





Abstract
Ubiquitous intramolecular aurophilic interactions in gold clusters have usually been assigned as the main determinants of their molecular geometry and various properties. By utilizing N-heterocyclic carbene (NHC) ligands instead of phosphine ligands which have been exclusively used, we have developed ligand-specific photophysical properties of C-centered hexagold(I) (C@Au6) clusters. This account describes the recent development of carbene-protected C@Au6 clusters. First of all, a strong intramolecular C–H⋯Au interaction was found as a key structural feature of these clusters. These are also present in the newly emerged NHC-gold nanoclusters, but are negligible in the phosphine counterparts. Interestingly, the use of NHC ligands also produces clusters with a nearly identical metal kernel, but their luminescence changes significantly. In addition, a simple extension of the π-conjugated system of NHCs from imidazolylidene to benzimidazolylidene is expected to produce significant changes in both absorption coefficient and emission wavelength. These results show the promising potential of carbene ligands in character modulation of metal clusters, and may help in the rational design of carbene ligands for constructing functional metal clusters.
1. Introduction
Inter- and intramolecular aurophilic interactions are the most attractive structural features of gold complexes and clusters. Such interactions are determinants of geometry of metal clusters, luminescence properties, external stimuli responsiveness, etc., and are generally considered to be the basis of further applications in biology and materials science.1–3 On the other hand, little attention has been paid to the organic parts of gold complexes and clusters. This is probably natural in the traditional view of chemistry, as the word “ligand”, derived from the Latin ligare, means only “to bind/tie”. However, some recent examples imply that the ligand shell of clusters cannot be ignored in terms of their photophysical and catalytic properties.4,5
To systematically investigate the ligand effects on the structures and properties of metal clusters, it is necessary to utilize a unified model cluster that maintains the metal cluster part including the charge state and to observe the changes when alternative ligands are used. The high designability of the steric and electronic structures of the organic ligands is a key to finely adjusting the functions of the metal clusters. These almost fix the other factors and allow us to focus on one variable. Therefore, the characteristics of the cluster can be directly correlated with the ligand shell. Ideally, the model cluster should be easy to synthesize and isolate, and stable in terms of quality and quantity.
Hexagold(I) clusters containing an internal carbon tetraanion (C@Au6) are one of the most classical models of gold clusters. In the 1980s, Schmidbaur et al. succeeded in constructing a series of C@Au6 clusters for the first time using phosphine ligands such as triphenylphosphine.6–13 These octahedral clusters are fairly robust to air and moisture, and have been reported to have relatively high synthetic yields (up to ∼50% based on gold). More interestingly, they emit strong phosphorescence under photoexcitation. For example, the yellow-green luminescence from the C@Au6 cluster protected by triphenylphosphine is visible to the naked eye in sunlight. These were further developed by the Wang group into a series of C@Au6-based heterometallic clusters.14–23 The octahedral core, or in some cases twisted into a triangular prism, was further capped by two, four, or six secondary metal ions. As a result, the clusters not only emit light in the solid state, but also emit strong phosphorescence in solution, resulting in a quantum yield of up to 92%. Therefore, it is promising to use C@Au6 cluster as a model for investigating ligand effects.
N-Heterocyclic carbenes (NHCs) were selected as ligand candidates as stronger σ-electron donors because they bind to coinage metals in a mode similar to those of phosphine ligands.24 Another advantage of NHC is that it is easy to handle. In the NHC ligands, the two wing-tip groups that determine the steric factor and the heterocyclic part that is responsible for donating electrons to the metal are factors that can rationally adjust the steric and electronic structures of the complex.25 Only a few examples have been reported on carbene-protected gold clusters,26–30 and the investigations mainly focused on the carbene-gold complexes and their catalytic activities,31 and carbene-stabilized particles/surfaces.32,33 Shortly after we reported the first C@Au6 cluster protected by carbene in 2018,34 this type of ligand was successfully introduced into gold nanoclusters by Crudden, Tsukuda, and Häkkinen et al.35,36 Since then, a series of carbene-protected gold nanoclusters have been extensively developed, and their stability and catalytic activities were reported more recently,37–44 indicating the great potential of carbene ligands for developing metal cluster chemistry.
This account summarizes our current results from the C@Au6 clusters. Although these clusters have been long protected only by phosphine ligands, they were successfully reconstructed using NHCs. An important structural feature of this series of structures was the strong intramolecular C(sp3)–H⋯Au interactions, where the shortest hydrogen-gold distance is 2.566 Å.45–49 These features were also found in the newly emerged NHC-protected Au11-, Au13-, Au25-based nanoclusters,35–44 but not observed with the C@Au6 clusters protected by phosphine ligands PPh3, PiPr3 and o-C6H4(CH2CH2PPh2)2.6–13 The hydrogen-gold distance above is much shorter than that of a similar carbene-gold complex (2.869 Å) found by Tsutsumi and Prabusankar,50 and that of a diphosphine-protected Au6 nanocluster with C(sp2)–H⋯Au interactions (2.722 Å) developed by Konishi,51–53 and is comparable to those reported by Balakrishna (2.495 Å),54 and Mobin (2.638 Å).55 More notably, the use of N,N′-diisopropylimidazolylidene (IiPr) instead of phosphine ligand PPh3 as the ligand for the carbon-centered gold cluster can significantly alter its luminescence properties while largely maintaining its cluster structure.34 Furthermore, extending the π-conjugated system from IiPr to N,N′-diisopropylbenzimidazolylidene (BIiPr) causes even greater changes in both absorption coefficient and emission wavelength.56 Although Schmidbaur and Wang et al. have reported a series of C@Au6 clusters with various phosphine ligands, here we select representative C@Au6 cluster with PPh3 and compare with those protected by NHC ligands.6–23
2. Molecular Structures and Intramolecular C(sp3)–H⋯Au interactions
In 1988, Schmidbaur et al. reported a novel gold cluster represented by the formula [(C)(Au-PPh3)6](CH3OBF3)2 and first introduced the term “aurophilicity” to gold chemistry.6 Single crystal X-ray diffraction analysis reveals that the dicationic C@Au6 moiety of this complex had an octahedral structure containing a total of twelve aurophilic bonds (2.910(1)–3.090(1) Å), identifying the aurophilicity. A μ6-C was encapsulated in an Au6 cage, each of the six PPh3 ligands linearly binds to one of the gold atoms, and the C-Au-P angles ranged from 172.4(1) to 175.7(1)°. After that, a series of chemically modified clusters including [(C)(Au2-LdP)3](BF4)2 (LdP = o-C6H4(CH2CH2PPh2)2),7 [(C)(Au-PiPr3)6](B3O3F4)2,10 and [(C)(Au-PPh3)6](BF4)2 (1)13 were reported by the same group. Although different phosphine ligands and counterions were employed, in each case the C@Au6 moiety preferentially formed an octahedral geometry. It was not until 2011 that Wang first reported a twisted structure in a triangular prism.19 There were only nine possible aurophilic bonds in this structure. However, theoretical calculations suggest that there is only very little energy difference between these two types of structures. The twist in this cluster moiety was due to the involvement of another metal ion (Cu+). Other external stimuli can also induce/stabilize such twists.20,21 It should be noted that all the precursors of these heterometallic species share a common octahedral C@Au6 structure, similar to the complexes reported by Schmidbaur and coworkers.
Figure 1 illustrates the structures of phosphine and NHC ligands successfully used for the C@Au6 clusters, and corresponding clusters such as [(C)(Au-PPh3)6](CH3OBF3)2 (P21/c), [(C)(Au2-LdP)3](BF4)2 (C2/c), [(C)(Au-PiPr3)6](B3O3F4)2 (P-1), [(C)(Au-PPh3)6](BF4)2 (1, P21/c), [(C)(Au-IiPr)6](BF4)2 (2, P-3),34 and [(C)(Au-BIiPr)6](BF4)2 (3, P-1).56 One can see that different components can change the space group of a single crystal of the cluster. However, this difference did not significantly affect the octahedral configuration of the C@Au6 moieties. In fact, cluster 1 shows a slightly distorted C@Au6 core compared to [(C)(Au-PPh3)6](CH3OBF3)2. It turned out that the gold-gold distances were in the range from 2.887(1) to 3.226(1) Å. However, cluster 1 and [(C)(Au-PPh3)6](CH3OBF3)2 exhibit identical 31P NMR chemical shifts in CD2Cl2. Therefore, a possible cause of distortion is the co-crystallization of CH2Cl2 molecules in the cells of 1. It was found that the octahedral core is also ubiquitous when using the NHC ligands. The main difference between phosphine and NHC-protected clusters is the metal-ligand distances due to the different ligand properties. For example, the Au-P distances of 1 are in the range of 2.254(4)–2.277(4) Å, while the Au-C (NHCs) distances of 2 and 3 are 2.020(5) and 2.026(5)–2.038(5) Å, respectively.
2 (b), [(C)(Au2-LdP)3](BF4)2 (c, LdP = o-C6H4(CH2CH2PPh2)2), [(C)(Au-PiPr3)6](B3O3F4)2 (d), [(C)(Au-PPh3)6](BF4)2 (e, 1), [(C)(Au-IiPr)6](BF4)2 (f, 2, 50% probability), and [(C)(Au-BIiPr)6](BF4)2 (g, 3, 50% probability). Color codes: C, gray; N, blue; P, magenta; Au, orange. Counterions, co-crystallized solvent molecules, and hydrogen atoms are omitted for clarity.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bcsj/94/4/10.1246_bcsj.20210060/2/m_20210060fig01cmyk.jpeg?Expires=1750380650&Signature=KEqwmeExyeO0vJOqB3WMpVctlh-pJe0SCWcwDEd6Cr1S8C-knWZDraXiuwdlZVolNocKZeGxWF4uJZdZU9Yv9VJisXtLJUdU484YjtF-i5WAdMrH2bJ2Hyot3~DXG20SXnCJjWK8s4dqECZPSp-fvd3XfzLp-m9LtvXjD2mjsc0pwuXgrUdAGo3MycrTL97qDnOQXiHGbaOfO05Wvv~CUQgbY8jEYpr9BcvNgNBioNedk921dd3R8Zt1msDcY2K2DtBCJ4Yb7xF9n8GkQBbTeGxXn~lPM7pJFmqWye4Fo9JzmqGYPUwbo3oyGxZrBzHKa2mKREIsQIVApjFn80pZIA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structures of phosphine and NHC ligands that were successfully used for C@Au6 clusters (a), and the corresponding clusters [(C)(Au-PPh3)6](CH3OBF3)2 (b), [(C)(Au2-LdP)3](BF4)2 (c, LdP = o-C6H4(CH2CH2PPh2)2), [(C)(Au-PiPr3)6](B3O3F4)2 (d), [(C)(Au-PPh3)6](BF4)2 (e, 1), [(C)(Au-IiPr)6](BF4)2 (f, 2, 50% probability), and [(C)(Au-BIiPr)6](BF4)2 (g, 3, 50% probability). Color codes: C, gray; N, blue; P, magenta; Au, orange. Counterions, co-crystallized solvent molecules, and hydrogen atoms are omitted for clarity.
A structurally unique intramolecular C(sp3)–H⋯Au interaction was identified at the outer edge of the NHC-stabilized C@Au6 clusters, but was absent in the phosphine-protected ones. As shown in Figure 2a, the distances between gold atoms and one of the o-H of phenyl groups of PPh3 in 1 were found in the range from 2.918 to 3.626 Å. As pointed out by Schmidbaur et al.,45 such C–H⋯Au interactions in triarylphosphine-gold systems may not have a significant influence on the molecular structures. In 2, all isopropyl groups of the ligand IiPr have almost the same orientation, and all the hydrogen atoms on secondary carbons point to the gold atoms in a roughly cis manner. The H⋯Au distances of 2.930 or 2.944 Å in the side-on coordination mode are very close to those in a similar mode found in cluster 1. However, we found even shorter H⋯Au distances in 3. As shown in Figure 2c, the isopropyl groups of the ligand BIiPr of 3 are in the form of cis or trans due to disorder. At the interface with BIiPr, which contains isopropyl groups in the trans form, the distances between the gold atoms and hydrogen atoms pointing to the metal core are in the range from 2.660 to 2.707 Å. Surprisingly, hydrogen atoms of the two opposite methyl groups also interact with gold, with a minimum distance of 2.755 Å. At the interface with BIiPr containing isopropyl groups in the cis form, the H⋯Au distance can drop to 2.566 Å, which is much shorter than the sum of van der Waals radii of gold and hydrogen (2.86 Å). In addition, similar C(sp3)–H⋯Au interactions were also observed on the surfaces of newly emerged gold nanoclusters, such as [Au11(PPh3)7(BIiPr)Cl2]Cl, [Au13(BIBn)9Cl3](PF6)2, and [Au25(BIiPr)10Br7](Cl)(NO3) etc. (Figure 2d–f).35–37,42,43 It should be noted that, in addition to the C–H⋯Au interaction in a side-on mode, modes close to end-on can also be found on the surface of Au25, with a minimum H⋯Au distance of 2.723 Å. These observations are not coincidental. Previously, C(sp3 or sp2)–H⋯Au interactions were reported. For example, Tsutsumi and Prabusankar synthesized a carbene-gold complex and found such an interaction from a wingtip group to a metal atom (H⋯Au: 2.869 Å),50 which is very similar to our case. Structures with much more shorter hydrogen-gold distances were also reported by Balakrishna (2.495 Å)54 and Mobin (2.638 Å).55 In particular, Konishi et al. assembled a diphosphine-protected Au6 nanocluster with a C(sp2)–H⋯Au interaction (2.722 Å), and characterized it with 1H NMR.51–53 Although no similar shifts were observed in the NMR spectra of 2 nor 3, based on this set of structural data, weak or relative strong C(sp3)–H⋯Au interactions in 2 and 3 were detected. The difference between 2 and 3, and the strong interactions in gold nanoclusters, also imply that benzimidazolylidene-based ligands may stabilize the clusters more efficiently by forming such intramolecular interactions.
![Structure motifs and the corresponding schemes showing intramolecular C(sp3 or sp2)–H⋯Au interactions in 1 (a), 2 (b), 3 (c), [Au11(PPh3)7(BIiPr)Cl2]Cl (d), [Au13(BIBn)9Cl3](PF6)2 (e, BIBn = N,N′-dibenzylbenzimidazolylidene), and [Au25(BIiPr)10Br7](Cl)(NO3) (f). The unit of distances is Å. A top view of structure motifs containing pairs of opposite ligands in 2 and 3 is also attached, showing the parallel arrangement of two opposite imidazolylidenes or benzimidazolylidenes in each structure.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bcsj/94/4/10.1246_bcsj.20210060/2/m_20210060fig02cmyk.jpeg?Expires=1750380650&Signature=z6cP7gH4pm58uYA71YsQ1NWu7PRAikApjoVj8Xa2WYZVAGwQ0Tcofbpo92mSDjll2hVw6SFt7Mp2YtuSt8qm12sCkjh7IDxMdgZLHbT93GeINEjREQDOosw8xGsi6dObIGrGYdQ0OFu1VdnccO6rjIco5kDBqRzhQ5bnczUmAzvwMOxHdiZ8dss0jV3Iog6FXjNjW26DHos36WuLqnNMFPafIz8gAdItIJ5J-mhqzIy42CXWG~-ISYONeM038xQDJnUIZxwkU8F9yxDVmV0J~VbRZLzif7Rj4cy5h3quWLtrTp4koGHMDLx68Xyfpt8S8cNlr9-51MtZFWbeR~2Z3Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structure motifs and the corresponding schemes showing intramolecular C(sp3 or sp2)–H⋯Au interactions in 1 (a), 2 (b), 3 (c), [Au11(PPh3)7(BIiPr)Cl2]Cl (d), [Au13(BIBn)9Cl3](PF6)2 (e, BIBn = N,N′-dibenzylbenzimidazolylidene), and [Au25(BIiPr)10Br7](Cl)(NO3) (f). The unit of distances is Å. A top view of structure motifs containing pairs of opposite ligands in 2 and 3 is also attached, showing the parallel arrangement of two opposite imidazolylidenes or benzimidazolylidenes in each structure.
Another notable structural feature is the orientation adopted by each benzimidazolylidene moiety in this series of clusters. Unlike phosphine ligands, which have three adjustable substituents arranged in a propeller-like shape, carbene ligands usually coordinate to a metal via a flat heterocycle. As a result, carbene ligands can be placed differently from each other on the surface of metal clusters. In 2 and 3, we found that each pair of opposite ligands was nearly parallel to each other. Each pair of them shares approximately the same plane with the CAu4 motif in the C@Au6 cores. (Figure 2b, c)
3. Optical Absorptions and Luminescence
Compared to phosphine ligands, NHCs exhibit a stronger σ-electron donating capacity, but they are poorer π-electron acceptors. On the other hand, theoretical calculations have confirmed that metal-to-ligand charge transfer (MLCT) processes are common in phosphine-protected C@Au6 clusters.14,56 As a result, the coordination of NHCs can affect the electronic structures of clusters and therefore the light absorption and luminescence. Luminescence is one of the most attractive properties of gold clusters. Compared to the emission of organic molecules and metal compounds, the emission of gold clusters usually exhibits relatively high emission efficiency, large Stokes shift, long lifetime, and/or high photostability. These properties favor practical applications such as bioimaging, OLED, and etc.57,58 It has long been thought that the luminescence of gold complexes/clusters is primarily associated with intermolecular and/or intramolecular aurophilicity. However, it was found that replacing phosphine on the cluster with an NHC ligand can result in significantly different emissions. Furthermore, even small changes in NHC ligands can significantly alter the emissions.
Figure 3a shows the UV-vis absorption spectra of clusters 1, 2, and 3 in CH2Cl2. Unexpectedly, they show fairly similar absorption patterns in the range of about 300 to 400 nm with respect to energy. There was a slight red shift in the quartet bands/shoulders. For example, the peaks at 320 and 334 nm for 1 shifted to 329 and 336 nm for 2, and 336 and 343 nm for 3. These were assigned to metal-to-ligand transition (HOMO-1 to LUMO+1) with the help of TD-DFT. On the contrary, their absorption coefficients in this region are significantly different. The ε values are calculated to be 2.1 × 104 M−1cm−1 at 334 nm for 1, 5.2 × 104 M−1cm−1 at 336 nm for 2, and 9.5 × 104 M−1cm−1 at 343 nm for 3. This is because the π* orbitals of imidazolylidenes of 2 and benzimidazolylidenes in 3 are largely involved in the unoccupied molecular orbitals (e.g. LUMO, LUMO+1 and LUMO+2 etc.), but the former shows a much smaller conjugated system than the latter.
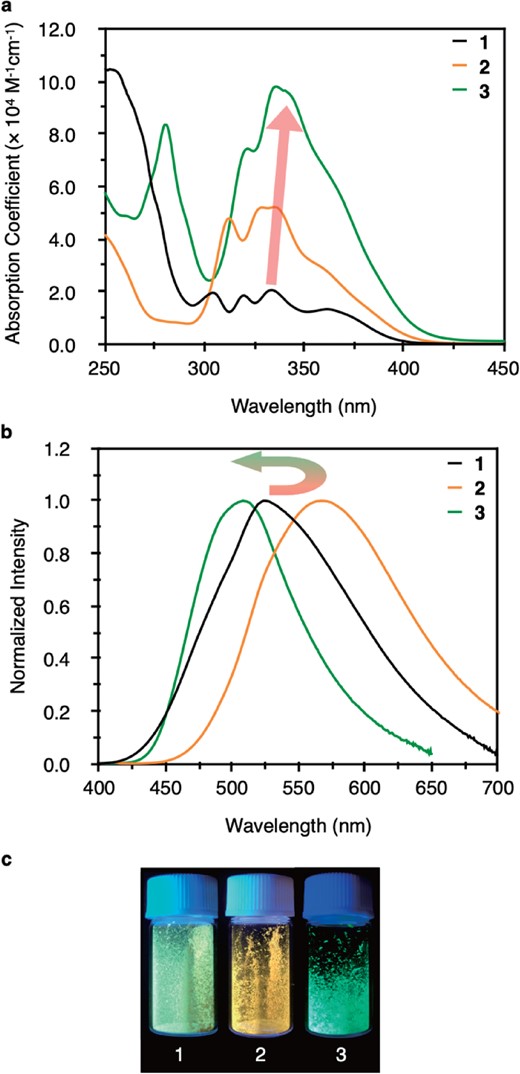
(a) UV-vis absorption spectra of clusters 1, 2, and 3 in CH2Cl2 at 293 K. (b) Emission spectra of 1, 2, and 3 in the solid state. (c) Photos of crystalline 1, 2, and 3 under 365 nm excitation.
Figure 3b, c shows the emission spectra of clusters 1, 2, and 3 in the solid state, and the photos showing the corresponding emission under 365 nm excitation. First of all, the apparent red shift involves the replacement of PPh3 with IiPr from 1 to 2. The maximum emission was found to be 525 and 567 nm, respectively. However, when a similar ligand BIiPr with a benzimidazolylidene moiety was used for 3, the emission shifted to deep green (λemmax = 509 nm). Clearly contrasting emission with a difference of about 60 nm between 2 and 3 suggests that the π-conjugated system of NHC ligands can significantly modulate the luminescence. We have long noted that the expanded π-system of NHC ligands may give different properties to the corresponding metal complexes, but for the first time, we could successfully adjust the emission of metal clusters based on the properties of ligands. These results indicate that not only different types of ligands (phosphine vs NHC) but also NHC ligands with different heterocyclic moieties (imidazolylidene vs benzimidazolylidene) can be used as a design factor to achieve fine-tuning of the emission of metal clusters.
4. Summary and Outlook
C@Au6 clusters are a series of gold clusters similar to diprotonized methane CH62+,59 but are experimentally separable and robust under ambient conditions. NHC ligands have been used to reconstitute hexagold clusters by forming a strong intramolecular C(sp3)–H⋯Au interaction on the surface. Interestingly and importantly, the unique photophysical properties of the metal clusters were found to change significantly maintaining the core structure, not only by changing the type of ligand (NHC or phosphine) but also by chemically modifying the homologous ligand (benzimidazolylidene or imidazolylidene). These results provide a new perspective on the study of metal clusters. Choosing the right organic ligands can be considered a common factor in application-oriented molecular design and synthesis of metal clusters.
For future directional aspects, promising opportunities lie in the engineering of carbene ligands. First of all, except for the classical NHCs, other types of carbenes including “abnormal” imidazolylidene (C4/5-imidazolylidene), triazolylidene, pyrazolylidene, oxazolylidene, thiazolylidene, isoxazolylidene, tetrazolylidene, and pyridylidene etc.60 can be employed to achieve fine-tuning of the photophysical properties of clusters, because they generally have less heteroatom stabilization of carbenic carbons, which gives them specific donor properties when coordinated to gold. Secondly, by introducing additional coordination sites, metal ions other than gold can be assembled in sequence. This leads to impeding the free rotation of the ligands, which causes nonradiative relaxation and emission quenching. The result is that clusters glow in solution, which may be useful for further applications such as bioimaging. Last but not the least, the functionalization of these homo and/or heterometallic clusters by a bottom-up approach or post-clustering modifications will have interesting and fruitful results. Future issues would include novel structures and coordination asymmetry,61 water-solubility, sensing/separation functions, and so on. Gold clusters with NHC ligands provide remarkable structural motifs that have emerged during the rapid development of the field of cluster chemistry.
Acknowledgment
This research was supported by JSPS KAKENHI Grant Nos. JP25810034 to H.U., and JP26248016 and JP16H06509 (Coordination Asymmetry) to M.S.
References

Zhen Lei
Zhen Lei graduated from Huazhong University of Science and Technology (HUST) with a B.S. in Applied Chemistry and B.A. in German in 2009. He got his Ph.D. of Science in Physical Chemistry from Xiamen University in 2015 under the supervision of Prof. Quan-Ming Wang. After a postdoc stay at Tsinghua University, he joined the Shionoya Lab at The University of Tokyo as a Project Researcher in Nov. 2018. He was promoted to Project Assistant Professor in Apr. 2020. His main research interests lie in ligand-protected coinage metal complexes/clusters/nanoclusters.

Xiao-Li Pei
Xiao-Li Pei was a PhD student in Prof. Quan-Ming Wang’s group at Xiamen University (2010–2015), China. She worked as a postdoctoral researcher in Prof. Antonio M. Echavarren group at the Institute of Chemical Research of Catalonia (ICIQ) (2016–2019), Spain. She continued her postdoctoral research in the Shionoya Lab since 2019 at The University of Tokyo, Japan. Activation of small molecules with metal clusters, catalytic and chiroptical properties of polynuclear coinage metal clusters are among her research pursuits.

Hitoshi Ube
Hitoshi UBE was born in 1980. He obtained his PhD from Tohoku University in 2008 under supervision of Prof. Masahiro Terada, and moved to the University of Tokyo as an assistant professor in the Shionoya Laboratory. His current research interests are metallo-architectures such as supramolecular metal complexes, metallo-molecular machines, metal clusters, and chiral-at-metal complexes.

Mitsuhiko Shionoya
Mitsuhiko Shionoya was born in 1958. He received his B.S. and M.S. from the University of Tokyo, and Ph.D. from Hiroshima University. In 1986, he accepted a position as Assistant Professor at Hiroshima University, and was promoted to Lecturer (1991) and Associate Professor (1994). In 1995, he was appointed a Professor at the Institute for Molecular Science in Okazaki, and in 1999, a Professor at the University of Tokyo. His research interests involve bio-inspired supramolecular array/space/motion and coordination asymmetry.

{kind=link}
{kind=link}
{kind=link}