles, including (A, B, C) HLA-A*02:01 (9, 10 and 11 mers), (D, E, F) HLA-A*02:04 (9, 10 and 11 mers), (G, H, I) HLA-B*27:01 (9, 10 and 11 mers) and (J, K, L) HLA-C*02:02 (9, 10 and 11 mers).
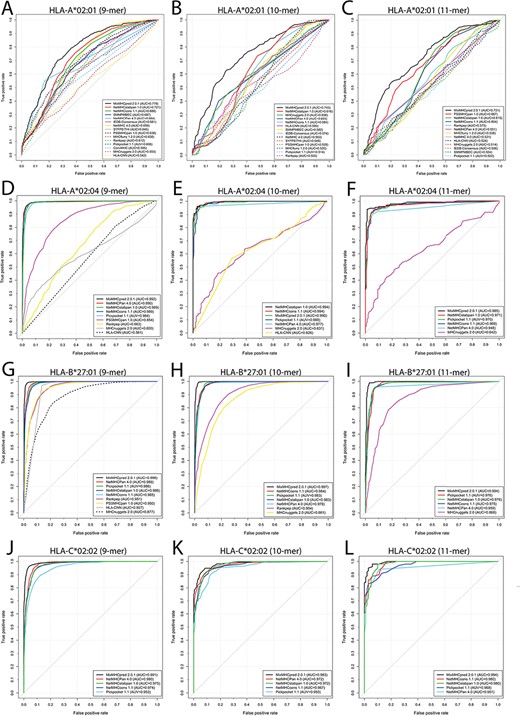
ROC curves and the corresponding AUC values of the reviewed predictors for peptides with lengths of 9, 10 and 11, binding to HLA-I molecules specific for (A, B, C) HLA-A*02:01 (9, 10 and 11 mers), (D, E, F) HLA-A*02:04 (9, 10 and 11mer), (G, H, I) HLA-B*27:01 (9, 10 and 11 mers) and (J, K, L) HLA-C*02:02 (9, 10 and 11 mers).
Performance evaluation of different tools for peptide-binding HLA-I prediction
The performance of different tools was assessed in terms of five commonly used metrics, namely AUC, Sn, Sp, Acc and MCC using the validation data set as an input. It should be noted that the training data sets of some reviewed tools are not available, while several other tools have been updated with an expanded training data set since their first release. Therefore, there might be some overlap between the data sets used for developing some tools and our validation data set. Whenever possible, we downloaded the training data sets of these tools and removed the overlapping entries from our validation data set. Then we submitted the 9-, 10- and 11-mer peptide sequences specific for each HLA-I allotype in the validation data set to the tools. For this evaluation, the tools’ parameters were set to the recommended configurations in the corresponding publications or to the default values if no recommendations were given. To illustrate the prediction performance of each tool, the ROC curves with the calculated AUC values were plotted and shown in Figure 3, Figure S2 and Table S2. Moreover, the performance results evaluated in terms of Sn, Sp, Acc and MCC for each tool are given in Table S2. Of note, the evaluation of prediction performance is dependent on the composition of the training data the reviewed predictors have provided.
MixMHCpred 2.0.1 achieved the best performance among all scoring function-based tools as it achieved the highest AUC values among nearly all 19 allotypes, while NetNHCpan 4.0 performed best among all machine learning-based tools, and NetMHCcons 1.0 achieved a better performance than IEDB-AR-Consensus in the consensus category. While no tool universally achieved the best performance for all HLA-I allotypes in the independent test data set, MixMHCpred 2.0.1 performed best for most HLA-I allotypes examined. We speculate that one reason for MixMHCpred 2.0.1 achieving the best performance is that it is a recently published tool trained with both public HLA-peptide data sources from 40 cell lines and also in-house data from immunoaffinity purification experiments involving 10 additional cell lines. In addition, MixMHCpred 2.0.1 applied both fully unsupervised and semi-supervised machine-learning strategies to identify a total of 52 HLA-I allomorph-specific binding motifs.
Among the machine learning-based tools, NetMHCpan 4.0 achieved the largest AUC values considering all HLA-I allotypes. This is possibly because NetMHCpan 4.0 represents the latest version of NetMHCpan series, which were trained using both experimental affinity measurements and MS-identified peptide ligands. Moreover, NetMHCpan 4.0 uses the pseudo-sequences of HLA-binding pockets to calculate the similarities in ligand binding between different HLA allotypes. Therefore, for HLA allotypes with little binding data NetMHCpan 4.0 is able to achieve a high-prediction performance compared to other machine learning-based tools, e.g. HLA-A*24:06 (9 mers) (AUC = 0.989; Figure S2A), HLA-A*24:13 (9 mers) (AUC = 0.974; Figure S2B), HLA-B*35:08 (9 mers) (AUC = 0.909; Figure S2L) and HLA-C*03:04 (10 mers) (AUC = 0.977; Figure S2T). As a comparison, other tools only obtained high AUC values when predicting peptide binding to well-studied HLA-I allotypes. For instance, MHCnuggets 2.0 achieved an AUC value of 0.877, 0.955, 0.942, 0.985 and 0.985 for HLA-B*27:01 (9 mers), -B*27:09 (10 mers), -B*27:09 (11 mers), -B*56:01(9 mers) and -C*03:04 (9 mers), respectively (Figure 3G; Figure S2J, K, O, S).
As for the consensus methods, NetMHCcons 1.1 achieved better AUC values compared with IEDB-AR-Consensus. The NetMHCcons 1.1 used different types of combinations of peptide-binding prediction tools according to the query pair of peptide and HLA-I allotype as discussed in the section of consensus methods. This might be the primary reason the NetMHCcons 1.1 achieved the best prediction performance. For instance, NetMHCcons 1.1 also utilizes pseudo-sequences of HLA-binding pockets used in NetMHCpan 4.0 to predict binding to HLA allotypes with little binding data, e.g. HLA-A*24:06 (9 mers) (AUC = 0.986; Figure S2A), HLA-A*24:13 (9 mers) (AUC = 0.971; Figure S2B), HLA-B*35:08 (9 mers) (AUC = 0.884; Figure S2L) and HLA-C*03:04 (10 mers) (AUC = 0.989; Figure S2T).
Prospective strategies for improving the prediction performance of antigenic peptides and developing next-generation bioinformatics tools
Based on our independent evaluation of prediction performance, we foresee that peptide-binding prediction will continue to be improved as the volume and quality of training data increases (such as observed for MixMHCPred 2.0.1). Similarly, machine-learning algorithms (such as NetMHCpan 4.0) also benefit from improved training data sets. Such ligand prediction tools have already facilitated the discovery of new epitopes in many diseases and cancers [99–102]. However, only a very small proportion of the predicted binders are naturally targeted by T cells (i.e. T-cell epitopes) [3]. In this context, we also used the IEDB T-cell assay data set, which contains experimentally verified immunogenic and non-immunogenic peptides, to evaluate the performance of the prediction tools reviewed here. As it turned out, independent of the predictor, those peptides that were predicted to be binders are mostly experimentally verified non-immunogenic according to IEDB (for peptides that are verified as non-immunogenic peptides and predicted as binders, more than 95% of are pathogen-derived peptides, data not shown). This result suggests that the prediction of immunogenicity cannot solely rely on the binding affinity between HLA allotypes and peptides. The immunogenicity of a peptide is associated with various properties. These include the available T-cell receptor repertoire, ability of the peptide to be effectively processed and liberated from the parental antigen, the duration and context of presentation and binding to HLA allotypes and so on [4, 103]. A recent study shows that more than 80% of MHC class I-bound peptides derived from virus can be immunogenic in virus-infected mouse. The study demonstrated that the major CD8+ T-cell responses are strongly associated with high affinity of peptide to MHC class I molecule. Besides that, the study also showed evidence and pointed out that the peptide abundance and the time of gene expression may play roles in the CD8+ T-cell immunity [104]. This finding suggests that most of presented viral MHC class I peptides may be immunogenic, but the T-cell response may be dominated by a few peptides. Thus, how to design a prediction method that can predict dominant T-cell epitopes is still a challenge. We believe that integrating peptide properties related to immunogenicity into an algorithm using cutting-edge machine-learning techniques is a promising direction for improving the prediction of immunogenicity. To this end, we provide several insights for further discussion and future directions.
Firstly, to identify potential epitopes efficiently, peptides that have been experimentally annotated as antigenic, i.e. being able to induce a T-cell response and peptides that are clearly presented yet fail to elicit a response, as high affinity of peptides is reported to be strongly associated with T-cell responses [104], are the ideal data source for extracting informative features and constructing accurate prediction models. Various features such as physicochemical properties of peptides, the stability of pHLAs complex, antigen processing and the structural contact between pHLAs and TCRs have been shown to influence the immunogenicity of peptides [48, 105–107]. More accurate epitope prediction may be achieved by considering these related features along with peptide binding. In addition to proteomic information, the discovery of immunogenic peptides may also be facilitated using genomic data generated from NGS techniques, which might provide potentially useful information such as identifying somatic mutations within tumour cells to aid the identification of neo-antigens for personalized cancer immunotherapy [108–110].
Secondly, the use of DL is also a promising direction in improving the performance of immunogenic peptide prediction. Compared to conventional machine-learning algorithms, DL-based methods can introduce innovative network architectures and regularization techniques, which allow the functions of the algorithms to be trained to simulate the complexity of peptide binding and T-cell recognition, while avoiding common issues faced by machine-learning methods such as overfitting and slow convergence [111, 112]. Despite the time-consuming model training process, DL-based methods have been shown to outperform other methods in a number of different research areas given sufficient training data [32, 111]. In addition to that, DL methods are particularly suitable for undertaking high-throughput prediction tasks, particularly because they do not need to involve and use feature encodings to represent the original data samples. Thus, DL methods are attractive and promising for analysing the rapidly increasing amount of available data generated by advanced high-throughput techniques.
With the rapid development in the field of machine learning, several novel algorithms have been proposed that exhibit promising prediction performance and are attractive for researchers. For instance, a novel incremental decision tree-learning algorithm, the Hoeffding Anytime Tree, has been recently developed. It is based on the conventional Hoeffding Tree [113] but with a minor modification that allows it to achieve a significantly improved predictive performance on most of the largest classification data sets [114]. Moreover, algorithms like reinforcement learning can improve the prediction accuracy further and autonomously by receiving reward or punishment according to the model performance [115, 116]. This property is also attractive for immunogenicity predictions because the results of experimental tests can be fed back to improve the efficiency and accuracy of the model.
Conclusion
Once liberated from the parental protein, the selection and binding of peptide antigens to available HLA allotypes is the first critical step in influencing the immune response and ultimately the survival of the host. For this reason, tremendous efforts and resources have been applied to accurately predict which pathogen-derived or neo-antigen-derived peptides are selected for antigen presentation. Available tools now do quite a respectable job for predicting a peptide’s ability to bind to a given HLA allotype, even in the absence of training data sets for a specific HLA allomorph. The development of these prediction tools enables immunologists to narrow down the search space of antigen candidates that need to be experimentally validated. However, the integration of other host-specific information will be essential to predict which of the peptides present on the cells surface will be targeted for T-cell responses. In addition, advanced machine-learning algorithms like DL are ideal for processing such extremely large information from the host and generating key features for predicting immunogenicity. Therefore, the combination of skills from immunologists and bioinformaticians will assist the development of immunogenicity prediction in a faster and more efficient way. In this review and survey work, we have introduced and comprehensively evaluated the currently available tools for the prediction of peptides binding to different HLA-I molecules. Additionally, we have discussed, reviewed and assessed all methods in terms of their calculation methods of the prediction score, prediction algorithm(s), functionality and performance evaluation strategy. To obtain a more objective performance evaluation, we constructed an independent test data set to benchmark all tools. Based on the assessment results, MixMHCpred 2.0.1 achieved the highest prediction performance across most of the HLA-I molecules and is the best predictor among scoring function-based tools. Apart from that, NetMHCpan 4.0 and NetMHCcons 1.1 generally achieved the best performance results in the machine learning-based and consensus-based tools, respectively. This study provides useful guidance to researchers who are interested in developing an antigen prediction model in future studies. Additionally, feedback of data on immunogenicity into such models will improve our understanding of the antigen-processing pathways and subsequent T-cell recognition patterns. Finally, we hope that more accurately predicted peptide binding will assist with the development of immunotherapy and vaccine design.
We conducted a comprehensive review and assessment of 15 currently available tools for predicting human leukocyte antigen (HLA) class I (HLA-I)-binding peptides, including 6 scoring function-based, 7 machine learning-based and 2 consensus methods.
This review and survey systematically analysed these tools with respect to the computational methods of the prediction score, employed algorithms, performance evaluation strategies and software functionality.
All tools underwent a comprehensive performance assessment based on 19 different HLA-I allotypes and up-to-date independent data set
sof experimentally verified HLA-I allotype-specific ligands.Extensive benchmarking tests show that MixMHCpred 2.0.1 performs best across most of HLA-I allotypes included in the validation data sets, while NetMHCpan 4.0 and NetMHCcons 1.1 achieve the overall best performance among machine learning-based and consensus-based tools, respectively.
This study provides a comprehensive analysis and benchmarking of currently available bioinformatics tools for HLA-I peptide-binding prediction and gives directions to the wider research community for developing the next generation of peptide-binding prediction tools.
Funding
This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) (APP1085018, APP1127948, APP1144652 and APP1084283), the Australian Research Council (ARC) (DP120104460, DP150104503) and the Collaborative Research Program of Institute for Chemical Research, Kyoto University (2018-28). J.L., A.W.P. and J.N.S. are supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965). J.R. is an ARC Australia Laureate fellow. J.L. and A.W.P are NHMRC principal research fellows. T.T.M.L. and A.L.’s work was supported in part by the Informatics Institute of the School of Medicine at UAB.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Shutao Mei is currently a PhD candidate in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Australia. His research interests are bioinformatics, machine learning, immunopeptidomics and immuno-informatics.
Fuyi Li received his BEng and MEng degrees in Software Engineering from Northwest A&F University, China. He is currently a PhD candidate in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
André Leier is an assistant professor and group leader in the Department of Genetics, School of Medicine, University of Alabama at Birmingham (UAB), USA. He is also an associate scientist in the UAB Comprehensive Cancer Centre. He received his PhD in Computer Science, University of Dortmund, Germany. He conducted postdoctoral research at Memorial University of Newfoundland, Canada, The University of Queensland, Australia and ETH Zürich, Switzerland. His research interests are in biomedical informatics and computational and systems biomedicine.
Tatiana T. Marquez-Lago is an associate professor and group leader in the Department of Genetics and Department of Cell, Developmental and Integrative Biology, School of Medicine, UAB, USA. Her research interests include multiscale modelling and simulations, artificial intelligence, bioengineering and systems biomedicine. Her interdisciplinary lab studies stochastic gene expression, chromatin organization, antibiotic resistance in bacteria and host–microbiota interactions in complex diseases.
Kailin Giam is a postdoctoral research associate at King’s College London, United Kingdom. She received her PhD from The University of Melbourne, Australia. Her research focuses on understanding the immunological aspects of juvenile onset diabetes (type 1 diabetes) using cross-disciplinary strategies.
Nathan P. Croft received his PhD from the University of Birmingham and is currently a postdoctoral research fellow at Monash University, Australia. He focuses on identifying and quantifying the peptides displayed by major histocompatibility complex (MHC) molecules on the surface of infected cells for scrutiny by CD8+ T cells. He is also interested in the kinetic profiling of virus and host proteins that are modulated upon infection.
Tatsuya Akutsu received his DEng degree in Information Engineering in 1989 from University of Tokyo, Japan. Since 2001, he has been a professor in the Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
A. Ian Smith completed his PhD at Prince Henry’s Institute, Melbourne and Monash University, Australia. He is currently the vice-provost (Research & Research Infrastructure) at Monash University. He is also a professorial fellow in the Department of Biochemistry and Molecular Biology at Monash University, where he runs his research group. His research applies proteomic technologies to study the proteases involved in the generation and metabolism of peptide regulators involved in both brain and cardiovascular function.
Jian Li is a professor and group leader in the Monash Biomedicine Discovery Institute and Department of Microbiology, Monash University, Australia. He is a Web of Science 2015–2017 Highly Cited Researcher in Pharmacology & Toxicology. He is currently a National Health and Medical Research Council of Australia (NHMRC) principal research fellow. His research interests include the pharmacology of polymyxins and the discovery of novel, safer polymyxins.
Jamie Rossjohn is a professor and group leader in the Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Australia. He is currently an Australian Research Council (ARC) Australian Laureate fellow. His research is focused on understanding the structural and biophysical basis of MHC restriction, T-cell receptor (TCR) engagement and the structural correlates of T-cell signaling.
Anthony W. Purcell is a professor and group leader in the Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Australia. He is currently an NHMRC principal research fellow. He specializes in targeted and global quantitative proteomics of complex biological samples, with a specific focus on identifying targets of the immune response and host–pathogen interactions.
Jiangning Song is an associate professor and group leader in the Monash Biomedicine Discovery Institute and Biochemistry and Molecular Biology, Monash University, Australia. He is a member of the Monash Centre for Data Science and also an associate investigator of the ARC Centre of Excellence in Advanced Molecular Imaging, Monash University. His research interests include artificial intelligence, bioinformatics, machine learning, big data analytics and pattern recognition.

{kind=link}
{kind=link}
{kind=link}