





Abstract
Polymer materials play a pivotal role in shaping modern society. The continuous development of polymer materials can be largely attributed to innovations in polymerization methods, especially in the development of new catalysts. When pursuing high-performance catalysts, it is crucial to consider their cost and safety for practical applications. Alkali metal carboxylates (AMCs) are widely available simple chemicals, and some of them are used as food additives. In the past few years, our group has demonstrated the ability of such simple compounds to catalyze the ring-opening polymerization of cyclic esters, cyclic carbonates, epoxides, and episulfides. Additionally, AMCs have demonstrated their efficacy in facilitating the ring-opening alternating copolymerization of cyclic anhydrides and cyclic ethers, resulting in the production of polyesters in a well-controlled manner. Moreover, AMCs can also catalyze the complicated self-switchable polymerization of a mixture of at least three monomers, yielding block copolymers with well-controlled block components and architectures. This account summarizes these successful examples, offering mechanistic insights and an outlook.
1. Introduction
Catalysis is the core of chemical and material production.1 Therefore, highly efficient green catalytic processes are required for sustainable human development.2,3 Synthetic polymers, an important type of material that supports modern human life, have been continuously renovated through the development of new synthetic methods, especially new catalysts.4
Metal-complex catalysts and enzymes have represented two distinct categories of catalysts, for quite a long time. Since around 2000, there has been a notable emergence and rapid development of small molecule organocatalysts, commonly called “the third” catalysts, for organic transformations.5,6 The “gold rush” of organocatalysis in organic chemistry quickly spread into the field of polymer chemistry. Since the seminal ring-opening polymerization (ROP) of lactides by 4-(dimethylamino)pyridine (DMAP) was reported by Hedrick et al. in 2001,7 tremendous efforts have been devoted to organocatalyzed polymerization. As shown in Scheme 1, numerous catalysts, including nucleophilic,8 basic,9,10 acidic,11 and multifunctional12 catalysts, have been developed and demonstrated to efficiently catalyze controlled polymerizations over a wide spectrum of monomers.13–16 Using the ROP of cyclic esters as an example, the polymerization mechanism using the above mentioned different types of organocatalysts can be drawn as Scheme 1a–e.
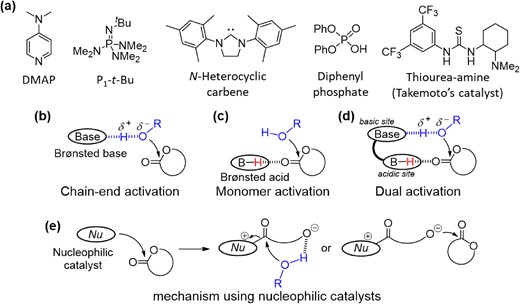
(a) Representative organocatalysts for controlled polymerization; polymerization mechanism of ROP of cyclic esters when using different types of organocatalysts: (b) chain-end activation using Brønsted base catalysts; (c) monomer activation using Brønsted acid catalysts; (d) dual activation using bifunctional catalysts; (e) mechanism using nucleophilic catalysts.
After the initial exploratory investigations into novel organocatalyzed polymerization systems, the focus gradually shifted towards industry-relevant factors such as cost-effectiveness, ease of handling, thermal stability, and low toxicity.17 Alkali metal carboxylates (AMCs), such as sodium acetate and sodium sorbate, are simple and widely available chemicals that find extensive application as food additives (Scheme 2a). These features meet industrial requirements. In terms of catalytic reactivity, careful selection of carboxylate moieties and alkali metal cations can endow a relatively broad spectrum of acidic/basic characteristics, which can adapt to different monomers types (Scheme 2b). With these considerations in mind, we successfully demonstrated that AMCs can function as good catalysts for the ROP of cyclic esters, cyclic carbonates, epoxides and episulfides, as well as the ring-opening alternating copolymerization (ROAC) of cyclic anhydrides and cyclic ethers. Moreover, we have also successfully developed self-switchable polymerization using a mixture of at least three monomers to yield block copolymers with well-controlled block components and architectures with AMC catalysts (Scheme 2c). This account provides a summary of these successful examples, predominantly from our group, along with our perspective on the subject.
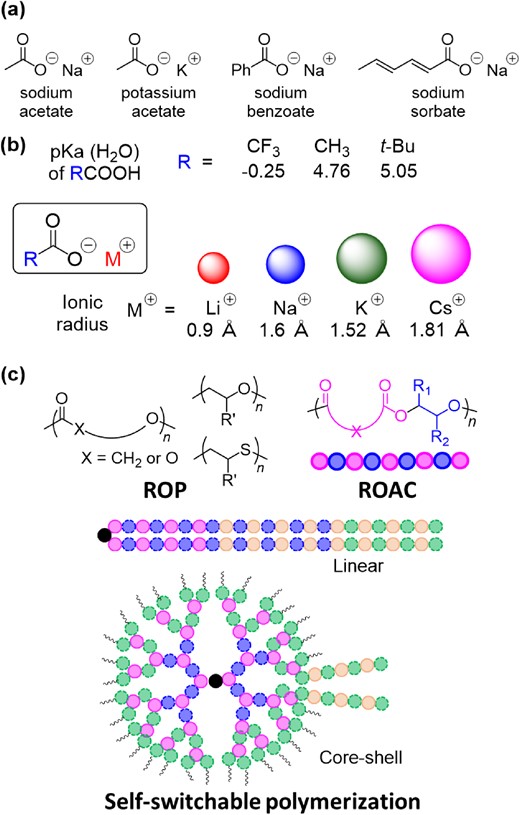
(a) FDA-approved AMC food additives; (b) general information regarding AMCs; (c) summary of polymers synthesized using AMC catalysts.
2. AMC-Catalyzed ROP
2.1 ROP of Cyclic Esters and Cyclic Carbonates
ROP of cyclic esters is an effective approach for the controlled synthesis of biodegradable and biocompatible aliphatic polyesters.18 In previous studies, AMCs were reported to initiate the ROP of four-membered β-butyrolactones (BLs), leading to the synthesis of poly(3-hydroxybutyrate).19,20 The proposed mechanism involves SN2 nucleophilic substitution at the sp3 carbon next to the ester oxygen atom, resulting in a carboxylic acid chain end in poly(3-hydroxybutyrate).
In 2018, our group first reported that AMCs could catalyze the ROP of cyclic esters with alcohols as initiators in an effective and controlled manner;21 this polymerization mechanism was different from that in previous reports on the ROP of BL. Polymerization was conducted in bulk at elevated temperatures, and l-lactide (LLA) was selected as the model monomer for detailed study. To confirm that the AMCs functioned as catalysts instead of as initiators, the polymer products were analyzed by nuclear magnetic resonance spectroscopy (NMR) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). This clearly shows the presence of alcohol instead of carboxylate in the polymer product. Furthermore, the polymer chain ends were identified as alcohols rather than carboxylic acids. Based on this experimental evidence, a dual-activation mechanism was proposed for the AMC-catalyzed ROP of cyclic esters, as shown in Scheme 3. The alkali metal cation interacts with the carbonyl oxygen of the cyclic ester to increase the electrophilicity of the carbonyl group, meanwhile the carboxylate forms a hydrogen bond with the hydroxy group of the initiator or polymer chain end and increases its nucleophilicity. By activating the cyclic ester monomer and hydroxy chain end maintaining them in proximity, AMCs effectively catalyze the ROP of cyclic esters.
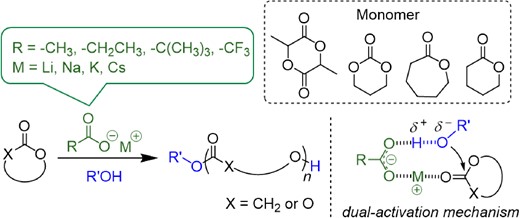
AMC-catalyzed ROP of cyclic esters.21
During the monitoring of the ROP of LLA, a linear increase in the molecular weight of the polymers was observed with an increase in monomer conversion. The dispersity remained low throughout the polymerization process (Ð < 1.2). First-order polymerization kinetics for the monomer concentration were also observed, which indicated that one molecule of the monomer was involved in the rate-determining step, and the catalytic activity of the AMC catalysts remained constant during the process (Figure 1).
![(a) Dependence of Mn,NMR (●), Ð (□), and Mn,th. (Dotted line) on the monomer conversion. (b) First-order kinetic plot for the ROP of LLA at the [LLA]0/[PPA]0/[CH3COONa]0 ratio of 50/1/0.5 at 100 °C in the bulk 3-phenyl-1-propanol (PPA). Reprinted with permission from ref 21. Copyright 2018 American Chemical Society.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bcsj/96/9/10.1246_bcsj.20230149/2/m_20230149fig01.jpeg?Expires=1747923207&Signature=Sh9r3uWKSHiUsYzLOoiS0Y-F8aDEyPiobYnZ-54YpznUTZ41pv0q0NjIg4yriuwYnDkILWXcgkb903ZU93feKlFtARBgFgSWkurREBkYYyDYDdtLGf4mQjPYPU~Kp-c06C87MvWYJQj27dZvFZxigq4b1E41THM~abNVeQU7shBsJLPmWzHweFWHIg7BehYVPuoZ8M4fUDrzDiI-H7s3RJZ-SF~PLolS3XEZNpbVmyKEs-yPJHm6KdEy-EOohQLowJmlOhf9WusT7K33033fUZa8A~TZ6NZa73InWO6q23obQOZLPPX4dnqwTJRJN0kcMMwhpmWkBlVvc44ZQnn0Lw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(a) Dependence of Mn,NMR (●), Ð (□), and Mn,th. (Dotted line) on the monomer conversion. (b) First-order kinetic plot for the ROP of LLA at the [LLA]0/[PPA]0/[CH3COONa]0 ratio of 50/1/0.5 at 100 °C in the bulk 3-phenyl-1-propanol (PPA). Reprinted with permission from ref 21. Copyright 2018 American Chemical Society.
Other AMCs with various alkali-metal cations and carboxylate structures were also investigated (Figure 2). When examining the acetates with other alkali metal ions, such as Li+, K+, and Cs+ cations, the turnover frequency (TOF) value was observed. The TOF value increased with an increase in the radius of the cation, with cesium acetate showing the highest value of 73.6 h−1 and lithium acetate showing the lowest value of 1.8 h−1. When the sodium metal ion was retained and carboxylate was changed to trifluoroacetate, propionate, and pivalate, sodium pivalate exhibited the highest catalytic efficiency with a TOF of 14.6 h−1. The catalytic efficiency of sodium trifluoroacetate was the lowest, with negligible monomer conversion after 24 h. This result demonstrates that both the metal cation and substitution of carboxylate can significantly influence the catalytic efficiency of the ROP of LLA. Alkali metal cations with large radii and electron-donating groups on the carboxylates could be beneficial for the ROP of LLA. Following this trend, cesium pivalate showed even higher catalytic efficiency for the ROP of LLA. However, the molecular weight distribution of the obtained poly-l-lactic acid (PLLA) became broader, probably because of the transesterification side reaction occurring simultaneously with the ROP of LLA.
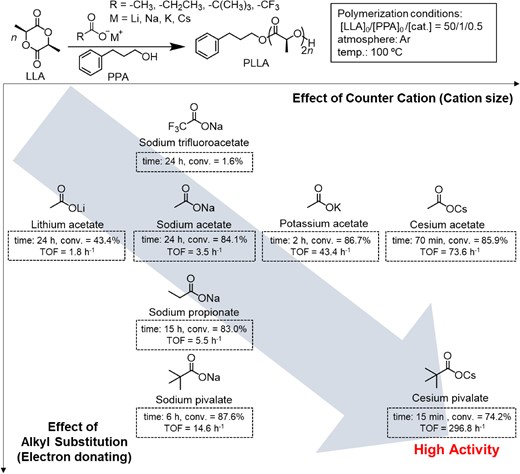
Catalytic performance of AMCs in the ROP of LLA. Reprinted with permission from ref 21. Copyright 2018 American Chemical Society.
A theoretical study was conducted to investigate the mechanism of PLLA polymerization using AMCs.22 After the initial calculation using the artificial force-induced reaction (AFIR) method to identify the most probable reaction pathway, the energy profiles of the initiation and propagation steps of the ROP of LLA were calculated. The calculation results support our proposal of a dual-activation mechanism. The relative reaction energy barrier (ΔG‡) when using cesium pivalate was calculated to be the lowest (11.4 kcal/mol), while that when using sodium trifluoroacetate was the highest (26.9 kcal/mol) (Figure 3). This tendency perfectly matched the experimental TOF results: the TOF of cesium pivalate was the highest and that of sodium trifluoroacetate was the lowest (Figure 2). In addition, carboxylates act as mediators of proton transfer, which is an important process in the ROP of LLA. The calculated free energy (ΔG) of the protonation reaction for AMC catalysts follows the same trend as ΔG‡, with the free energy of the protonation reaction using cesium pivalate being the lowest (51.4 kcal/mol).
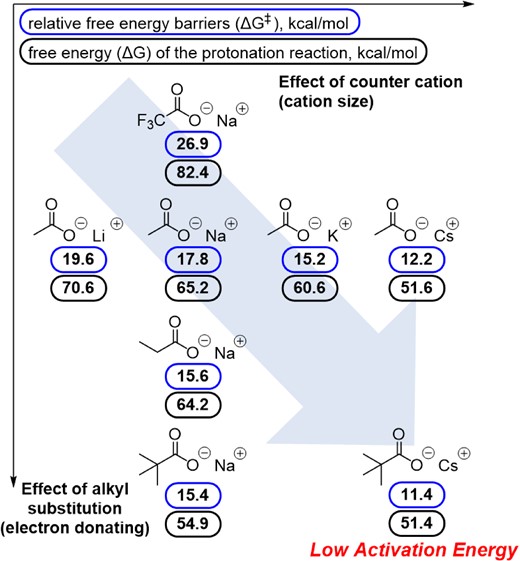
Calculated relative free energy barriers (ΔG‡) of for the ROP of LLA by each AMC catalyst (blue box) and calculated free energy (ΔG) of the protonation reaction for each AMC catalyst in which a H atom is transferred from the oxygen atom in alcohol initiator to the oxygen atom in the carboxylate moiety (black box). Adapted with permission from ref 22. Copyright 2020 Wiley-VCH.
Aside from polyesters such as PLLA, aliphatic polycarbonates are attractive biodegradable and bioresorbable polymer materials.23–25 The ROP of cyclic carbonates is undoubtedly one of the most controllable methods for controlling the molecular weight, dispersity, and chain-end functionalities.26 Therefore, after our initial success with the ROP of LLA, AMC catalysts were applied to the ROP of trimethylene carbonate (TMC).27 The catalytic efficiency of the AMCs was found to be very high for the ROP of the TMC. Polymerization proceeded smoothly when the catalyst loading of sodium acetate was reduced to 0.0001 mol% or 1 ppm. Analysis of the PTMC product revealed the absence of any ether linkages, proving that decarboxylation, which is a common side reaction in the ROP of cyclic carbonates, was effectively suppressed in the AMC-catalyzed ROP of TMC. This clean reaction was attributed to the mild basicity of the AMCs. Various functional groups were tolerated under mild polymerization conditions. As shown in Scheme 4a, alcohols with functional groups such as azido, alkyne, and methacrylate successfully initiated the polymerization. These functional groups remained at the α-end and could be utilized for further chain-end modifications. The countercation effect was examined using potassium acetate and cesium acetate as the catalysts. However, when comparing the TOF of polymerization using sodium acetate, no significant difference was observed when the cation was changed to potassium or cesium (Scheme 4b). Therefore, the cation had little effect on the ROP of TMC. This phenomenon can be attributed to the highly polarized nature of the carbonyl group of TMC compared with that of LLA. Because the highly polarized carbonyl groups could strongly interact with the alkali metal cation species and even solvate them in the polymerization medium, the effect of the cations was significantly reduced.
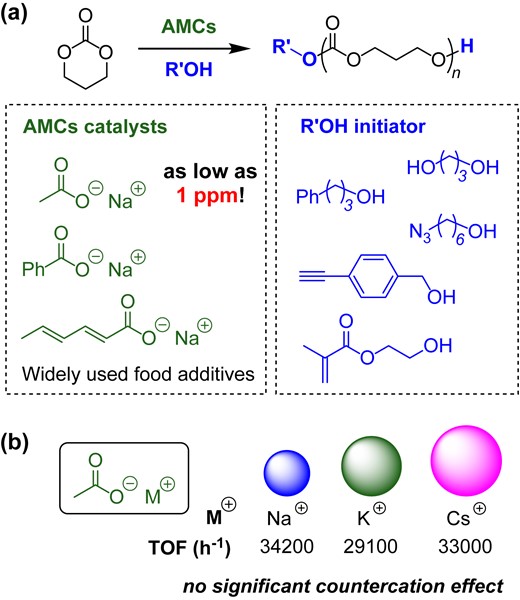
(a) AMC-catalyzed ROP of TMC; (b) TOF values when using different acetate alkali metal salts. Redrawn based on data from ref 27.
Utilizing the sodium acetate-catalyzed ROP of TMC and LLA, a biodegradable thermoplastic elastomer, PLLA-b-PTMC-b-PLLA,28 was synthesized in a one-pot process using a 1,3-propanediol initiator (Scheme 5). The initial ROP of TMC was conducted at 70 °C for 2 h with merely 10 ppm of sodium acetate catalyst. Subsequently, the LLA monomer and sodium acetate catalyst were added. The ROP of LLA was conducted at 100 °C for 24 hours with 1 mol% of sodium acetate. Both polymerization steps were well controlled, with a narrow distribution.
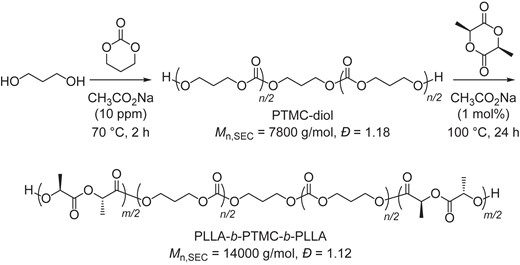
One-pot synthesis of PLLA-b-PTMC-b-PLLA triblock copolymer by CH3CO2Na catalyst. Redrawn based on the data from ref 27.
2.2 ROP of Epoxides and Episulfides
Aliphatic polyethers generated by the ROP of epoxides are an important class of polymers with widespread industrial, medical, and daily life applications. Anionic ROP with strongly basic catalysts or highly nucleophilic initiators is a conventional method for polyether synthesis.29,30 However, owing to the strong basicity of catalysts and propagation chain ends, chain-transfer side reactions, which undermine the synthesis of high-molecular-weight polymers often occur. These side reactions result in unwanted α-chain ends of the produced polyethers.31 Compared with alkoxides and phosphazenes, the basicity of carboxylates, circumventing the chain-transfer side reactions, is much weaker. Hence, AMCs were investigated as catalysts for the ROP of epoxides.32 A comprehensive study was conducted using 2-ethylhexyl glycidyl ether (EHGE) as a model monomer. Catalyst screening revealed that cesium pivalate exhibited the highest catalytic activity. The trend of the catalyst activity was the same as that of the ROP of LLA. Specifically, AMCs bearing large cations and carboxylates with electron-donating groups were beneficial (Figure 4).
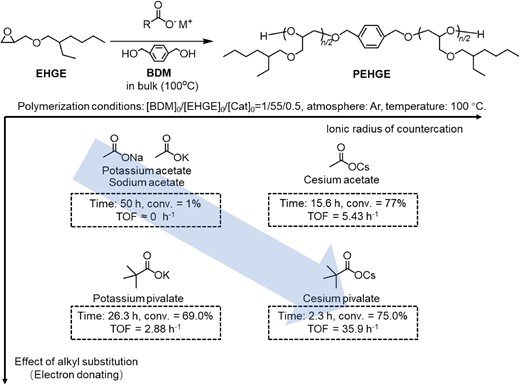
Catalytic performance of alkali metal carboxylates for the ROP of EHGE. Adapted with permission from ref 32. Copyright 2022 American Chemical Society.
A polymerization kinetics study revealed a linear increase in the polymer molecular weight with increasing monomer consumption, with the dispersity remaining narrow during the polymerization process (Ð < 1.07) (Figures 5a and b). The linear relationship in the plot of the logarithm of monomer concentration versus time indicated that polymerization follows first-order kinetics of the epoxide monomer (Figure 5c). The generated poly(2-ethylhexyl glycidyl ether) (PEHGE) showed excellent chain-end fidelity in MALDI-TOF MS measurements. Regarding the polymerization mechanism, a dual activation model, in which carboxylate anions and cesium cations activate the hydroxy chain end and epoxide monomer, respectively, was proposed based on a mechanistic study via infrared (IR) measurements (Figure 5d).
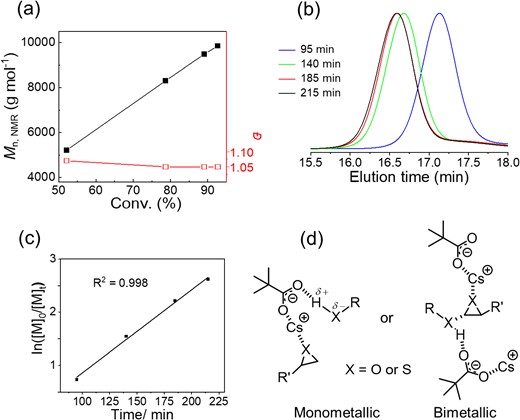
(a) Dependence of Mn,NMR (■) and Đ (□) on 2-ethylhexyl glycidyl ether conversion. (b) Size exclusion chromatography (SEC) traces of PEHGE obtained at different polymerization times. (c) First-order kinetic plot for the cesium pivalate-catalyzed ROP of EHGE. (d) Proposed catalytic mechanism: dual activation. Adapted with permission from ref 32. Copyright 2022 American Chemical Society.
This polymerization exhibited a broad scope of monomers and initiators. However, attempts to polymerize cyclohexene oxide and epichlorohydrin were unsuccessful. In addition to epoxide monomers, their sulfur analogs (episulfides) have also been examined for ROPs using AMC catalysts. As a result, cesium pivalate exhibited high catalytic activity and polythioethers were generated with controlled molecular weight and narrow dispersity (Ð = 1.06–1.35).
3. ROAC of Cyclic Anhydrides and Cyclic Ethers and Self-Switchable Polymerization
3.1 ROAC of Cyclic Anhydrides and Cyclic Ethers
The ROAC of cyclic anhydrides and cyclic ethers, especially epoxides, is an effective method for synthesizing polyesters because of its living polymerization nature and versatility in tuning polymer properties by independently changing the components of cyclic anhydrides and cyclic ethers (Scheme 6).33,34 Various transition-metal catalysts and organocatalysts35 have been reported to successfully catalyze this polymerization reaction in a controlled manner. The use of simple and safe AMCs as catalysts is advantageous for industrial applications.36
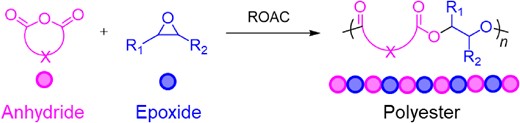
A general scheme of ROAC of cyclic anhydrides and epoxides.
When screening AMC catalysts for the ROAC of phthalic anhydride (PA) and ethyl glycidyl ether (EGE) in the presence of 1,4-benzenedimethanol (BDM) as an initiator, cesium pivalate showed the highest TOF among all the AMCs tested. This indicates that increasing the ionic radius of the cation and electron-donating ability of the substituent on the carboxylate could increase the catalytic reactivity of the AMCs (Figure 6). This tendency was the same for the ROPs of LLA and epoxides (Figures 2 and 4). When cesium pivalate was used as the catalyst, the polymer product P(PA-alt-EGE) was obtained with a narrow dispersity (Ð = 1.06). Moreover, MALDI-TOF MS of P(PA-alt-EGE) showed that all polymers contained the alcohol initiator BDM. However, no species with pivalate groups at the chain ends were observed. This demonstrates that cesium pivalate functions as a catalyst instead of an initiator.
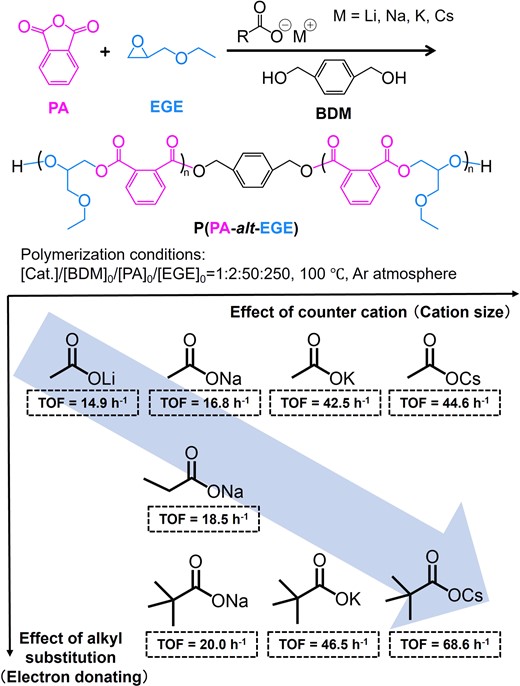
Catalytic performance of alkali metal carboxylates for the ROAC of PA and EGE. Adapted with permission from ref 36. Copyright 2021 American Chemical Society.
As shown in Scheme 7, the ROAC propagation involves two steps based on the polymerization mechanism. The first is the fast reaction between the hydroxy chain end and the cyclic anhydride monomer. The second is the slow reaction between the carboxylic acid chain ends with the epoxide monomers, which is the rate-determining step. Therefore, in theory, both alcohols and carboxylic acids (or carboxylates) can function as initiators of polymerization. There are reports on the use of carboxylic acids or carboxylates as initiators for ROACs.37,38 When alkali metal carboxylates (e.g., potassium acetate) were used as initiators without alcohol, polyesters with poor chain-end fidelity were obtained. To understand the reason for the perfect chain-end fidelity in our reported ROAC catalyzed by cesium pivalate, the proposed mechanism of the rate-determining step, viz. the insertion of the epoxide monomer into the polymer chain, is shown in Scheme 8. The carboxylic acid chain end formed a 1:1 complex with cesium pivalate, which is believed to be a stable intermediate in the reaction energy profile. Then, the epoxide monomer enters and interacts with this intermediate; the ring-opening of the epoxide via nucleophilic attack from both the chain-end carboxylate and pivalate (Paths A and B) could be considered in theory. Nucleophilic attack from the chain-end carboxylate (Path A) generates a propagated polymer chain, whereas nucleophilic attack from pivalate (Path B) results in a chain-transferred product with the pivalate structure at one chain end. In our experimental results, the ROAC polymer product with a pivalate structure was not observed by MALDI-TOF MS. Although two different carboxylates are involved in the transition state of the ring opening of the epoxide, nucleophilic attack from the pivalate is unfavored (Path B), because of the bulkiness of the tert-butyl groups on the pivalate.
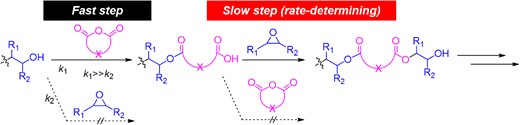
General scheme of the polymerization mechanism of ROAC.

Proposed reaction mechanism for the rate-determining step of the ROAC catalyzed by cesium pivalate. Adapted with permission from ref 36. Copyright 2021 American Chemical Society.
To expand this AMC-catalyzed copolymerization system, the synthesis of hyperbranched polyesters via ROACs of anhydrides and epoxides was investigated.39 Hyperbranched polymers (HBPs) are known to have characteristic properties, such as low viscosity, high solubility, internal voids, and large amounts of chain-end functional groups.40–42 Therefore, HBPs have the potential to be used as lubricants and drug delivery carriers.43 The ring-opening multibranching polymerization (ROMBP) of hydroxy-substituted cyclic lactones is a well-known approach for synthesizing hyperbranched polyesters (Scheme 9a).44,45 We assumed that a similar concept could be applied to the ROAC of anhydrides and epoxides by introducing an initiation site on either the anhydride or epoxide monomers. Specifically, we hypothesized that HBPs can easily be obtained by ROACs by choosing epoxide or anhydride monomer as a “latent trifunctional monomer” (Scheme 9b).
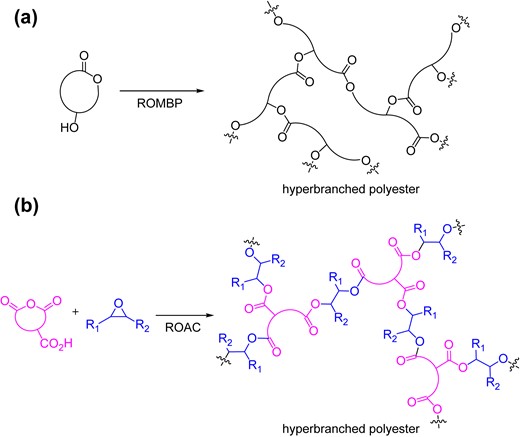
Hyperbranched polyesters by (a) ROMBP and (b) ROAC.
We focused on trimellitic anhydride (TA), which has a carboxyl substitution, as a cyclic anhydride monomer in ROAC. Coates et al. have used TA as a ROAC-chain-transfer agent, resulting in a hyperbranched polyester; however, a detailed mechanism and polymer properties have not been investigated.46 Hence, we selected TA as the anhydride monomer in ROAC with epoxides to synthesize hyperbranched polyesters using AMC catalysts (Scheme 10). The polymerization was conducted at 80 °C in the presence of an alcohol initiator. The 1H and 13C NMR spectra suggested that ring-opening reactions occurred at both the anhydride and carboxylic acid groups of TA. By comparing the NMR spectra with those of their linear counterparts (poly(PA-alt-EGE)) and a possible byproduct (polyEGE), we concluded that the hyperbranched polyesters were selectively synthesized by ROAC. The versatility of this synthetic method was demonstrated by applying a range of alcohol initiators and epoxide monomers, resulting in the formation of hyperbranched polyesters with various material properties or block copolymers. The obtained hyperbranched polyesters showed lower intrinsic viscosities and smaller hydrodynamic radii than their linear counterparts (Figure 7). Potential applications of these hyperbranched polyesters are currently being investigated by our group.
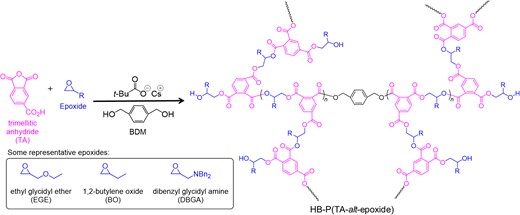
Hyperbranched polyester synthesis via the ROAC of trimellitic anhydride with epoxide. Adapted with the permission from ref 39. Copyright 2022 Royal Society of Chemistry.
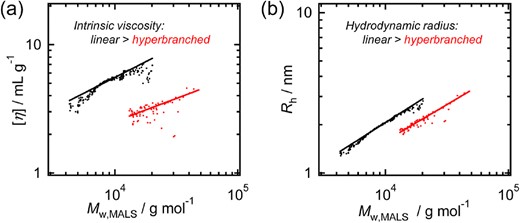
(a) Mark–Houwink–Sakurada plots and (b) conformation plots for hyperbranched-poly(TA-alt-EGE) (Mn,MALS = 16 900 g mol−1, red) and linear-poly(PA-alt-EGE) (Mn,MALS = 8 000 g mol−1, black). Reprinted with permission from ref 39. Copyright 2022 Royal Society of Chemistry.
3.2 Block Copolymer Synthesis by Self-Switchable Polymerization with AMC Catalysts
Block copolymers consist of at least two homopolymer subunits linked by covalent bonds. Compared with blends of the corresponding homopolymers or random copolymers, block copolymers show distinctly different properties, especially because of their separated monomer domains.47,48 Block copolymers have wide applications as elastomers,49,50 nanolithography materials in the semiconducting industry,51 and nanocarriers in medicines.52,53 Conventionally, the synthesis of block copolymers requires the stepwise addition of monomers or connection of different pre-synthesized polymers via chemical reactions. In recent years, one-pot and one-step synthesis of block copolymers from monomer mixtures have attracted significant attention. This process is known as self-switchable polymerization.54–64 The self-switchable polymerization of the monomer mixtures of cyclic anhydrides, epoxides, and cyclic esters using transition-metal catalysts or organocatalysts has been reported.56,58,60
After successfully realizing the ROP of cyclic esters, ROP of epoxides, and ROAC of cyclic anhydrides and epoxides using AMC catalysts, we combined them in one pot and checked the possibility of self-switchable polymerization to produce (multi)block copolymers.
Initially, a ternary monomer mixture of cyclic anhydrides (PA), epoxides (EGE), and cyclic esters (LLA) was tested with cesium pivalate as the catalyst and alcohol (BDM) as the initiator (Figure 8).36 As a result, the ROAC of PA and EGE proceeds first; when PA reaches almost full conversion, the ROP of the LLA begins. PLLA-b-P(PA-alt-EGE)-b-PLLA was obtained in a one-pot, one-step process by using the difunctional initiator BDM. To determine the monomer scope of the AMC-catalyzed self-switchable polymerization, nine cyclic anhydrides, six epoxides, and four cyclic esters were successfully polymerized (Scheme 11). This demonstrates the wide monomer scope of the simple AMC-catalyzed self-switchable polymerization. Similar to the synthesis of the hyperbranched polyesters introduced in Section 3.1, a core–shell block copolymer structure could be generated, when using TA for ROAC with epoxides. By changing the alcohol initiator to a trifunctional initiator or macroinitiator, the corresponding three-armed star-block copolymer or triblock terpolymer can be easily obtained.
![Plots of monomer conversion vs time, as monitored by 1H NMR spectroscopy. Polymerization conditions: the bulk was at 50 °C under an Ar atmosphere with a [PA]0/[EGE]0/[LLA]0/[BDM]0/[t-BuCO2Cs] feed ratio of 50/250/100/2/1. Adapted with permission from ref 36. Copyright 2021 American Chemical Society.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bcsj/96/9/10.1246_bcsj.20230149/2/m_20230149fig08rgb.jpeg?Expires=1747923207&Signature=1gHo7oV8eWyARml2No1fQu4XA2ta0CuK-dRO4fJS4APwVHsJd1CDd4jy-ezD56GzSKRaLY10OT5EQKQFwq5DCwemWhtSXlphriqAdKIXgPCmqKwJcJWMSAQ7xl9kb4tfNOqCDEWtaF45zOcwDjsrUVQzs3VOcx4R86l~C6rn0vmgDPaRUhN19s8k3LbJyFVrhk1fakhFXGr7kBKGzvjSKUgY39DiazT5kZrnp~JqR5bPxWbP72adswF3Zh6c8kuQBNzE8d7rLsuops0GlITfwlElAAERKVEVSZXtAeOfInpE4N~M-a2BKgJAJpgHQM3F1Ikp-i3uLwq3USZixUC7ng__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Plots of monomer conversion vs time, as monitored by 1H NMR spectroscopy. Polymerization conditions: the bulk was at 50 °C under an Ar atmosphere with a [PA]0/[EGE]0/[LLA]0/[BDM]0/[t-BuCO2Cs] feed ratio of 50/250/100/2/1. Adapted with permission from ref 36. Copyright 2021 American Chemical Society.
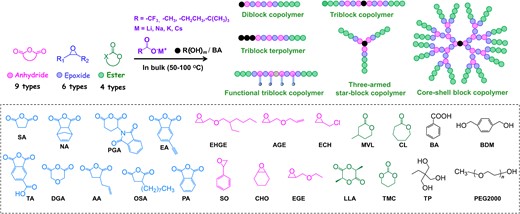
Self-switchable polymerization of mixed anhydrides, epoxides, and cyclic esters with alkali metal carboxylates as catalysts and carboxylic acids/alcohols as initiators. Adapted with permission from ref 36. Copyright 2021 American Chemical Society.
Taking advantage of this facile synthetic method, a polyester-based ABA type block copolymer with “hard” A block and “soft” B block was synthesized using a mixture of n-octylsuccinic anhydride (OSA), EHGE, and LLA. Two Tg values (Tg,P(OSA-alt-EHGE)block ≅ −41 °C and Tg,PLLA block ≅ 26 °C) were observed because of microphase separation by these two blocks with largely different polarity. This microphase separation enables the design of thermoplastic elastomers and adhesives based on ABA block copolymers by tuning the weight fractions of the blocks.65 PLLA-b-P(OSA-alt-EHGE)-b-PLLA with a 50/50 (w/w) block ratio exhibited elastomeric behavior and recovered once the stress was removed (Figure 9). No obvious yield point was observed, and it reached approximately 220% of the elongation at break. When the weight percentage of PLLA was decreased to 40%, a viscous and sticky material was obtained that could potentially be utilized as an adhesive.
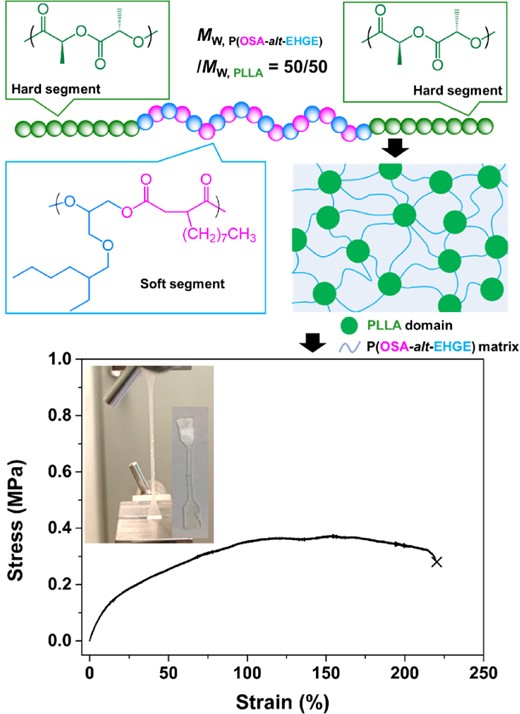
Structure–property relationship for PLLA-b-P(OSA-alt-EHGE)-b-PLLA with a 50:50 block ratio (w/w). Adapted with permission from ref 36. Copyright 2021 American Chemical Society.
In previous studies on self-switchable polymerization, no more than three monomer components were usually involved. They resulted in di- or triblock copolymers, at most ABCBA-type pentablock terpolymers, in a one-step synthesis.66 When the AMC-catalyzed self-switchable polymerization of a ternary mixture of cyclic anhydrides, epoxides, and cyclic esters was investigated, a significant difference in reactivity was observed when changing the cyclic anhydrides. Therefore, we assumed that when a mixture of more than one cyclic anhydride was utilized, the difference between the reactivity of the anhydrides could lead to self-switchable polymerization between different ROAC cycles. This, in turn, enables the incorporation of an increasing number of monomer components in the self-switchable polymerization, ultimately yielding multiblock copolymers with more block numbers. With this concept, we successfully developed a self-switchable polymerization system with up to six different monomers, including four kinds of cyclic anhydrides, one type of epoxide, and LLA (Scheme 12).67 A macroinitiator with alcohol functionality at both chain ends, such as PEG-diol, was utilized as a starting material. By using this macroinitiator, a one-pot, one-step polymerization approach was employed to synthesize sequence-controlled multiblock polymers with 11 segments. The catalytic cycle of the ROP of LLA lies between two different ROAC cycles using 5-norbornene-endo-2,3-dicarboxylic anhydride (NA) and rac-cis-endo-1-isopropyl-4-methyl-bicyclo[2.2.2]oct-5-ene-2,3-dicarboxylic anhydride (DPMA) as cyclic anhydride components.
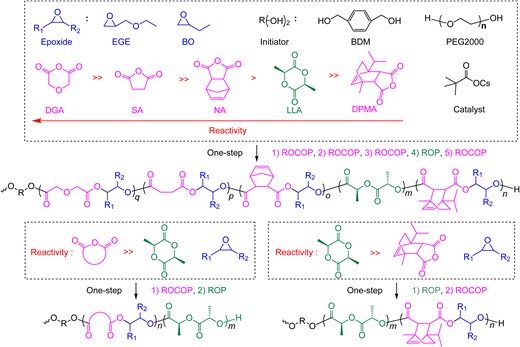
One-step synthesis of multiblock polymers from a monomer mixture: multiblock polymers from six-component monomer mixture. Reprinted from ref 67. Under the terms of the Creative Commons CC BY license, 2022 Springer Nature.
In general, the material properties of copolymers are largely influenced by the monomer sequence and composition, which can be quantified using reactivity ratios during copolymerization. Self-switchable polymerization, which produces nearly perfect block copolymers from monomer mixtures, can be considered as a special case of copolymerization with reactivity ratios of rmonomer 1 ≫ 1 and rmonomer 2 ≅ 0, for the two competing monomers. In our investigation of self-switchable polymerization using the ROAC of multiple cyclic anhydrides and one epoxide, the switching behavior varied among different pairs of cyclic anhydrides. Therefore, it is important to quantify the switching behavior by determining the reactivity ratio and establishing a reactivity gradient with an expanded monomer scope (Scheme 13).68
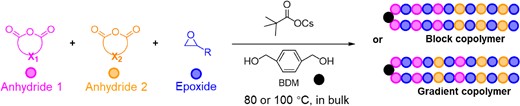
Copolymerization of the mixture of two cyclic anhydrides and one epoxide. Adapted with permission from ref 68. Copyright 2023 American Chemical Society.
For the two competitive anhydrides/one epoxide three-component copolymerizations, the selectivity-determining step was the nucleophilic attack of the hydroxy chain end on the cyclic anhydrides. The reactivity strongly depended on the nature of the cyclic anhydride, instead of the terminal structure of the propagating polymer chain (Scheme 14). Therefore, a nonterminal copolymerization kinetic model (Beckingham–Sanoja–Lynd) was applied to determine the reactivity ratio and compositional drift.69 After monitoring the copolymerization kinetics, calculating the fraction of the tapered region, and determining the reactivity ratio, the following conclusions were drawn:
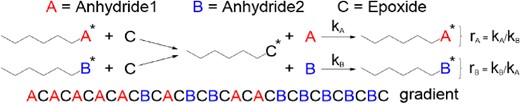
Non-terminal model to determine the reactivity ratio of two cyclic anhydrides in ROAC with epoxide. Reprinted with permission from ref 68. Copyright 2023 American Chemical Society.
(1) for the reactivity ratios of 2 < ranhydride 1 < 800 and 0.7 > ranhydride 2 > 0.0049, the competitive copolymerization produced copolymers with various gradient strengths;
(2) copolymerization at higher reactivity ratios (ranhydride 1 > 800 and ranhydride 2 < 0.0049) provided real block polymers with a very sharp boundary, showing real self-switchable behavior.
Four examples are shown in Figure 10. Copolymerization of EGE with a PA/succinic anhydride (SA) mixture (rPA = 4.59 and rSA = 0.63; Figure 10a) produced copolymers with medium gradient strengths (80.4% tapered region). Copolymers with hard gradient strengths (49.2% tapered region) were produced by the copolymerization of diglycolic anhydride (DGA)/PA/EGE mixtures (rDGA = 10.98 and rPA = 0.098; Figure 10b). The copolymerization of the PA/NA/EGE mixture (rPA = 739.83 and rNA = 0.0055; Figure 10c) resulted in a block-like copolymer with a marginally tapered region of 1.6%. The “real” block copolymer without a tapered region was achieved using a monomer mixture of 2,2,3,3,4,4-hexafluoropentanedioic anhydride (HFA)/DGA/EGE, whose reactivity ratio differences were extremely large (rHFA = 816.39 and rDGA = 0.0036; Figure 10d).
![Total polymerization conversion plotted against monomer conversion. The data were obtained from time-resolved 1H NMR spectra of a cesium pivalate-catalyzed copolymerization at 100 °C: (a) PA/SA/EGE; (b) DGA/PA/EGE; (c) PA/NA/EGE; (d) HFA/DGA/EGE. Polymerization conditions: [anhydride 1]0/[anhydride 2]0/[EGE]0/[BDM]0/[CsOPiv]0 = 25/25/150/2/1, in the bulk at 100 °C under Ar atmosphere. Adapted with permission from ref 68. Copyright 2023 American Chemical Society.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bcsj/96/9/10.1246_bcsj.20230149/2/m_20230149fig10rgb.jpeg?Expires=1747923207&Signature=GH8wYWbwlt9cbHkq566~WuvJpiHtQJTtBDhtwBGm6rhXau8h-1bP01X9VIOUQOjBo8x9S8OP1slB0YvAR2zBmqUw0cgzfTVefzYNavT-cDGYQjc18LaFWNSvSK0BX~L3uVokYhYancIGzk2SY-rh-C8nBdXfD1heweptk8AKOA1qSspE2Dfz-6xHcmwm-X7p8VE3iiu5TrYXFxuBzQu7qlF8sdCpryYgP7sqWtZ4M~EU6nYQ66qMgEDi-geG~nzxRGrjxhBwcHOyj8PYXGX0Lp~XbLVYlSfVNmsT236xz-l-90TxQi0c0AipEXCuMUm-1DcEuqsQWRlGpuhTQQciMw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Total polymerization conversion plotted against monomer conversion. The data were obtained from time-resolved 1H NMR spectra of a cesium pivalate-catalyzed copolymerization at 100 °C: (a) PA/SA/EGE; (b) DGA/PA/EGE; (c) PA/NA/EGE; (d) HFA/DGA/EGE. Polymerization conditions: [anhydride 1]0/[anhydride 2]0/[EGE]0/[BDM]0/[CsOPiv]0 = 25/25/150/2/1, in the bulk at 100 °C under Ar atmosphere. Adapted with permission from ref 68. Copyright 2023 American Chemical Society.
In addition to measuring the reactivity ratios for various monomer pairs, we summarized the rule of thumb based on 13C NMR analysis to roughly predict the reactivity of cyclic anhydrides in the ROAC with epoxides. By comparing the results of the competitive copolymerization and chemical shift of the carbonyl group of cyclic anhydrides in 13C NMR, the following tendency was observed: cyclic anhydrides with lower chemical shift (higher field) of the carbonyl group exhibited higher reactivity in competitive copolymerization (Figure 11). Although the mechanism behind this phenomenon requires further investigation, this rule of thumb will be useful for roughly predicting the reactivity of new cyclic anhydrides. Hence, this rule will be beneficial for designing polyester materials using self-switchable polymerization.
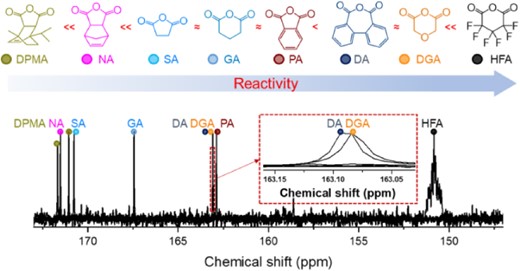
13C NMR spectroscopic signals of the carbonyl groups of the cyclic anhydrides and their relative reactivities determined by competitive copolymerization experiments. Reprinted with permission from ref 68. Copyright 2023, American Chemical Society.
The reactivity ratio of the two cyclic anhydrides can vary when the reaction conditions, especially the catalyst, are changed.70 In addition, the sequence of reactivity of the cyclic anhydrides determined by competitive copolymerization was not related to the polymerization rate difference when different cyclic anhydrides were used in the binary ROAC of one cyclic anhydride/one epoxide. This is because the polymerization-rate-determining step in the ROAC of cyclic anhydride/epoxide is the insertion of the epoxide, whereas the selectivity-determining step in the competitive copolymerization is the insertion of the cyclic anhydride.
Utilizing the reactivity difference between cyclic anhydrides, we successfully demonstrated multiblock copolymer synthesis by connecting up to four cyclic anhydride/epoxide ROAC catalytic cycles in a one-pot, one-step fashion.67 Despite this tremendous advancement, only one of the two monomers underwent self-switchable polymerization between the two ROAC catalytic cycles. This is also the case in other reports; for example, the switch between the ROACs of the epoxides/cyclic anhydrides and N-sulfonyl aziridines/cyclic anhydrides was reported by Hadjichristidis et al.,71 and the self-switchable polymerization that connects the ROACs of the epoxides/cyclic anhydrides and epoxides/thioanhydrides was reported by Li et al.72 To overcome this limitation and further advance the synthetic capabilities, achieving complete switching of both components in the ROAC becomes a challenging but important task.73 After the establishment of the reactivity sequence for the different cyclic anhydrides in the ROAC with epoxides in the presence of AMC catalysts, we investigated other possible surrogates for epoxides. These surrogates should meet two requirements: 1) they should undergo ROACs with cyclic anhydride catalyzed by AMC catalysts, and 2) they should show distinct reactivity differences with epoxides. Oxetanes, which are 4-membered ring cyclic ethers, emerged as promising candidates. Oxetanes can undergo nucleophilic ring-opening reactions similar to epoxides; however, they have a much higher energy barrier owing to their lower ring strain. There are only a few previous reports on the ROAC of cyclic anhydrides and oxetanes.74–77 In these cases, transition-metal or onium-salt catalysts have been used. However, the previous methods suffered from imperfectly alternating polymer structures containing ether linkages or poor polymer dispersity control, probably because of the low reactivity of oxetanes.
We tested the ROAC of cyclic anhydride/oxetane using a cesium pivalate catalyst (Scheme 15). Various cyclic anhydrides and oxetanes were copolymerized in a well-controlled manner (Figure 12), and strictly alternating sequences and polyester products with narrow dispersities are obtained. As expected, the reactivity of oxetanes was much lower than that of the epoxides in the ROAC with cyclic anhydrides in this cesium pivalate-catalyzed system. The rate-determining step of the cyclic anhydride/oxetane ROAC is the insertion of oxetanes, specifically the nucleophilic attack of the carboxylic acid chain end on the oxetanes.
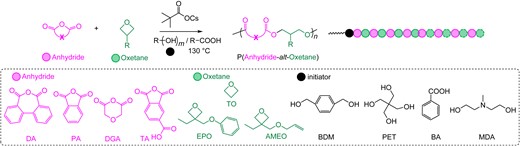
ROAC of anhydrides and oxetanes catalyzed by cesium pivalate. Reprinted with permission from ref 73. Copyright 2022 American Chemical Society.
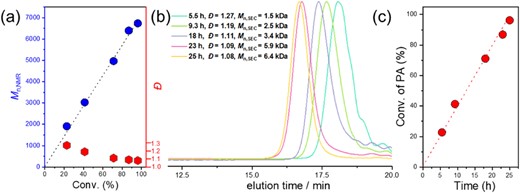
Kinetics analysis of PA/3-ethyl-3-(phenoxymethyl)-oxetane (EPO) ROAC: (a) dependence of Mn,NMR and Đ on the monomer conversion, (b) evolution of SEC traces (THF), and (c) zero-order kinetic plot for PA/EPO ROAC. Reprinted with permission from ref 73. Copyright 2022 American Chemical Society.
By combining the established reactivity gradients among different cyclic anhydrides, the self-switchable polymerization of the four-component monomer mixtures (two cyclic anhydrides, epoxides, and oxetanes) was successfully realized (Scheme 16). Tri-/penta-block tetrapolymers were obtained using a difunctional alcohol initiator by tuning the monomer ratio. When TA was used as the more active cyclic anhydride in this mixture, core-shell type tetrapolymers were obtained.
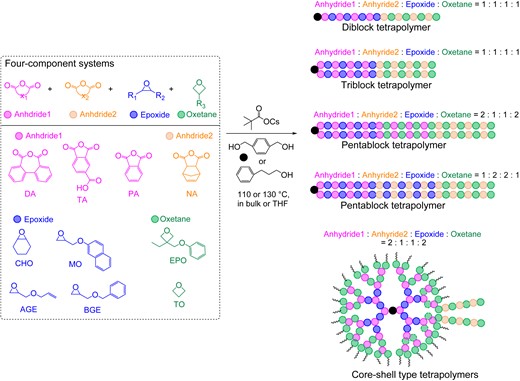
One-step synthesis of multiblock copolymers from a four-component monomer mixture of two cyclic anhydrides, epoxide, and oxetane. Adapted with permission from ref 73. Copyright 2022 American Chemical Society.
4. Conclusion and Outlook
AMCs, which are simple and widely used chemicals, with some approved as safe food additives, have been discovered to effectively catalyze a variety of polymerization reactions, including the ROP of cyclic esters, cyclic carbonates, and cyclic (thio) ethers; ROAC of cyclic anhydrides and cyclic ethers; and self-switchable polymerization that combines various polymerization catalytic cycles from complex monomer mixtures. Their thermal stability allows them to withstand the most industrially favored bulk polymerization and subsequent thermal processing of polymer products. The low toxicity of many AMCs eliminates the need for extra expensive catalyst removal from the obtained polymer products using conventional purification methods. In most reported cases, the catalytic activity of AMC catalysts was relatively low compared with some other well-established organocatalytic systems. Further improvement of the catalytic activity while keeping the advantages of AMC catalysts, especially in the safety aspect, could be a prominent challenge in future development. Recently, sodium ascorbate (the sodium salt of vitamin C)78 and alkali metal carbonates (e.g., K2CO3)79 have been identified to be effective catalysts for polyester synthesis. Modifying the structures of the anions and changing the alkali metal cations to organic onium cations could provide new opportunities for catalyst designs. Hence, our group had previously demonstrated that trimethylglycine, a natural betaine from sugar beets, can effectively catalyze the ROP of TMC.80
In the pursuit of ideal catalysts for polymer synthesis, numerous excellent catalyst designs have been reported over the past decades. However, the catalytic abilities of simple compounds such as AMCs have long been overlooked. Recent studies by us and other researchers have clearly shown the untapped potentials that lie within these unassuming substances, challenging our assumptions and opening new avenues for exploration.
Acknowledgment
This work was financially supported by the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (B) (grant no. 19H02769), Ministry of Education, Culture, Sports, Science, and Technology of Japan Grants-in-Aid for Scientific Research on Innovative Areas (Hybrid Catalysis for Enabling Molecular Synthesis on Demand; grant nos. 18H04639 and 20H04798), Frontier Chemistry Center (Hokkaido University), Photo-excitonic Project (Hokkaido University), Creative Research Institution (CRIS, Hokkaido University), List Sustainable Digital Transformation Catalyst Collaboration Research Platform (Hokkaido University), Hokkaido University DX Doctoral Fellowship (Grant JPMJSP2119), and Inamori Foundation.
References
M. North, Sustainable Catalysis: With Non-endangered Metals, Part1 and 2, The Royal Society of Chemistry, Cambridge, 2015.
M. North, Sustainable Catalysis: Without Metals or Other Endangered Elements, Part1 and 2, The Royal Society of Chemistry, Cambridge, 2015.
Organic Catalysis for Polymerisation, ed. by A. Dove, H. Sardon, S. Naumann, The Royal Society of Chemistry, 2018.
P. Lecomte, C. Jérôme, Ring-opening Polymerization of Lactones. in Organic Catalysis for Polymerisation, ed. by A. Dove, H. Sardon, S. Naumann, The Royal Society of Chemistry, 2018, pp. 198–223.
K. Fukushima, ROP of Cyclic Carbonates. in Organic Catalysis for Polymerisation, ed. by A. Dove, H. Sardon, S. Naumann, The Royal Society of Chemistry, 2018, pp. 274–327.
I. W. Hamely, The Physics of Block Copolymers, ed. by I. W. Hamely, Oxford University Press, Oxford, 1988.
M. Lazzari, G. Liu, S. Lecommandoux, Block Copolymers in Nanoscience, Wiley-VCH Press, Weinheim, 2006.

Feng Li
Feng Li received his Ph.D. from Tohoku University in 2016 under the supervision of Prof. Masahiro Terada. After postdoctoral research at Yale University (2017–2019 with Prof. Mingjiang Zhong) and École Polytechnique Fédérale de Lausanne (EPFL) (2019–2020 with Prof. Harm-Anton Klok), he moved to Kanazawa University as a Specially Appointed Assistant Professor at the end of 2020. In 2022, he began his current position as an Assistant Professor at Hokkaido University. His current research interests are centered on sustainable polymer synthesis, which includes utilizing renewable biomass materials, developing environmentally friendly catalysts, and chemical recycling.

Ryota Suzuki
Ryota Suzuki received his B.E. degree at National Institute of Technology, Asahikawa College in 2020. He received his master’s degree at Hokkaido University in 2022 under the supervision of Prof. Toshifumi Satoh. His current research interests focus on ring-opening alternating copolymerization of cyclic anhydrides and epoxides.

Tianle Gao
Tianle Gao graduated with a bachelor’s degree from Harbin Engineering University, China, in 2019. Later that year, he pursued a master's degree at Hokkaido University, where he conducted research on the ring-opening polymerization of epoxides and lactones under the supervision of Prof. Toshifumi Satoh. Upon completing his master's degree in 2021, he continued his research in the same laboratory as a doctoral student. His current research interests include organocatalyzed self-switchable polymerization for block copolymer synthesis.

Xiaochao Xia
Xiaochao Xia received his Ph.D. from Sichuan University in 2017 under the supervision of Prof. Mingbo Yang. He then worked at the Chongqing University of Technology as a lecturer (2017–2023). During this period, he moved to Hokkaido University as a postdoctoral researcher (2019–2022, Prof. Toshifumi Satoh). In 2023, he worked as a postdoctoral research associate at Washington University in St. Louis. His current research interests include the development of biomaterial platforms for angiogenesis and vascularization, stem cell engineering, and tissue regeneration by synthesizing biodegradable and biocompatible polymers.

Takuya Isono
Takuya Isono earned his Ph.D. in polymer chemistry from the Graduate School of Chemical Sciences and Engineering at Hokkaido University in 2014. He was a JSPS Research Fellow (DC1). After completing his Ph.D., he began his research career as an Assistant Professor at the Faculty of Engineering at Hokkaido University in 2014. Since April 2021, he has been an Associate Professor at Hokkaido University. His expertise is in precise polymer synthesis and his research interests are currently centered on organocatalytic polymerization, bio-based polymers, block copolymers, and topological polymers.

Toshifumi Satoh
Toshifumi Satoh received his Ph.D. in Polymer Chemistry from Hokkaido University under the supervision of Prof. Kazuaki Yokota and Prof. Toyoji Kakuchi in 1996. He was subsequently appointed as an Instructor in the Faculty of Engineering at Hokkaido University in 1996. During this period, he studied in the group of Prof. Bruce M. Novak at the University of Massachusetts Amherst and at North Carolina State University, USA, from 1998–2000 as a post-doctoral fellow. He became an Associate Professor in 2007 and a professor in 2013 at Hokkaido University. His current fields of interest include topological polymers and controlled polymerization using organocatalysts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}