





Abstract
The scope of cross-coupling reactions using tertiary carbon-centered radicals has expanded rapidly over the past decade. In this review, we outline the development of the cross-coupling reactions that involve persistent tertiary carbon-centered radicals as a powerful toolbox to synthesize molecules containing quaternary carbon(s) and/or tetra-substituted carbon(s). In particular, we focus on persistent tertiary carbon-centered radicals derived from carbonyl- or related compounds. We first describe the historical background and structural characterization of these radicals, and their reactivity/selectivity relationships. We then present selected recent examples of cross-coupling reactions involving tertiary carbon-centered radicals, categorized according to the originally proposed reaction mechanism, to showcase their versatile synthetic utility for structural diversification of small molecules.
1. Introduction
The use of carbon-centered radicals began more than a century ago, when Gomberg proposed that the reaction of trityl chloride 1 with zinc would produce a triphenyl methyl radical 2, leading to hexaphenyl ethane 3 (Scheme 1).1 Although the structure of the product was corrected to 4 in 1968,2 Gomberg’s report served as a foundation stone of free radical chemistry, showing that carbon can exist in a trivalent state.3 However, for many years, the reactivity and selectivity of free radicals were considered to be unpredictable and difficult to control. But, in the past decade, radical-based cross-coupling reactions have become increasingly important in general synthetic strategies.
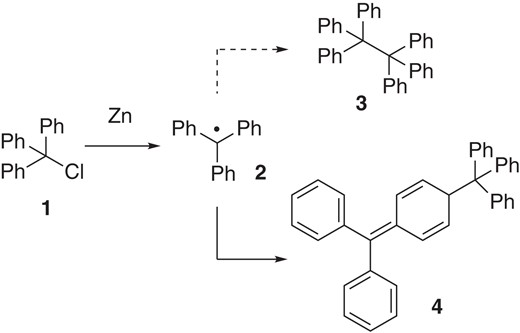
Discovery of triphenyl methyl radical.
A key factor underpinning the successful development of cross-coupling reactions involving tertiary carbon radicals has been the classification of radicals4 and the conceptualization of the persistent radical effect.5,6
Griller and Ingold first clearly differentiated “stable radicals”, “persistent radicals” and “transient radicals”.4 They proposed that the term “stable” should be used to specify a radical that is inert to air and moisture. Thus, a stable radical is isolable. The term “persistent” should be used to describe a radical that is long-lived relative to the methyl radical (t1/2 > 10−3 s). Short-lived radicals (t1/2 < 10−3 s), such as the methyl radical, are classified as “transient” radicals.
Another critical factor to understand the cross-selectivity in radical-based coupling using two different radicals is the equilibrium (i.e., radical recombination) between the persistent radicals and the corresponding dimer.6 The equilibrium between triphenylmethyl radical 2 and dimer 4 is an early example (Scheme 1).1 Since Fischer first rationalized the unprecedented cross-selectivity in radical-radical cross-coupling of a persistent radical and a transient radical,7 the persistent radical effect has been recognized as an underlying principle accounting for selective product formation in free-radical cross-reactions (Figure 1).5 As noted above, a transient radical (RT) is so reactive that it should be readily terminated in situ. Accordingly, the concentration of a transient radical in solution remains consistently lower than that of a persistent radical. In contrast, persistent tertiary carbon radicals can exist at a relatively high concentration, owing to their persistent nature. Even though persistent tertiary radicals inherently re-combine to form the corresponding dimer or radical-precursor, irreversible bond-formation with the transient radical proceeds kinetically, eventually leading to the generation of the cross-adduct with high selectivity.
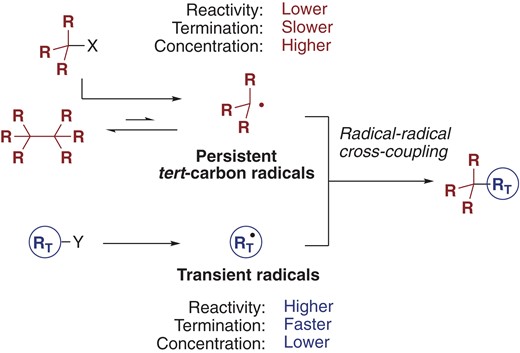
The persistent radical effect in the radical-radical cross-coupling of persistent tert-carbon radicals and transient radicals.
Historically, radical precursors (X or Y in Figure 1: halides, Barton esters, or xanthates) are activated with a stoichiometric amount of reagent for in situ preparation of radicals of interest.3 Currently, many synthetic strategies to generate radical species under mild conditions are available to achieve radical-based cross-coupling. Representative approaches include the use of (i) redox-active functional groups (esters, halides, carboxylic acids, alcohols, etc.) in the presence of a photoredox catalyst;8 (ii) reaction of epoxides with titanium reductants;9 (iii) metal-catalyzed hydrogen atom transfer of alkenes;10 and (iv) α-alkoxy- and acyltellurides.11 Recent reviews summarize how cross-coupling with tert-carbon radical is effective to construct all-carbon quaternary centers and/or tetra-substituted carbon centers in the total synthesis of complex molecules.5c,12,13 This reversibility between the dimer and the corresponding monomer radical is also the basis of dynamic covalent chemistry.14
With this background, we focus in this review on recent progress in the development of cross-coupling reactions starting from dimers or the corresponding hydrocarbons (X = H in Figure 1), with particular emphasis on the dimer-monomer radical equilibrium, as a strategy that offers superior atom economy.15 We will begin by briefly summarizing the characterization of the structure and persistent nature of persistent tert-carbon radicals (Section 2). In the early 1900s, the triphenyl methyl radical 2 was to some extent viewed more as a special case than as an example of a general class. Since then, however, the number of persistent radicals available for cross-coupling has increased dramatically (Figure 2). These radicals are not restricted to terphenyl radicals;16 currently, several classes of persistent radicals are known (Figure 2). These include radicals derived from oxindoles,17 benzofuranones,18 fluorenes,19 malononitriles,20 cyanoacetates,20 azlactones,21 and 1,4-oxazin-2-one.22 Most of them are stabilized by resonance and/or captdative effects. But, we should note that persistence is also associated with diamondoids, whose curious dimer structures arise from London dispersion forces (attractive Van der Waals interactions).23 In the following sections, we highlight the mechanistic differences between radical reaction (single-electron manifold) and ionic reaction (two-electron manifold), by focusing mainly on selected recent examples of cross-couplings starting from carbonyl or related compounds. Reactions are grouped according to the originally proposed mode of bond formation, i.e., radical-radical cross-coupling (Section 3), anion-radical coupling (Section 4), radical addition to olefins (Section 5), reductive coupling (Section 6), and umpolung (Section 7). We have used new compound numbers for each figure and scheme, even if similar structures had appeared previously, in order to make it easier for readers to follow the main text without referring back to previous sections.
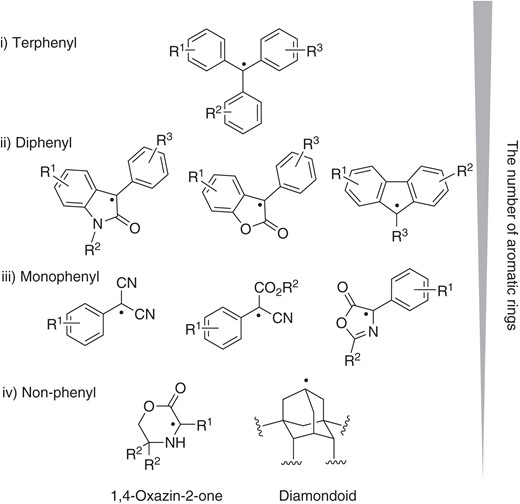
The structures of some persistent radicals.
2. Structure and Persistence of Tert-Carbon Radicals
2.1 Dynamic Equilibrium of Persistent Radical Derived from a Benzofuranone (HP-136)
Since synthetic chemists are mainly interested in the discovery of new substrate combinations, radical-based bond-forming processes have often been discussed without classifying the radicals or considering the persistent radical effect. One reason for this may have been that radicals were considered to be undetectable as active species due to their short lifetimes, though this is not the case for persistent radicals. Even though the lifetimes of tert-carbon radicals are generally short (only milliseconds), the generation of these radicals can be readily confirmed by EPR22 or UV/vis spectroscopic analyses,18 or NMR analyses.23c The detectability of persistent tert-carbon radicals stems from the elongated C(sp3)–C(sp3) bond in the homo-dimer, which is often longer than the typical C(sp3)–C(sp3) bond [1.530(15) Å].24 Since the energetic barrier to recombination is only slightly higher than that of homolysis, the dynamic equilibrium between the dimer and the monomer radical can be altered by changing the temperature of the solution.
In 2000, Scaiano and co-workers characterized the persistence of the radical 6 derived from commercially available benzofuranone 5 (HP-136),18a which was originally developed for the protection of polymers (Scheme 2). They detected the existence of the persistent radical by laser-flash photolysis (LFP) upon exposure of 5 to a stoichiometric amount of di-tert-butyl peroxide (DTBP). Importantly, little decay of the UV/Vis absorption (500–600 nm region), which may be associated with generation of 7, was observed in the millisecond time range, even in O2-saturated benzene under neutral conditions. In contrast, in similar experiments using diphenylmethyl radical (Ph2C•H) prepared from diphenylmethane (Ph2CH2) and a stoichiometric amount of di-tert-butyl peroxide, the Ph2C•H signal was totally quenched. Considering that triphenylmethyl radical reacts with O2 to form an isolable peroxide, as Gomberg reported,1 these results highlight that 6 is considerably more stable than Ph2C•H. The structure of 6 resembles that of triphenylmethyl radical 2, but insights into the dynamic behavior of the persistent radical 6, containing the carbonyl group, have considerably influenced exploration of the cross-couplings described in the following sections. In particular, the inertness of 6 to O2 permits simple operation without the need for a glovebox.
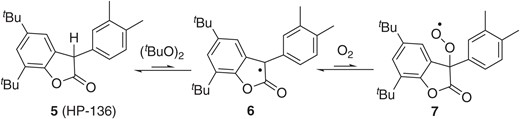
Persistence of 6.
2.2 Substituent Effect on Cleavability of the σ-Bond in Dimers
As already mentioned, the dimers prepared by oxidative dimerization of monomers (Scheme 3) serve as precursors to generate the corresponding persistent monomer radicals. The CCDC (The Cambridge Crystallographic Data Centre) database includes dimers derived from 2-oxindoles, and we found that the σ-bond (1.565–1.597 Å) in 3-alkyl-substituted dimers is shorter than that in 3-aryl-substituted dimers (1.586–1.610 Å). It is noteworthy that in the oxidative dimerization of 3-alkyl-substituted monomers 8, the diastereoselectivity is low (dl-9/meso-10 = ∼1/1). On the other hand, meso-10 is a major product in the case of the oxidative dimerization starting from 3-aryl-substituted monomers. Sohtome, Sodeoka and co-workers recently uncovered the reason why the 3-aryl-substituted monomers give the corresponding meso-dimers, based on structural characterization of a dimer (P = Boc, R1 = Ph, R2 = Me) in both the solid and solution states, together with DFT (density functional theory) calculations.26a They revealed that the dimer bearing a Ph group at the C(3) position has a lower BDE (bond dissociation energy) of the C(3)–C(3′) bond than does the Me-substituted dimer. The phenyl group at the C(3) position is crucial to stabilize the radical. They also concluded that the C(3)–C(3′) bonds in dimers having an aromatic group at the C(3) position are readily cleavable even at ambient temperature, and meso-10 is energetically more stable in the equilibrium due to CH/π interactions (Figure 3).
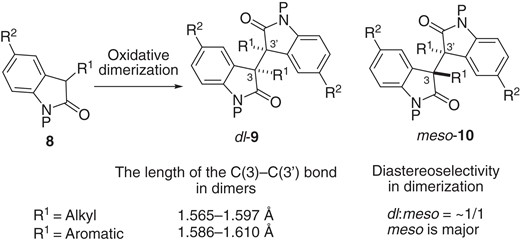
Oxidative dimerization of 8: difference between 3-alkyl-substituted dimers and 3-aryl-substituted dimers.
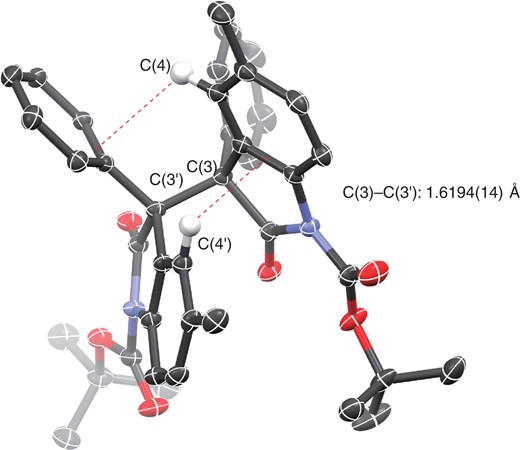
CCDC 1499431. For the sake of clarity, only the two hydrogens at C(4) and C(4′) contributing to CH/π interactions are shown. Red broken lines show CH/π interaction.
2.3 Bond Dissociation Energies of Dimer and Monomer
The cross-coupling reactions from the dimer and monomer exhibit inherently different outcomes. Recently, Kozlowski and co-workers systematically obtained the BDE values of a set of dimers and monomers by means of DFT calculations (Figure 4), aiming to understand their availability in oxidative cross-coupling with alkylarenes in the presence of catalytic Pd(OAc)2/stoichiometric oxidant (see Schemes 20 and 21 in Section 6).20 They found a good correlation between the BDE values and availability as substrates under the conditions that they developed. The substrates possessing BDE values of C–H in the monomer in the range of 60–70 kcal/mol (highlighted blue in Figure 4) were effective in their reactions. BDE values may be a good indicator for the initial screening of substrates for other classes of reactions as well.
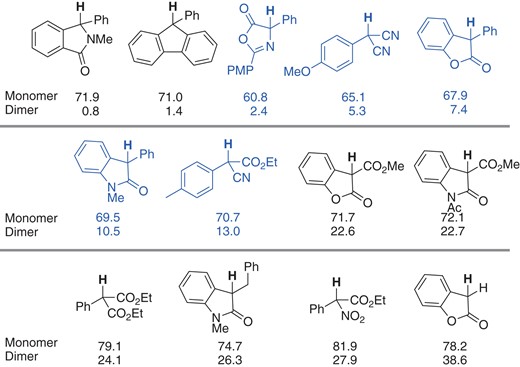
BDE values (kcal/mol) of C–H in monomers and C–C in meso-dimers calculated at the UB3LYP/6-31G(d) level.
3. Radical-Radical Cross-Coupling
3.1 Homolysis under Thermal Conditions
In addition to tert-carbon radicals, the corresponding tert-carbon anions (Section 6) or tert-carbon cations (Section 7) can participate in bond-forming processes, depending upon the reaction conditions, as we will discuss later. Accordingly, careful mechanistic control experiments are required in order to determine which active species is the key to achieve the bond-forming process of interest. The radical clock approach has been widely used to obtain indirect evidence as to whether a putative radical participates in the bond-forming process.25 Radical trapping to capture the hypothetical radical species is also recognized as an alternative approach. Addition of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl), which is a stable radical according to Ingold’s classification,4 has been often considered as a first choice. But, we should note that the combination of a persistent radical and a stable radical is not a good pairing for radical-radical cross-coupling, though a persistent radical can react with a transient radical, as illustrated in Figure 1. In 1975, Koch and co-workers clearly demonstrated that dimer 11, derived from 5,6-dihydro-3,5,5-trimethyl-1,4-oxazin-2-one, can serve as a radical precursor in a pioneering study entitled “An Unusually Weak Carbon–Carbon Single Bond”.22 They reported radical-radical cross-coupling between 11 and AIBN (azobisisobutyronitrile). Although only one substrate combination (11 and AIBN) was used and the persistent radical effect was not considered, the result showcases that the persistent radical can be efficiently captured by the transient radical derived from AIBN, providing a synthetic route to 12 having adjacent tetra-substituted carbon and quaternary centers (Scheme 4).
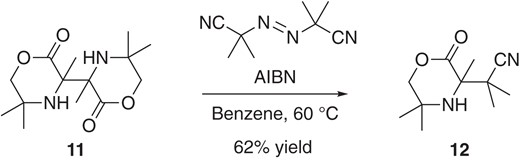
Cross-coupling strategy using a dimeric substrate.
The utility of the radical-radical cross-coupling reaction with azo-compounds was significantly expanded by Sohtome and Sodeoka, on the basis of detailed mechanistic studies as described in Section 2.2.26a They synthesized a series of dimers 13 (X = NBoc, NMe, NPh and NH) and 14 (X = O), and applied them in radical-radical cross-coupling reactions with a range of azo compounds 15, facilitating the synthesis of congested heterocycles 16 and 17 having vicinal all-carbon quaternary centers.26b
The two examples shown in Schemes 4 and 5 required a stoichiometric amount of chemical oxidant, such as K3[Fe(CN)6] under basic conditions at high temperature for preparation of the requisite dimers. Sohtome, Sodeoka and co-workers recently developed a formal aerobic oxidative cross-coupling reaction starting from monomers 18 for the synthesis of 19 (Scheme 6).26c The reaction sequence involves aerobic Pd(II)-catalyzed oxidative dimerization of 18,27 followed by cross-coupling of 20 with azo compounds 21. The key to achieving this single-flask sequential transformation is the chemoselectivity of their Pd(II) catalyst, which effectively promotes the catalytic dimerization of 18 under aerobic conditions, but does not participate in radical-radical cross-coupling of 20 and 21.
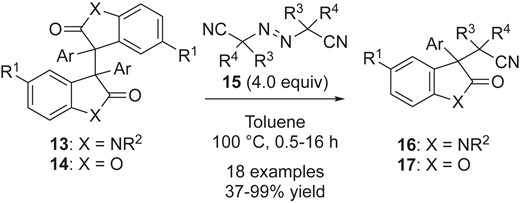
Radical-radical cross-coupling of various oxindoles 13 and benzofuranones 14 with azo compounds.
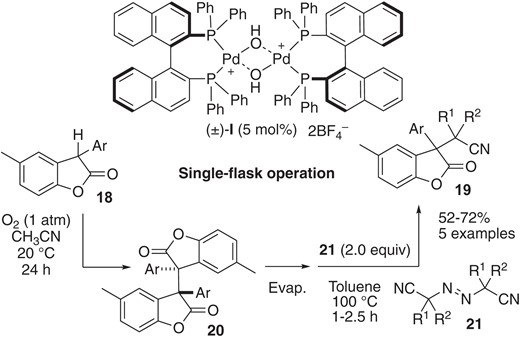
Sequential catalytic oxidative dimerization and cross-coupling starting from 18 using (±)-Pd(II)-µ-hydroxo complex I.
3.2 Hydrogen Atom Transfer
Persistent tert-carbon radicals can be generated from not only dimers, but also the corresponding monomers through HAT (hydrogen atom transfer) processes with the use of oxidant. For example, Wei and co-workers reported peroxidation reactions of oxindoles 22 using tert-butyl hydroperoxide (TBHP) to synthesize the desymmetric peroxide 23 (Scheme 7).28 This could be categorized as a cross-dehydrogenative coupling (CDC) reaction.29 In their proposed mechanism, TBHP serves as both a HAT (hydrogen atom transfer) mediator and a peroxidation reagent. The reaction would be initiated by thermal homolysis of TBHP to generate two distinct radicals: tBuO• radical and •OH radical. They proposed that the tBuO• radical abstracts the hydrogen atom from oxindoles 22 to form the persistent tert-carbon radicals 24, while the •OH radical would react with another TBHP to form the transient tBuOO• radical. Subsequent cross-coupling between 24 and the tBuOO• radical would afford the peroxides 23. The use of transition metal catalysts such as Co30a and Cu30b provides alternatives to design much milder reaction conditions, wherein the transition metals could be a key driver of catalytic redox cycles to enhance the generation of tBuO• and tBuOO• radicals.
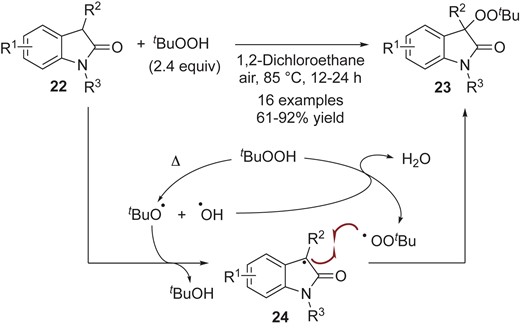
Metal-free thermal peroxidation of oxindoles by TBHP.
Persistent tert-carbon radicals were also coupled with carbon-centered transient radicals generated via HAT. Kozlowski and co-workers reported the CDC reaction of oxindoles 25 with acetonitrile having a relatively high BDE using di-tert-butyl peroxide (DTBP), giving the corresponding cross-adduct 26 (Scheme 8-i). In the CDC reaction of fluorenes 27 with alkyl nitriles 28 (Scheme 8-ii), they found that addition of pivalic acid (PivOH) slightly improves the yield of 29.19 In this reaction, the tBuO• radical may abstract hydrogen from fluorenes 27 and alkyl nitriles 28 to generate a pair of persistent and transient radicals. Subsequent radical-radical cross-coupling reaction affords the cyanoalkyl fluorenes 29. The reaction was completely inhibited by addition of radical scavengers such as TEMPO and 2,6-di-tert-butyl-4-hydroxytoluene (BHT). The corresponding adducts of the scavengers to acetonitrile were observed by ESI-MS analysis, while no cross-adduct from fluorene 27a to TEMPO was detected (Scheme 8-iii). These results again highlight that the combination of persistent and transient radicals is favorable to promote radical-radical cross-coupling. The authors suggest that the persistent fluorene radicals are in equilibrium with their dimeric forms.
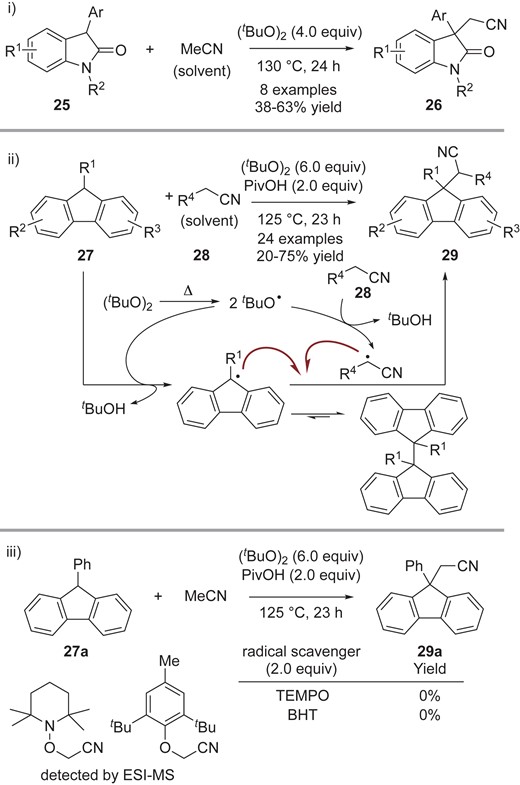
Cyanomethylation of i) oxindoles and ii) fluorenes; iii) control experiments with radical scavengers.
Cheng, Liu, Li and co-workers developed an oxidative cross-coupling reaction of oxindoles 30 and benzofuranones 31 with iodo-acetylenes 32 under basic conditions, giving the corresponding cross-coupling adducts 33 and 34 with an alkynyl group at the C(3) position (Scheme 9).31 The proposed reaction sequence is initiated by homolysis of iodoalkyne 24 to generate the transient alkynyl radical and iodine radical species. If the resulting iodine radical can abstract the hydrogen at the C(3) position in the starting monomer (30 or 31), the persistent radicals should be generated. Subsequent radical-radical cross-coupling forms the C(sp3)–C(sp) bond, giving the corresponding cross-coupling products 33 and 34.
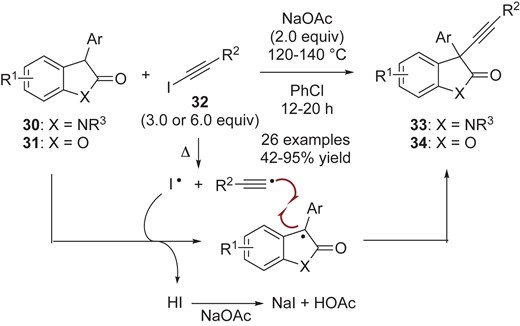
Oxidative alkynylation of oxindoles 30 and benzofuranones 31.
3.3 Single-Electron Transfer under Basic Conditions
Oxindoles 35 and benzofuranones 36 are prone to form enolates under basic conditions. These enolates can be oxidized to form persistent tert-carbon radicals, which undergo cross-coupling reactions with transient radicals. Li, Cheng and co-workers presented an example of this class of reaction. They developed an amination reaction of oxindoles 35 and benzofuranones 36 with sulfonic carbamate 37 bearing a dinitrophenylsulfonyloxy (ODNs) group (Scheme 10).32 On the basis of cyclic voltammetry (CV) measurement of substrates [benzofuranone enolate (Ar = Ph, R1 = H, X = O): −0.54 V and 37a (R2 = Bn): −1.0 V in acetonitrile/0.10 M n-Bu4NPF6], they proposed that the radical anion would be generated via single-electron oxidation of enolates generated from 35 and 36 with sulfonic carbamate 37. The putative radical anion would collapse to generate a transient nitrogen-centered radical as a key radical intermediate with the release of ODNs anion, as MacMillan had proposed.33 Finally, radical-radical cross-coupling reaction would result in C(sp3)–N bond formation to give 38 and 39. The authors also mentioned a chain-propagation mechanism involving reaction of the nitrogen-centered radical with another enolate as an alternative mechanism.
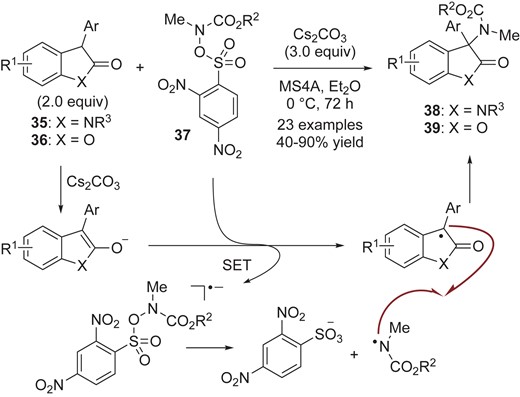
Amination of oxindoles 35 and benzofuranones 36 with sulfonic carbamate 37 via single-electron transfer.
3.4 Aerobic Conditions
As discussed in Section 2.1, the inertness of the persistent tert-carbon radicals derived from benzofuranones to O218a makes it easy to explore reactions under aerobic conditions. Wei and coworkers reported a nitration reaction of oxindoles 40, using •NO2 as a transient radical (Scheme 11).34 The key NO2• radical could be obtained by treatment of tBuONO under aerobic conditions; after homolysis of tBuONO, the tBuO• radical would abstract a hydrogen atom from oxindole 40 to generate persistent tert-carbon radical 41, while •NO would be oxidized by aerobic oxygen to generate transient •NO2.35 Subsequent radical-radical cross-coupling would afford the oxindole 42 containing the nitro group at the C(3) position.

Nitration of oxindoles 40 under aerobic conditions.
3.5 Catalytic Conditions
3.5.1 TEMPO
As we illustrated in the case of the catalytic dimerization of benzofuranones in Scheme 6, the combination of catalytic technology with the use of persistent radicals is important not only to further expand the available substrate scope, but also to achieve catalyst-controlled reaction outcomes. Wei and co-workers reported a TEMPO-catalyzed aerobic hydroxylation reaction of oxindoles 43 to give the alcohol 44 (Scheme 12).36 Although the precise mechanism was not established, the generation of a persistent oxindole radical and subsequent radical-radical coupling with O2 seem likely to be key steps in this reaction. Interestingly, the O–O bond derived from O2 is cleaved even under base- and reductant-free conditions.
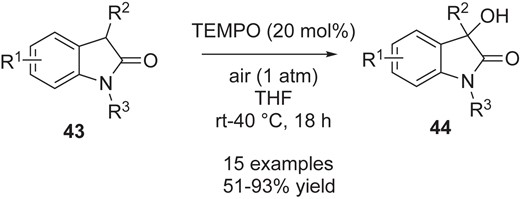
TEMPO-catalyzed hydroxylation of oxindoles by molecular oxygen.
3.5.2 Cooperative Photoredox and Chiral Brønsted Acid Catalysis
As described in the previous sections, the range of transient radicals capable of coupling with persistent radicals has been significantly extended in recent years. However, absolute stereo-control in radical-radical cross-coupling has remained elusive.37 The lack of enantioselective variants in this reaction class might be due to the relatively short lifetime of the radical species. In 2018, Knowles and co-workers reported an enantioselective cross-coupling of tryptamine 45 for the synthesis of various substituted pyrroloindolines 46,38 using an Ir photoredox catalyst and chiral phosphate (Scheme 13).39 By systematic optimization, they established that the Cbz group in 45 and chiral H8-TRIP phosphate (47) are essential to obtain high enantioselectivity under their conditions. In terms of reactivity, the addition of TIPS-EBX (48), an iodonium oxidant, is critical to reduce the formation of TEMPO-H, which interferes with the desired cross-coupling, perhaps owing to weak BDE. They proposed that TIPS-EBX (−1.12 V vs SCE) may associate with the excited state of the Ir(ppy)3 photocatalyst (*−1.73 V vs SCE) to enhance the redox cycle, reducing the generation of TEMPO-H. Thus, in their proposed mechanism, Ir(IV) species, which could be generated from photoexcited Ir(ppy)3 by quenching with TIPS-EBX, would promote single-electron oxidation of tryptamine 45 in combination with phosphate anion via proton-coupled electron transfer (PCET). The generated indole radical cation 49 would be trapped by TEMPO, enabling the radical-radical cross-coupling reaction to proceed in the chiral environment constructed by the phosphate anion. Subsequent diastereoselective cyclization would finally afford pyrroloindoline 46. The authors showed that the lone pair on the nitrogen atom of the TEMPO unit in 46 is readily oxidized, and the C–O bond of the resulting radical cation is significantly weakened to undergo mesolytic cleavage. They applied this principle to a wide range of nucleophilic substitution reactions starting from 46.

Enantioselective synthesis of pyrroloindolines 46 via PCET activation of tryptamines 45.
In 2018, Jiang and co-workers reported the development of a redox-neutral, enantioselective, radical-radical cross-coupling reaction between activated ketones (isatins 50 or 1,2-diketones 51) and N-aryl glycines 52 by combining organophotoredox and chiral Brønsted acid catalysis (Scheme 14).8,40a In the developed procedure, two distinct catalysts, dicyanopyrazine (53: DPZ)41 and a chiral phosphoric acid (CPA) 54, are synergistically used to control the reactivity and selectivity, enabling access to a wide range of chiral 1,2-amino tertiary alcohols (55 and 56) with high enantioselectivity. The conditions for isatins 50 are rather simple, while TBPB (tetra-n-butylphosphonium bromide) and Na2S2O4 are added in the cross-coupling of 1,2-diketones 51. However, these two additives only slightly affect the enantioselectivity. In the proposed redox-neutral cycle, photoexcited dicyanopyradine (DPZ*) catalyst would oxidize N-aryl glycine 52 in a SET process to generate α-amino radical 57 with concomitant release of CO2 and a proton. Meanwhile, the radical anion DPZ•− thus formed would reduce 1,2-diketones 51 to form ketyl radical 58. The success of these enantioselective reactions may be associated with the stronger basicity of the ketyl radicals rather than the corresponding ketone, as originally suggested by Knowles.42 Following the widely accepted dual activation mechanism,39 the author also proposed a plausible hydrogen-bonding network with CPA 54, in which 57 and 58 are fixed in close proximity, accounting for the observed high enantioselectivity. Jiang and co-workers further expanded this DPZ/CPA combined catalysis to achieve enantioconvergent aminomethylation of 3-chlorooxindoles.40b
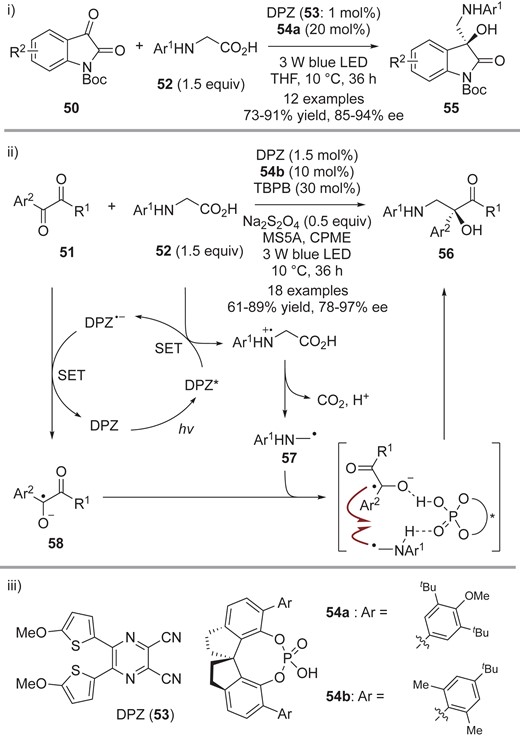
Enantioselective aminomethylation of i) oxindoles 50 and ii) 1,2-diketones 51. iii) Structures of DPZ (53) and chiral phosphoric acids 54.
In 2019, Jiang and co-workers further developed a chiral Lewis acid-catalyzed cross-coupling of isatins 59 and tolyl derivatives 60, driven by visible light (Scheme 15-i), affording chiral tert-alcohols 61.43 In their report, they described their working hypothesis, which involves i) photo-irradiation of isatins 59 to generate the triplet excited n–π* state 62 upon irradiation with visible light (as occurs in the Peterno-Büchi reaction),44 ii) SET oxidation of tolyl derivatives 6045 by the excited isatins 62, which have a high redox potential, to generate the dioxindolyl-like radicals 63 and benzyl radicals 64, and iii) radical-radical cross-coupling between metal-coordinated persistent radicals 63 and transient benzylic radicals 64. Extensive screening revealed that the combination of La(OTf)3 and a PyBox 58 gave the best yield and enantioselectivity. The authors also showed that dimer 65a, which is a by-product in the alkylation reaction, afforded the alkylated product 61a in 73% yield with 90% ee under the developed reaction conditions with stoichiometric amounts of La(OTf)3 and a PyBox 58 (Scheme 15-ii).
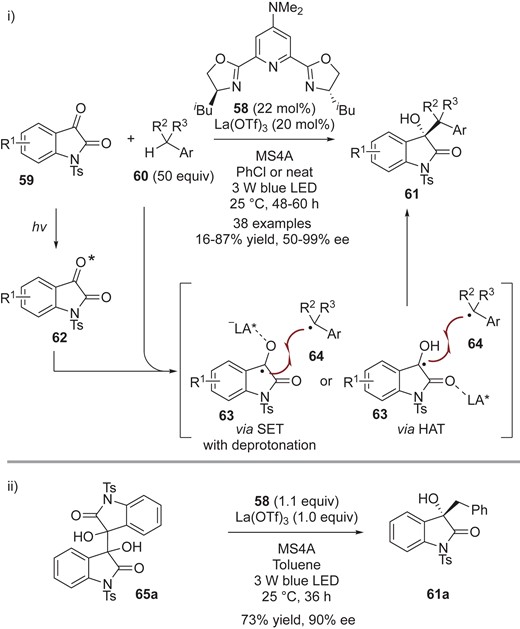
i) Enantioselective cross-coupling of isatins 59 with tolyl analogs 60. ii) Enantioselective cross-coupling of dimers 65a with toluene.
3.5.3 Copper-Enabled Cross-Coupling of Two Transient Radicals
While highly selective radical-radical cross-coupling reactions between persistent radicals and transient radicals have been developed, driven by the persistent radical effect, cross-coupling reactions between two different transient radicals are still quite challenging. To achieve this, Huang, Gao, Lei and co-workers discovered an attractive synthetic strategy to stabilize one transient radical (i.e., prolong its lifetime) in the course of development of catalytic aerobic cross-coupling of thiols 66 and azo-compounds 67 to synthesize α-thionitriles 68 (Scheme 16).46 They reported that treatment of thiol 66 with Cu(OTf)2 gave the complex 69, which could be detected by means of X-ray absorption fine structure (XAFS) and electron paramagnetic resonance (EPR) spectroscopy. These investigations provided evidence that the thiyl radical coordinates to the Cu(I) center, and its oxidation potential is increased. This formal persistent radical 69 would then undergo selective radical-radical cross-coupling with the transient α-nitrile radical in line with the persistent radical effect, affording α-thionitriles 68. Cu(I) should be oxidized to Cu(II) by O2.
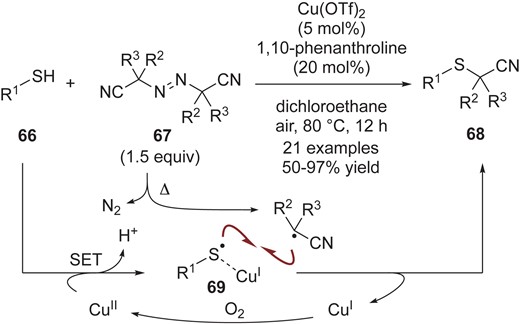
Cross-coupling of thiophenols 66 and azo-compounds 67.
4. Anion-Radical Coupling
4.1 Homolytic Aromatic Substitution under Thermal Conditions
In Section 4, we focus on anion-radical coupling, in which interaction between the SOMO of the radical and the HOMO of the nucleophilic substrate is a key driver of cross-selectivity.47 The anion-radical mechanism has been widely discussed in connection with the oxidative cross-coupling of two different phenols.48 However, application of this strategy to radical species other than phenoxyl radicals is rare. Quite recently, Sohtome, Sodeoka and co-workers developed regiodivergent oxidative cross-coupling reaction of dimers 70 (derived from oxindoles and benzofuranones) and 4-substituted catechols 71 (Scheme 17).49 The C(6)-selective oxidative adducts 72 are the major product under catalyst-free conditions (Scheme 17-i), while the C(5) products 73 were selectively obtained when binuclear (±)-Pd(II)-BINAP complex I was used (Scheme 17-ii; the structure of I is shown in Scheme 6). On the basis of mechanistic control experiments, they concluded that the homolytic aromatic substitution mechanism,50 in which the persistent radical reacts directly with catechols or Pd(II)-catecholates, is the regio-determining step. Subsequent oxidative aromatization affords the oxidative cross-coupling adducts 73. The proposed reaction sequence is significantly different from previously reported oxidative cross-coupling reactions of phenols; typically, the reaction is initiated by the generation of phenoxyl radical from the corresponding phenol regardless of the coupling partner. They also presented a unique energetic profile for the Pd(II)-catalyzed pathway, involving catalyst-enabled endothermic carbon–carbon bond formation (energetically uphill) and exothermic aerobic oxidative aromatization (energetically downhill), leading to a net exothermic process that affords the kinetically controlled C(5) adducts 73.
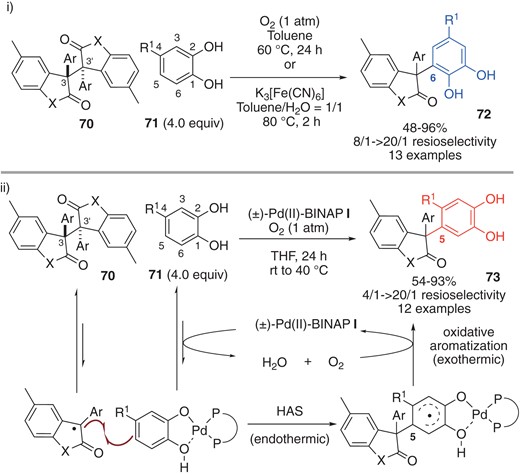
Regiodivergent oxidative cross-coupling of 4-substituted-catechos.
4.2 SET of the Electron–Donor–Acceptor Complex
Xia and co-workers reported a new procedure for the cross-coupling of 74 with aryl halides 75 to synthesize 3,3-disubstituted oxindoles 76, featuring the formation of electron–donor–acceptor (EDA) complexes without the use of transition metals or photoredox catalysts (Scheme 18).51 The reaction was designed based on their working hypothesis that oxindole enolates 77, which are strong electron donors, could serve as donors for the formation of EDA complexes with aryl halides 75. Indeed, they successfully captured the generation of the putative EDA complexes 79 by means of UV-vis analysis. The absorption spectra of a mixture of 74a (R1 = H, R2 = Me and R3 = Me), iodobenzene (75a) and CsOH showed a drastic red shift. Importantly, when they examined the reaction of 74a with 75a in the presence of CsOH by using a 45 W white LED lamp, equipped with a band-pass filter at 532 nm, the reaction efficiency was not altered. Based on the spectroscopic evidence that the enolate generated from 74a with CsOH is unable to absorb at this wavelength, they proposed that the EDA complex 79 acts as a photosensitizer.52 SET induced with a household compact fluorescent light (CFL) then generates key intermediates, the persistent radical of oxindole 80 and the aryl radical anion 81. The radical anion 81 should be transformed into aryl radical 82 via fragmentation with concomitantly release of halide anion (X−). In their mechanistic scenario, the electron-rich enolate (anion) 77 is a good coupling partner with electrophilic aryl radical 79 for cross carbon–carbon bond formation. Based on the CV analysis, they proposed that the ketyl radical 83 participates in the SET process with aryl halide 75 to give the cross-coupling adduct 76, with concomitant generation of the radical anion 81. The possibility of cross radical-radical coupling reaction between the persistent tert-carbon radicals 80 and aryl radical 82 cannot be ruled out.
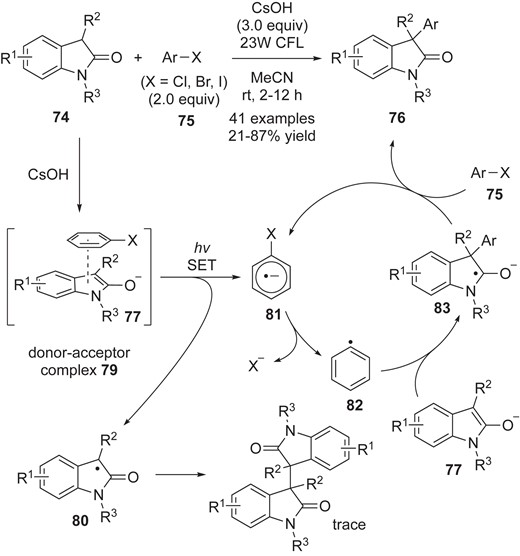
Arylation of oxindoles via visible-light-promoted electron transfer.
5. Radical Addition to Olefins
Gade and co-workers reported allylation of oxindoles 84 with allyl halide 85 for the synthesis of 86, using a transition metal and (S)-boxmi [bis(oxazolinmethylidene)isoindolate] (87) (Scheme 19).53 Intriguingly, they showed that the reaction pathways can be tuned by suitably selecting the transition metal. Upon treatment with Cu(OTf)2, the persistent tert-carbon radical 88 could be oxidatively generated from oxindole 84 with concomitant formation of Cu(I). The resulting radical would react with 85, and then the radical 89 should be terminated by halogen abstraction by Cu(I). They also confirmed that the oxindole dimer is converted to the product 86 at ambient temperature. On the other hand, the use of Zn(OTf)2 results in a switch to the ionic pathway to give the products 86 with high enantioselectivity (94–98% ee). These results suggest that the copper–87 complex could not coordinate to the persistent tert-carbon radical 88 even though Cu(II) should be a trigger for the generation of 88.
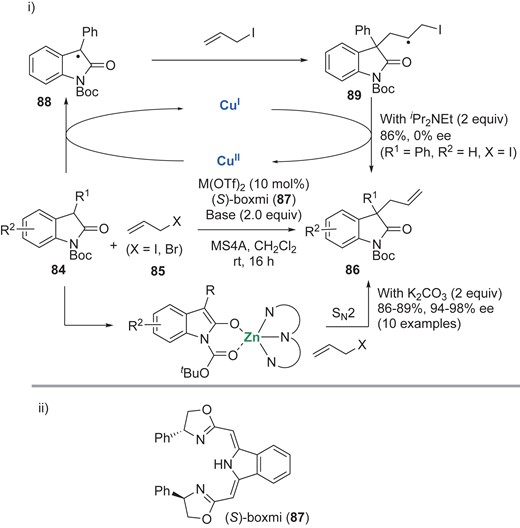
i) Catalytic allylation of oxindoles 84 with allyl halides 85, using Cu and Zn. ii) The structure of (S)-boxmi (87).
6. Reductive Coupling
As we described in Sections 3, 4 and 5, radicals provide a complementary strategy to ionic reactions. Another powerful approach that has recently been developed is the merger of persistent radicals and the reductive carbon–carbon bond-forming process (two-electron manifolds) by employing transition metal catalysis.
In 2015, Curto and Kozlowski reported Pd-catalyzed CDC reactions with azlactones 89, featuring C(sp3) C–H activation of the terminal position of the tolyl analogs 90 (Scheme 20).21a This reaction is synthetically attractive, generating the masked α,α-disubstituted α-amino acids 91. Extensive optimization studies led to the identification of two distinct reaction conditions, with a stoichiometric amount of Pd(OAc)2 (Scheme 20-i, Conditions A) and with a catalytic amount of Pd(OAc)2 (Scheme 20-i, Conditions B). By adding 2,6-dimethylbenzoquinone (2,6-DMBQ) as an oxidant, together with PivOH and activated carbon, they successfully reduced the necessary loading of Pd(OAc)2 to 10 mol%. Importantly, the dimer 92, which was detected in their investigations, serves as the substrate in the reaction with toluene under Pd-catalyzed conditions even without adding 2,6-DMBQ (Scheme 20-ii). These results indicate that once the dimer 92 is generated in situ, the redox-neutral cross-coupling takes place. Considering that deuterium scrambling occurred in the reaction of deuterated ethylbenzene with Pd(OAc)2 under the developed conditions, they proposed that the key intermediate 93 should be sequentially generated by Pd-metalation, β-hydride elimination, and migration of Pd to the terminal position of the ethylbenzene (Scheme 20-iii). There are two mechanisms that can account for the carbon–carbon bond-forming reaction, with two putative intermediates, 92 and 93. One is the oxidative addition/reductive coupling pathway that involves the Pd(IV) intermediate 94. The other is the metathesis path from 92 and 93. In both pathways, in addition to the cross-coupling adduct 91b, 95 having a Pd–carbon bond should be generated. Acetic acid may enhance the regeneration of Pd(OAc)2 from 95 with concomitant release of the monomer 89, which could be oxidatively converted into the corresponding dimer 92 in the presence of oxidant.
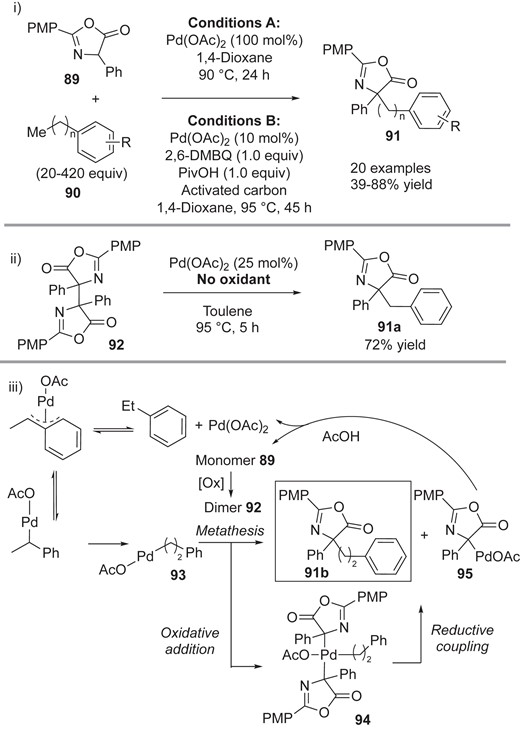
i) Pd-catalyzed CDC reaction of azlactones 89 with tolyl derivatives 90 (n = 0–3). ii) The reaction of dimer 92 and toluene. iii) Plausible pathways.
Kozlowski and co-workers successfully expanded the scope of radical precursors, including malononitriles 96/cyanoacetates 97 and cyclic oxindoles 98/benzofuranones 99, in the CDC reaction of tolyl analogs 100 (Scheme 21).20 For malonitrile 96, similar conditions to those used in their previous work (conditions B, Scheme 20-i) are available. On the other hand, in the reaction with cyanoesters 97, oxindoles 98, benzofuranones 99, the selection of a suitable oxidant is important to attain high yield; they found that K2S2O8 is a better oxidant than 2,6-DMBQ. With this slight modification, they obtained a range of cross-coupling adducts 101–104. Together with these extensive experimental investigations, they also computed the BDE values of dimers and monomers, as shown in Figure 4, aiming to examine the correlation of BDE value with reactivity. In terms of monomer BDE values, the available substrates for the CDC reactions have BDE values in the range of 60–70 kcal/mol. Those BDE values are smaller than that of toluene (90 kcal/mol), supporting the idea that Pd(OAc)2 plays an important role in the C–H activation shown in Scheme 20-iii. As regards dimer BDE values, they found that substrates with BDE values outside the range of 2–15 kcal/mol are not suitable for cross-coupling with tolyl analogs. The dimers with higher BDE values did not afford cross-adducts, even though dimer formation occurs. These results suggest that such rather stable dimers could not form the proposed Pd(IV) intermediate 94 shown in Scheme 20-iii.
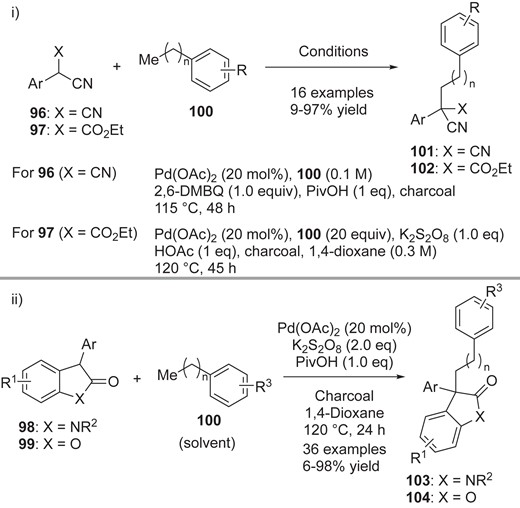
Pd-catalyzed cross-dehydrogenative coupling of i) malononitriles 96/cyanoacetates 97 and ii) oxindoles 98/benzofuranones 99.
In 2019, Qiu, Kambe and co-workers reported Cu-catalyzed CDC reaction of benzofuranones 105 with a variety of heteroarenes 106, leading to 3,3-diaryl benzofuranones 107 (Scheme 22).54 On the basis of a series of mechanistic investigations, including the kinetic isotope effect, radical trapping experiments, and control experiments involving removal of the catalyst or a reagent from the optimized conditions, they concluded that dimer 108 is the key intermediate or a resting state in the developed reaction. Their proposed mechanism involves a single-electron Cu(I)/Cu(II)/Cu(III) catalytic cycle. Cu(I)–Br should react with both benzofuranones 105 and Ar2–H 106; benzofuranones could be cooperatively oxidized to persistent tert-carbon radical 109 by Cu(I)–Br and K2S2O8, while Ar2–H 106 reacts with Cu(I)–Br and oxidant to generate Ar2Cu(II)Br. The resulting benzofuranone radical 109, which is in equilibrium with the corresponding dimer 108, would react with Ar2Cu(II)Br to form a Cu(III)-state intermediate 110. Then reductive elimination would afford the product 107 with regeneration of Cu(I)–Br.
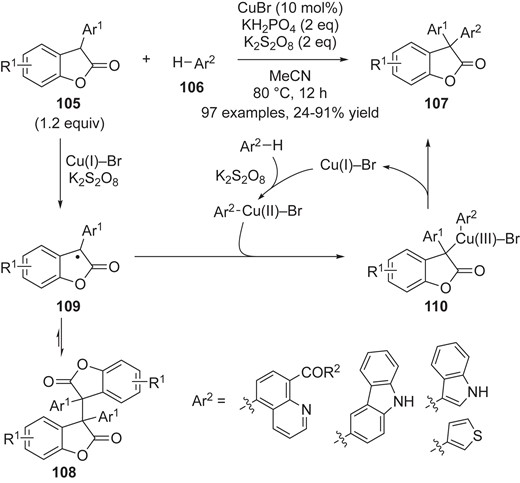
Cu-catalyzed catalytic dehydrogenative arylation of benzofuranones.
7. Umpolung
Yazaki, Ohshima and co-workers reported an aerobic Fe-catalyzed CDC reaction of azlactones 111.21b,21c Scheme 23-i shows a recent example of aerobic cross-coupling between 111 and indoles 11221b with FeCl3/(±)-113 for the preparation of indole-substituted α,α-disubstituted amino acid derivatives 114. The developed procedure under base-free conditions is applicable to a wide variety of substrates with high functional group compatibility, giving the CDC adducts with high regioselectivity (C4/C2: 10/1 → 30/1). The dimer 115 also undergoes the CDC reaction under the optimized conditions (Scheme 23-ii). Interestingly, the silyl enol ether 116 also serves as a substrate in the cross-coupling with 111a, selectively giving the adduct 117 (Scheme 23-iii). These results suggest a different mechanism from that reported by Kozlowski (Scheme 20-iii).21a In their proposed mechanism, azlactone dimer 115 would be formed in the presence of Fe catalyst via azlactone radical 118. Subsequent Fe-catalyzed heterolytic cleavage of the dimer 115 would provide reactive azaallyl intermediate 119, which would then react with nucleophilic indole 112 to form the product 114. Oxindoles and benzofuranones also afforded the indolylated products under similar reaction conditions. The authors also developed the Fe-catalyzed CDC reaction of azlactones with 2-acylimidazoles, which was again proposed to proceed via the dimerized azlactone intermediate.21c Although the authors did not rule out the possibility of anion-radical coupling, their results raise the possibility of future applications using other nucleophiles, in place of indoles or 2-acylimidazoles.
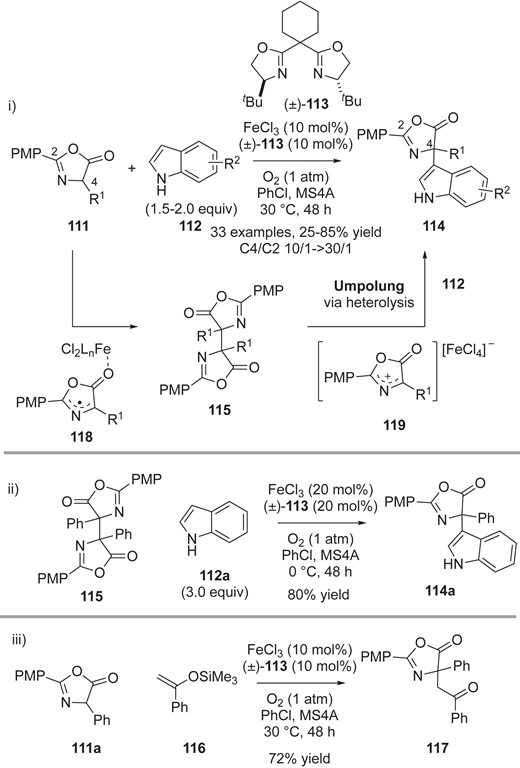
i) Fe-catalyzed catalytic CDC reaction of azlactones 111 with indoles 112. ii) Fe-catalyzed catalytic oxidative cross-coupling of azlactone dimer 115 with indole 112a. iii) Fe-catalyzed catalytic oxidative cross-coupling of azlactone dimer 111a with silyl enol ether 116.
8. Conclusion
In this review, we have provided an overview of cross-coupling reactions initiated by the generation of tertiary carbon-centered radicals, with selected examples grouped according to the originally proposed mode of bond formation. Although it is still difficult to experimentally trace the kinetic phenomenon associated with the persistent radical effect,5d,7,55 the classification of radicals based on their lifetimes is quite useful to design the radical-based cross-coupling. High cross-selectivity is feasible if one radical has a significantly longer lifetime than the coupling partner. The astonishing array of reactivities, including radical-radical cross-coupling (Section 3), anion-radical coupling (Section 4), radical addition to olefins (Section 5), reductive coupling (Section 6), and umpolung (Section 7), holds vast promise for further expanding the utility of persistent tertiary carbon radicals in organic synthesis, even though some of the mechanisms remain controversial. Given the importance of compounds containing quaternary carbon(s) and/or tetra-substituted carbon(s), the development of a new class of reaction would be very exciting.56 Mechanistic investigations have highlighted that the reactivity and selectivity of tert-carbon radicals can be tuned not only by modifying the inherent nature of the naked persistent radicals, but also by varying the organocatalysts and metal catalysts used. However, methods for controlling the (enantio-, diastereo-, regio-, and site-) selectivity are still limited, and offer exciting challenges for further research. A better understanding of the mechanisms controlling the reactivity and selectivity of persistent radicals will require considerable work, even though the conceptual framework for the persistent radical effect has been established. Collaboration among synthetic organic chemists, physical organic chemists, and computational chemists is likely to have a critical role. We hope that this review will be helpful in the discovery and optimization of new synthetic methodologies utilizing persistent radicals.
Acknowledgment
Financial support was provided by RIKEN’s Strategic Programs and KAKENHI (Grant Numbers JP17H02213, 18K19156 and JP18H04277 in Precisely Designed Catalysts with Customized Scaffolding) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology (MEXT). KK gratefully acknowledges the SPDR program in RIKEN. We also thank all co-workers listed in our original papers cited in this Review for their tremendous efforts.
References
d)Radicals in Organic Synthesis, 1st ed.; P. Renaud, M. Sibi, Eds.; Wiley-VCH: Weinheim, 2001.
e)H. Togo, Advanced Free Radical Reactions for Organic Synthesis; Elsevier: Amsterdam, 2003.
f)S. Z. Zard, Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, 2003.
g)Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons: Chichester, U.K., 2012; Vol. 1 and 2.
b)
c)
a)
b)
d)
e)
f)
g)
h)
i)
b)
c)
c)
d)
For selected recent reviews, see; a)W. Zhang, Y. Jin, Dynamic Covalent Chemistry: Principles, Reactions, and Applications, John Wiley & Sons, New York City, USA, 2017.
b)
b)
b)
c)
c)
‘International Tables for Crystallography’, C, Table 9.5.1.1. ed. by A. J. C. Wilson, Kluwer Academic Publishers, Dordrecht, 1992.
a)
b)
c)
b)
c)
e)
f)
g)
a)
b)
b)
c)
e)
f)
g)
a)
a)
b)
b)
b)

Yoshihiro Sohtome
Yoshihiro Sohtome received his Ph.D. from the University of Tokyo under the supervision of Professors Nagasawa and Hashimoto in 2006. He joined Professor Shibasaki’s group at the University of Tokyo as an assistant professor. Following a postdoctoral study with Professor Hamilton’s group at Yale University, he returned to Professor Nagasawa’s group at TUAT as an assistant professor in 2009. He joined Professor Sodeoka’s group at RIKEN in 2011 as a Research Scientist and was promoted to Senior Research Scientist in 2018.

Kyohei Kanomata
Kyohei Kanomata received his Ph. D. from Tohoku University under the supervision of Professor Terada in 2015. After working at Kyoto University and Kyushu University as a postdoctoral researcher, he joined Professor Sodeoka’s group at RIKEN in 2020 as a Special Postdoctoral Researcher (SPDR), and then moved to Osaka University as an assistant professor.

Mikiko Sodeoka
Mikiko Sodeoka received her B.Pharm., M.Pharm., and Ph.D. from Chiba University. After working at the Sagami Chemical Research Center, Hokkaido University, Harvard University, and the University of Tokyo, she became a Group Leader at the Sagami Chemical Research Center in 1996. She subsequently moved to the University of Tokyo as an associate professor and then to Tohoku University as a full professor in 2000. Since 2006, she has been a Chief Scientist at RIKEN.
Author notes
Present address: Graduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan.
Dedicated to Professor Eiichi Nakamura on the occasion of his 70th birthday.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}