





Abstract
Metal clusters composed of no more than one hundred metal atoms have the potential to exhibit novel catalysis that is unexpected from the corresponding bulk and nanoparticles. The emergence of cluster-specific properties is ascribed to the quantized electronic structures, unique geometrical packing, and structural fluxionality. Polymer-stabilized metal clusters provide ideal platforms for exploring new catalysis based on the chemical properties of naked model clusters and studying the correlation between structural parameters and intrinsic catalytic properties of metal clusters. In this article, we describe our recent efforts to explore novel catalysts using polymer-stabilized metal clusters.
1. Introduction
Metal nanoparticles (MNPs)1–6 and their alloys7,8 have been widely used in heterogeneous and homogeneous catalysts for efficient usage of metal elements by taking advantage of their high surface-to-volume ratio. The catalytic performances (activity and selectivity) of a MNP can be designed based on the intrinsic reactivity of model surfaces and interfaces since they are governed by the chemical properties of the MNP’s surface and interface with the support. In contrast, metal clusters (MCs)9–16 and their alloys17,18 whose diameters are smaller than a critical dimension of ∼2 nm exhibit novel catalysis that cannot be predicted from those of the bulk metal and MNPs. The emergence of peculiar catalytic properties of MCs is ascribed to the quantized electronic structures, specific surface structures, and structural fluxionality. The electronic structures of MCs are significantly modulated by changing the sizes, compositions, and mixing modes of multi-elements (Figure 1), according to previous studies on MNPs.19–25 In addition to the increased population of low coordination sites, unique active sites such as twin defects appear due to the formation of non-bulk atomic packing: icosahedral (Ih) and decahedral (Dh) motifs (Figure 1).26
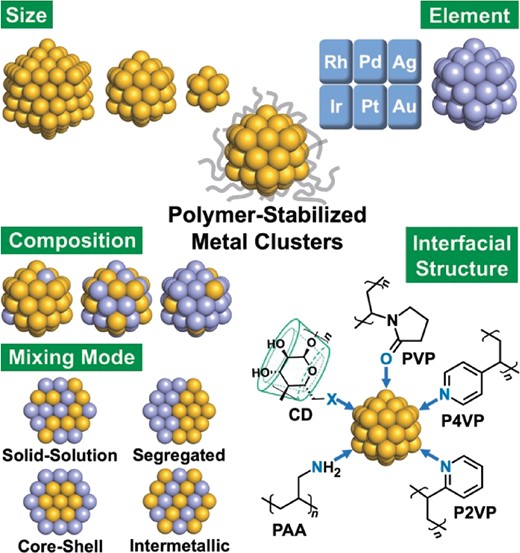
Structural parameters of polymer-stabilized metal clusters correlating with the catalytic performance. Abbreviations of stabilizers are summarized in Figure 2.
In order to apply the MCs for catalysis, it is necessary to stabilize them against aggregation while exposing the active surface. There are several methods of the stabilization; ligand protection,11,14 stabilization by polymers9,10,12,15 and immobilization on a solid support.10,12,13,16 Each method has benefits and drawbacks. First, ligand protection allows us to control the size and composition of MCs with atomic precision and further to determine the atomic structures by X-ray crystallography.27–29 Although ligand-protected MCs show catalysis for some reactions11 and ligation promotes some reactions,30 their catalytic activities are generally suppressed by the complete poisoning of the surface metal atoms with the ligands. Second, immobilization on a solid support is suitable for practical application in catalysis because it improves the robustness and ease of recovery. Although atomically-precise preparation of supported MCs is possible by calcination of ligand-protected MCs deposited on the support while suppressing the thermal-induced aggregation,10–12,31 the atomic structures of the resulting MCs are polydisperse and cannot be determined by X-ray crystallography. Third, stabilization by polymers through weak interaction allows us to develop MC catalysts based on knowledge of the fundamental chemical properties of naked MCs in the gas phase.32 The drawbacks of polymer-stabilized MCs are that X-ray crystallography cannot be used, the MCs are fragile under harsh conditions, and recovery for reuse is difficult. Nonetheless, we have used polymer-stabilized MCs as platforms to explore new catalysts and to study the fundamental correlation between structural parameters and catalysis. In this article, we describe our recent achievements toward precision synthesis of the MCs stabilized by polymers listed in Figure 2 and exploration of novel catalysis. We studied the impacts of structural parameters such as sizes, compositions, mixing modes of elements and interfacial structures on the catalytic properties of polymer-stabilized MCs. We hope that the results reported here will contribute to the development of MC catalysts with catalytic properties designed in accordance with rational guiding principles.
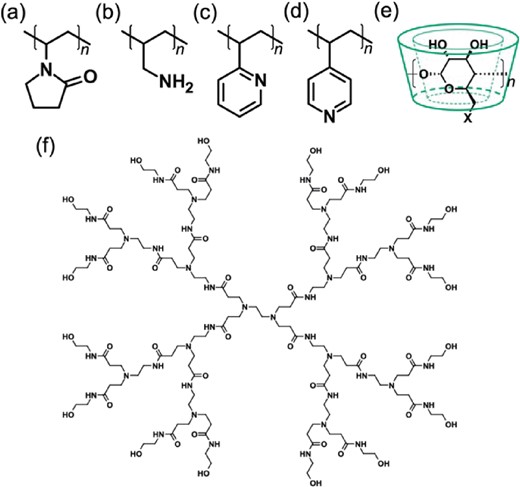
Structures of polymers used for stabilization of metal clusters: (a) poly(N-vinyl-2-pyrrolidone) (PVP); (b) poly(allylamine) (PAA); (c) poly(2-vinylpyridine) (P2VP); (d) poly(4-vinylpyridine) (P4VP); (e) cyclodextrin derivatives (X-(α, β or γ)-CD) with X = OH, SH or NH2 (n = 6, 7, and 8 for α, β, and γ); (f) OH-terminated poly(amidoamine) dendrimer of generation 2 (PAMAM-G2).
2. Effects of Size
2.1 Oxidation by Au:PVP
Since Haruta’s discovery of CO oxidation catalyzed by Au NPs on metal oxides,33,34 the catalytic properties of nano-sized Au have attracted intense interest in nanoscience.2,5,35 In 2005, we found that Au clusters stabilized by poly(N-vinyl-2-pyrrolidone) with average molecular weight (Mw) of 40 kDa (composed of ∼360 monomer units) (Figure 2a) (Au:PVP) exhibited size-specific catalysis for aerobic oxidation of alcohols:36 the oxidation catalysis emerged only when the particle diameter was smaller than ∼3 nm and the catalytic activity dramatically increased with the decrease of diameter.37,38 It was also found that the small Au clusters were efficient catalysts for various types of organic reactions under aerobic conditions.39 The size-specific oxidation catalysis was ascribed to the negative charge on Au clusters donated by PVP, based on spectroscopic results and the high reactivity of small bare Au cluster anions to molecular oxygen observed in the gas phase.40–43 It was proposed that adsorbed oxygen molecules are activated by electron transfer from the negatively charged Au clusters and that the resulting superoxo-like species facilitate the catalytic oxidation reactions. The above reaction mechanism was supported by theoretical studies on alcohol oxidation on model Au clusters.44,45
For clarifying the mechanism of the size-specific catalysis of Au:PVP and optimizing the catalysis, we attempted to control the cluster size with atomic precision. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry was successfully used to evaluate the cluster size and revealed magic numbers such as 35 ± 1, 43 ± 1, and 58 ± 1.46 Agreement with the magic numbers of naked Au34 and Au58 clusters47 suggests that the closure of the electronic shell contributes also to the stability of the Au clusters stabilized by PVP. In order to better control the cluster size, we mixed two solutions containing Au ions and BH4− homogeneously by using a micromixer.48 MALDI mass analysis under minimal laser fluence indicated that Au34Clm and Au43Clm were preferentially formed in Au:PVP prepared under optimized conditions (Figure 3).49 It was found that the Cl ligands were easily released during the oxidation reactions and did not hinder catalytic processes.
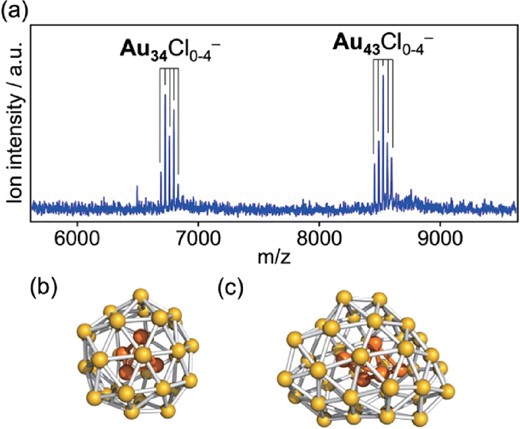
(a) MALDI mass spectrum of Au:PVP. Proposed structures of isolated (b) Au34 and (c) Au43 based on gas phase experiments and DFT calculations.50–52 The Au4 and Au5 cores are highlighted in orange.
2.2 Hydrogenation by Cu@PAMAM
Motivated by the theoretical prediction that the H2 molecule undergoes dissociative adsorption on small Cu clusters,53 hydrogenation catalysis of Cu clusters was investigated by using the OH-terminated poly(amidoamine) dendrimer of generation 6 (PAMAM-G6) (Figure 2f) as a stabilizer.54 Cun clusters encapsulated within PAMAM-G6 (Cun@PAMAM-G6) were prepared by the rapid reduction of nCu2+@PAMAM-G6 (n = 10, 20, 30, 40, 50, 60) complexes by NaBH4 at room temperature.
Catalytic activity of Cu30@PAMAM-G6 for the hydrogenation of different functional groups such as C=O, C=C and NO2 was examined. The hydrogenation reactions were conducted under 3 MPa of H2 at room temperature (298 K) in water. Clear chemoselectivity was observed: the activity was increased in the order of NO2 < C=C < C=O. Notably, this selectivity is significantly different from those of other clusters of precious metals (Ir, Pd, Pt, Rh) as shown later (Figure 7). By taking advantage of the high selectivity toward the C=O group, we demonstrated that Cu30@PAMAM-G6 catalyzed the selective hydrogenation of cinnamaldehyde (1a) and 4-nitrobenzaldehyde (1b) to cinnamyl alcohol (2a) and 4-nitrobenzyl alcohol (2b), respectively (Scheme 1). The selective hydrogenation of 1a to 2a, which is also catalyzed by finely tuned Pt-based catalysts,55,56 is appealing given that the hydrogenation of the C=C bond is thermodynamically preferable over that of the C=O bond.

Selective hydrogenation reactions catalyzed by Cu30@PAMAM-G6 dispersed in water. Values in parentheses correspond to product selectivity.
Furthermore, the catalytic activities of Cun (n = 10, 20, 30, 40, 50, 60) were compared by using the hydrogenation of 2-hexanone (1c) to 2-hexanol (2c) as a model reaction (Figure 4). Conversion of 1c in 30 min showed a monotonic increase with the decrease of cluster size, whereas TOF based on surface Cu atoms exhibited a volcano-shaped behavior with the maximum value at Cu30. Further study is needed to understand the origin of the size-specific catalysis of the Cu clusters.
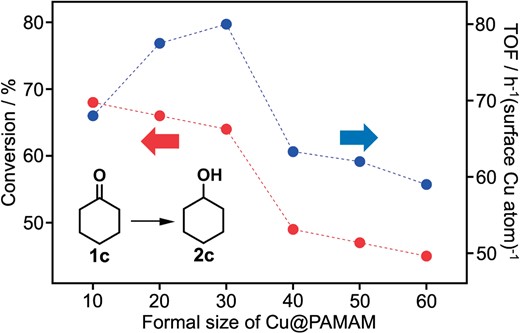
Effect of size on the catalytic activity of Cun@PAMAM-G6 for the hydrogenation of cyclohexanone (1c) under the same conditions as Scheme 1 (reaction time: 30 min).
The major problem of using Cu(0) clusters is their air sensitivity. For example, Cu:PVP dispersed in aerated water irreversibly oxidized to CuO or Cu2O,57 while Cu30@PAMAM-G6 quickly oxidized into 30Cu2+@PAMAM-G6 in air. It was demonstrated that Cu30@PAMAM-G6 were regenerated by the NaBH4 reduction of 30Cu2+@PAMAM-G6 and that the regenerated clusters showed catalytic activity comparable to the original one. This result suggests that PAMAM is a promising stabilizer of air-sensitive MCs.
2.3 Hydrogenation by Ir@PAMAM
Irn@PAMAM-G6 (n = 15, 30, 60) supported on SiO2 was applied to the catalytic hydrogenation of 2-nitrobenzaldehyde (1d) under mild reaction conditions (0.1 MPa H2; 303 K).58 All clusters efficiently catalyzed the selective hydrogenation of the NO2 group (Figure 5), indicating that the Ir clusters can activate H2 molecules under mild conditions. High selectivity toward the NO2 group is ascribed to the attractive interaction between the NO2 group and oxidized Ir surface. It was found that the branching ratio between 2-aminobenzaldehyde (2d) and anthranil (3d) was dependent on the size. Structural analysis by powder X-ray diffraction (PXRD), X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) revealed that Ir clusters of Irn@PAMAM-G6 were partially oxidized. According to XPS, the population of Ir(0) is higher for smaller Ir clusters. Given that 2d and 3d were obtained from the same hydroxylamine intermediate by hydrogenation and heterocyclization-dehydration, respectively,59 the high selectivity to 2d was ascribed to more efficient hydrogenation of the hydroxylamine intermediate over the cyclization on smaller clusters having a larger proportion of Ir(0) phase. Enhancement of the selectivity to 2d by increased H2 pressure also supported this conjecture.
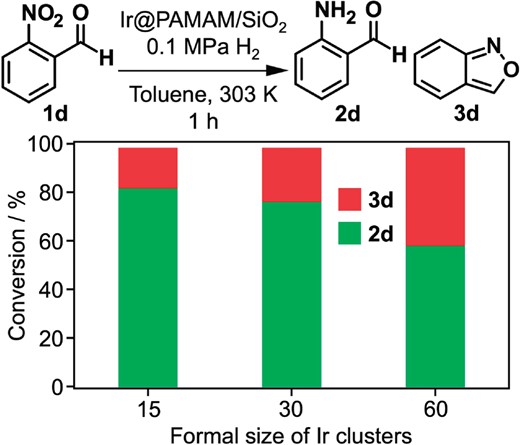
Effect of size of Irn@PAMAM-G6/SiO2 on the selectivity of hydrogenation of 2-nitrobenzaldehyde (1d).
3. Effects of Compositions
3.1 Oxidation by Ag or Pd Mixed Au Clusters
It has been demonstrated that PVP-stabilized Au-based alloy clusters show improved catalytic performance compared to monometallic counterparts.60–63 We prepared Ag-doped Au:PVP by the co-reduction method in a batch system; an aqueous solution of NaBH4 was injected to an aqueous solution of HAuCl4, AgNO3 and PVP with 5–30% of Ag.60 The rate constant of catalytic oxidation of p-hydroxybenzyl alcohol normalized by unit surface area was maximized at the Ag content of 10%. The enhanced catalytic activity was ascribed to the increased electron density on Au atoms, which is consistent with the proposed reaction mechanism where negatively charged Au sites act as active sites.38 However, the effect of size cannot be excluded because the size of alloy clusters also increased from 1.3 to 2.2 nm with increasing the Ag ratio from 0 to 30%.
Nakajima demonstrated that Pd could be doped to Au:PVP while retaining the size of the alloy clusters by using a microfluidic mixer.62 AuPd:PVP clusters were prepared by the co-reduction of HAuCl4 and H2PdCl4 by NaBH4 in the presence of PVP with the Au/Pd ratio ranging from 100/0 to 0/100. The AuPd:PVP clusters exhibited solid solution structures and narrow size distribution (1.0–1.7 nm). It was found that the catalytic activity of AuPd:PVP was higher than that of undoped Au:PVP for aerobic oxidation of benzyl alcohol, which was explained by the transfer of electronic charge from Pd to Au.
Toshima conducted precise doping of Au atoms to Pd:PVP by galvanic reaction to produce alloy clusters.61 High-resolution transmission electron microscopy (HRTEM) revealed that the Au atoms thus introduced were located at the corner sites of cuboctahedral Pd147 clusters. The resulting AuPd:PVP clusters with a crown jewel (CJ) structure showed remarkably high TOF for aerobic glucose oxidation. Based on XPS and DFT calculations, the Au dopants with negative charge were considered to be responsible for the prominent catalytic activity. Furthermore, CJ-structured AuPdIr trimetallic clusters with Au atoms at the corner sites were successfully synthesized by a similar method.63 It was demonstrated that the introduction of Ir significantly enhanced the catalytic activity for the glucose oxidation.
Sakurai demonstrated that AuPd:PVP clusters showed novel catalysis which is not observed in monometallic counterparts, as a result of the synergistic effect between Au and Pd.64–67 Ullmann coupling reactions of various chloroarenes (ArCl) were catalyzed by AuPd:PVP under mild reaction conditions, whereas Au:PVP and Pd:PVP and their physical mixture showed no catalytic activity.64,65 The large kinetic isotope effect (kH/kD = 7.0) in DMF-d7 indicated that the rate-determining step was the hydrogen transfer from DMF to the surface of AuPd:PVP. Theoretical investigation of the detailed reaction mechanism was conducted by Ehara.68 DFT calculations indicated that the key step was oxidative addition of ArCl: a pure Au cluster required a too large activation energy for the oxidative addition, while a pure Pd cluster formed a too stable intermediate as a product of the oxidative addition due to large structural relaxation. Thus, only AuPd alloy clusters could catalyze the coupling reaction. In the bimetallic system, the active center was Pd and the important role of Au was to promote the migration of a Cl atom and to suppress deactivation processes including Pd leaching.66
3.2 Hydrogenation by Single Pd or Rh Atom Doped Au Clusters
Kinetically controlled synthesis by microfluidic mixing48 and mass spectrometric analysis46 have enabled us to study the effects of doping a single atom to Au:PVP.69,70 Pd or Rh doped Au:PVP was prepared by the co-reduction of metal ions using a micromixer with the mixing ratios of Au/M = 97/3 (M = Pd, Rh). MALDI mass spectrometry revealed the selective formation of Au33Pd1 and Au34Rh1 (Figures 6a, b). Extended X-ray absorption fine structure (EXAFS) analysis showed that the coordination number (CN) of the Rh-Au bond (3.2 ± 0.7) was significantly smaller than that of the Pd-Au bond (7.3 ± 0.8). The results suggested that a Rh atom is attached on the surface of Au34, while a Pd atom is incorporated in the surface of Au34. As a consequence of the single atom doping, hydrogenation catalysis for alkenes emerged and Au34Rh1:PVP showed much higher catalytic activity than Au33Pd1:PVP (Figure 6c). It was proposed that heterometal atoms act as active sites for H2 and the lower coordination state of the Rh atom results in the remarkable activity. In a recent theoretical study, the reaction mechanism of ethylene hydrogenation on Au8, Au20 and their Rh-doped forms (Au8Rh1 and Au20Rh1) was investigated.71 Each elementary step showed a much lower activation barrier for Rh-doped clusters than for pure Au clusters. In the most energetically favorable reaction pathway on both Au8Rh1 and Au20Rh1, ethylene and hydrogen molecules were co-adsorbed on the Rh dopant. The H2 molecule adsorbed on the Rh dopant dissociated with very low activation barrier. Finally, hydrogen atoms sequentially formed C(sp3)-H bonds. These studies demonstrate that single atom doping is enough to enable the hydrogenation catalysis to sub-nm Au clusters.
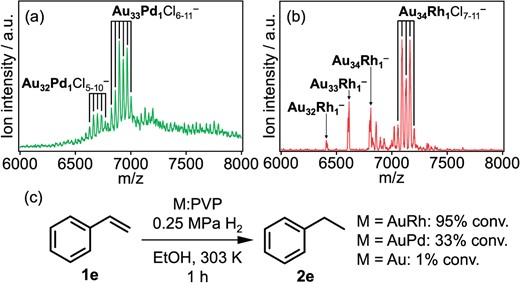
MALDI mass spectra of (a) Pd-doped and (b) Rh-doped Au:PVP. (c) Hydrogenation reaction of styrene (1e) catalyzed by the clusters dispersed in ethanol.
3.3 Hydrogenation by Partially Oxidized Ir Clusters
We examined the selective hydrogenation of nitroarenes catalyzed by PVP-stabilized noble metal clusters and found that Ir clusters efficiently catalyzed the selective hydrogenation reactions.72 Ir:PVP clusters with the diameter of 2.2 ± 0.5 nm were prepared by the polyol reduction of H2IrCl6 using ethylene glycol. According to XPS, 11% of surface Ir atoms formed the IrO2 phase while the other atoms were metallic and slightly negatively charged by electron donation from PVP. Ir:PVP catalyzed the selective hydrogenation of 4-nitrobenzaldehyde (1b) to 4-aminobenzaldehyde (3b) under mild reaction conditions (0.1 MPa H2; 298 K) with the selectivity of 99% at the conversion of 99% (Figure 7). This selectivity is much higher than those by Pt and Rh:PVP (1.8 and 2.0 nm) and contrasts to those by Pd:PVP (1.9 nm) and Cu30@PAMAM-G6 (Scheme 1).53 Moreover, Ir:PVP catalyzed the selective hydrogenation of nitroarenes having C=O, CN or Cl groups to the corresponding aniline derivatives.
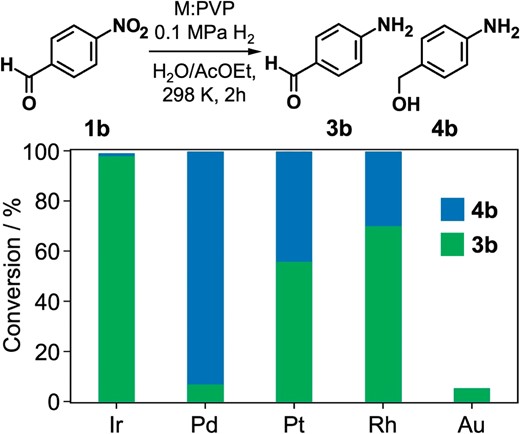
Hydrogenation reaction of 4-nitrobenzaldehyde (1b) dissolved in ethyl acetate catalyzed by PVP-stabilized metal clusters dispersed in water (biphasic condition).
According to studies on the selective hydrogenation of polarized functional groups (e.g. NO2 and CHO) catalyzed by MNPs on metal oxides, the oxide support plays a pivotal role in the selective adsorption and activation of such functional groups.73–77 Thus, the high selectivity in the hydrogenation reactions catalyzed by Ir:PVP was ascribed to the coexistence of metallic and oxidized phases; the nitro group was preferentially adsorbed on oxidized Ir, whereas H2 was activated by metallic Ir.
The partially oxidized Ir clusters can be viewed as an ultrasmall nanocomposite consisting of Ir and IrO2. Kaneda and co-workers prepared nanocomposite catalysts composed of the MNP core and surrounding metal oxide NPs to maximize the active metal-oxide interface.78,79 For instance, Au@CeO2 catalyst showed excellent selectivity for the semi-hydrogenation of alkynes, since the dissociation of hydrogen molecules selectively occurred at the Au-CeO2 interface in a heterolytic manner.79
3.4 Hydrogenation by Cu Mixed Rh Clusters
The coexistence of two phases (IrO2 and Ir) in Ir:PVP facilitated the selective hydrogenation of the nitro group. Motivated by the role of the IrO2 phase in Ir:PVP, we attempted to adjust the selectivity of the hydrogenation by modifying the surface of Rh:PVP by alloying. Cu was chosen as a dopant with the expectation of forming Cu oxide phase on the Rh cluster surface.80 RhCu alloy clusters were prepared by the rapid co-reduction of Rh(OAc)3 and CuSO4 by NaBH4 in the presence of PVP with different Rh/Cu ratios (100/0, 75/25 and 50/50). The particle diameter of Rh:PVP, RhCu(75/25):PVP and RhCu(50/50):PVP was estimated by TEM to be 2.2 ± 0.4, 2.5 ± 0.5 and 2.6 ± 0.6 nm, respectively. PXRD measurements revealed that the lattice constants of the clusters followed Vegard’s law, indicating the formation of random alloy structures even though Rh and Cu are immiscible in the bulk state at room temperature according to the phase diagram. It was found that the selectivity was improved from 70% to 97% by increasing the Cu doping amount from 0% to 50% (Figure 8). Considering the selective hydrogenation catalysis of partially oxidized Ir clusters and the reactivity of Cu with oxygen, the surface Cu atoms may be oxidized to improve chemoselectivity by the preferential adsorption of the nitro group.
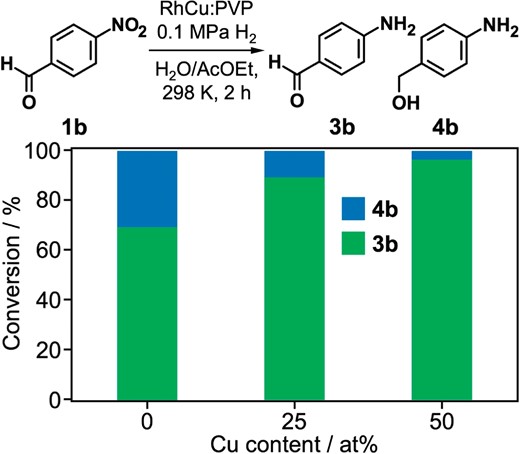
Effect of Cu doping on the catalysis of Rh:PVP for the selective hydrogenation of 4-nitrobenzaldehyde (1b).
4 Effects of Surface Modification
4.1 Activities
As described in 2.2, PVP plays a crucial role in activation of Au clusters toward O2 by donating electronic charge. Thus the catalytic activities are regulated not only by the steric hinderance by the polymers but also by the chemical nature of the coordination sites of the polymers to the Au NCs.
Sakurai found that the diameter dependence of the catalytic activity of Au:PVP for aerobic alcohol oxidation was dependent on the Mw values of PVP: Au NPs with the diameter of ∼7 nm showed the highest activity when using long PVP (Mw = 360 kDa) as a stabilizer,81 whereas the activity increased with decrease in diameter when using PVP (Mw = 40 kDa).36–38 This unexpected dependence of Mw on the catalytic activity was ascribed to the high negative charge density on the NP surface derived from the densely packed PVP network. Moreover, the importance of surface coverage by PVP was pointed out by theoretical studies based on DFT and molecular dynamics.82,83
Small (<2 nm) Au clusters stabilized by poly(allylamine) (PAA; Mw = 17 kDa) (Figure 2b) (Au:PAA) showed much poorer catalytic activity than Au:PVP (Mw = 40 kDa) with comparable size.38 XPS and theoretical calculations84 suggested that the poorer activity of Au:PAA than that of Au:PVP is ascribed to smaller negative charge and higher surface coverage of the Au NCs.
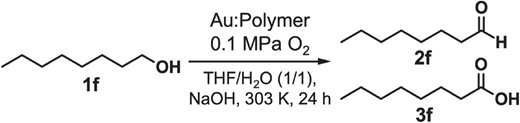
Monodisperse ∼1 nm Au clusters stabilized by poly(n-vinylpyridine) (PnVP, n = 2, 4) (Figure 2c, 2d) showed higher catalytic activities than Au:PVP for the aerobic oxidation of 1-octanol (1f) (Table 1),85 whereas Au:PVP was more active for benzyl alcohol oxidation. XPS, optical spectroscopy, and DFT calculations on the model systems consisting of Au34 and monomer units of the corresponding polymers indicated that the extent of electron transfer to the Au core was in the order of P2VP > P4VP ∼ PVP. This trend is consistent with our hypothesis that the oxidation catalysis is enhanced by donation of electronic charge. Nonetheless, the activity of Au:P2VP was slightly lower than that of Au:P4VP, which is opposite to what would be expected based on the higher electron-donating ability of P2VP. This is probably due to the higher surface coverage derived from the closer distance between the cluster surface and the polymer main chain of P2VP. The dependence on substrates was ascribed to the hydrophilicity of polymers: hydrophilic PVP promoted the access of hydrophilic benzyl alcohol to the cluster surface whereas hydrophobic PnVP allowed the access of hydrophobic 1f. TEM observation revealed that Au:PnVP were more robust than Au:PVP during catalytic reactions (Table 1). This enhanced robustness was attributed to stronger binding affinity of the pyridine ligands than that of the pyrrolidone ligand, according to the DFT calculations.
Catalytic performance of Au:PnVP (n = 2, 4) and Au:PVP for the aerobic oxidation of 1-octanol (1f).
| Polymer | Conversion/% (2f:3f) | DTEM/nm (before) | DTEM/nm (after) |
|---|---|---|---|
| P2VP | 21 (18:82) | 1.1 ± 0.2 | 1.5 ± 0.5 |
| P4VP | 32 (12:88) | 1.1 ± 0.2 | 1.6 ± 0.6 |
| PVP | 17 (0:100) | 1.0 ± 0.2 | 2.0 ± 0.7 |
| Polymer | Conversion/% (2f:3f) | DTEM/nm (before) | DTEM/nm (after) |
|---|---|---|---|
| P2VP | 21 (18:82) | 1.1 ± 0.2 | 1.5 ± 0.5 |
| P4VP | 32 (12:88) | 1.1 ± 0.2 | 1.6 ± 0.6 |
| PVP | 17 (0:100) | 1.0 ± 0.2 | 2.0 ± 0.7 |
Catalytic performance of Au:PnVP (n = 2, 4) and Au:PVP for the aerobic oxidation of 1-octanol (1f).
| Polymer | Conversion/% (2f:3f) | DTEM/nm (before) | DTEM/nm (after) |
|---|---|---|---|
| P2VP | 21 (18:82) | 1.1 ± 0.2 | 1.5 ± 0.5 |
| P4VP | 32 (12:88) | 1.1 ± 0.2 | 1.6 ± 0.6 |
| PVP | 17 (0:100) | 1.0 ± 0.2 | 2.0 ± 0.7 |
| Polymer | Conversion/% (2f:3f) | DTEM/nm (before) | DTEM/nm (after) |
|---|---|---|---|
| P2VP | 21 (18:82) | 1.1 ± 0.2 | 1.5 ± 0.5 |
| P4VP | 32 (12:88) | 1.1 ± 0.2 | 1.6 ± 0.6 |
| PVP | 17 (0:100) | 1.0 ± 0.2 | 2.0 ± 0.7 |
The effect of the generation of the PAMAM dendrimer on the catalytic activity was also investigated by employing Cu10@PAMAM-Gx (x = 4–7).54 The catalytic activity for the hydrogenation of 1c was increased by the generation. This generation dependence has been ascribed to the promotion of access of hydrophobic substrate 1c to the Cu surface by the enhanced hydrophobicity of the dendrimer cavity. The trend observed in Cu10@PAMAM-Gx was opposite to that reported in the hydrogenation of allylic alcohols by Pd40@PAMAM-Gx:86 the decrease of TOF with the generation has been attributed to suppression of access of substrates to Pd40 due to the steric crowding on the dendrimer periphery.
4.2 Selectivity
CD derivatives, X-β-CD (X = OH, SH, NH2) (Figure 2e), were introduced as additional modifiers of Au:PVP for asymmetric aerobic oxidation of secondary alcohols by taking advantage of unique features of the CD derivatives:87 (1) the ability to include organic guest molecules into their hydrophobic cavities and (2) enantioselectivity of the inclusion derived from the stereocenters of glucopyranose units. The effect of the structure of CD derivatives was examined for the aerobic oxidative kinetic resolution of racemic 1-(2-naphthyl)ethanol (1g) (Figure 9) by Au:PVP (1.8 ± 0.6 nm). Since the reaction product is an achiral ketone, the enantioselectivity was evaluated based on the selectivity factor defined as the ratio of rate constants for the conversion of chiral alcohols (kfast/kslow). HO-β-CDs did not affect the catalysis while HS-β-CDs suppressed the activity due to a strong poisoning effect. In contrast, H2N-β-CD improved the enantioselectivity without affecting the activity. In addition, H2N-β-CD showed higher selectivity than H2N-(α and γ)-CDs as expected from the equilibrium constants for the inclusion of 1g, and the presence of a competitive guest molecule, 1-adamantanol, appreciably decreased the enantioselectivity. Based on these results, it was proposed that the local concentration of (S)-1g around Au:PVP is higher than that of (R)-1g owing to the selective inclusion by H2N-β-CD coordinating to Au clusters.

Plots of selectivity factor (red) and conversion (blue) for the oxidation of racemic 1-(2-naphthyl)ethanol (1g) catalyzed by Au:PVP in the presence of CD derivatives.
5. Summary and Prospects
5.1 Summary
Sections 2–4 described how the catalytic performances of MCs are modulated by their key structural parameters: size, composition, mixing mode of metal elements, and interfacial structure. We summarize three unique MC catalysts as follows.
(1) Au clusters can be catalytically activated for aerobic oxidation reactions by donating electronic charge. This is achieved by doping more electropositive metals (Ag, Pd) and/or stabilizing by PVP or PnVP having pyrrolidone or pyridine moieties, respectively. Pure Au clusters are inert for hydrogenation, but doping a single atom of Rh can activate the hydrogenation of C=C bonds. It was proposed that the doped Pd or Rh atom exposed on the surface acted as an active site for hydrogenation reactions.
(2) Cu clusters encapsulated by PAMAM-G6 exhibit chemoselective hydrogenation catalysis: the activity is increased in the order of NO2 < C=C < C=O. Interestingly, clusters of precious metals (Ir, Pd, Pt, Rh) showed much higher selectivity to the NO2 group than the C=O group. The Cun@PAMAM-G6 are not oxidized to Cu2O precipitates, but to the nCu2+@PAMAM-G6 precursor complexes under an ambient environment. This unique oxidation behavior allows us to regenerate Cun@PAMAM-G6 by simple chemical reduction. The selectivity for hydrogenation of the NO2 group by Rh:PVP could be improved by doping Cu. These findings may facilitate the effective use of cheap and abundant elements.
(3) Ir clusters stabilized by PVP and PAMAM facilitated chemoselective hydrogenation reactions of the NO2 group. Selectivity to the NO2 group is ascribed to preferential adsorption onto the partially oxidized Ir surface.
5.2 Prospects
Several challenges remain to be overcome for establishing the correlation between structural parameters and catalysis and for the further development of new MC-based catalysts.
(1) Atomic-level precision is required for the synthesis to take full advantage of the tunability of catalysis of MCs. At present, kinetic control of the formation process of MCs using a microfluidic device48,62,88 is a promising method to control the cluster size and the number of dopant atoms. Synthesis using a dendrimer template is also a versatile approach to achieve atomically-precise synthesis.9,15 Yamamoto and co-workers have reported the synthesis of size-selected MCs and composition-controlled alloyed MCs by using a designed conjugated dendrimer, phenylazomethine dendrimer of generation 4 (DPA-G4).15 Mass spectral (MS) characterization is indispensable to confirm the atomically defined sizes and compositions of the synthesized polymer-stabilized MCs. Versatile soft ionization methods are required to expand the scope of MS. The mixing mode of more than two elements within a single MC (Figure 1) is difficult to control since mixing behavior on the nanoscale does not necessarily follow the phase diagram of the bulk metal, as Kitagawa and co-workers demonstrated.89 Establishment of a phase diagram on the nanoscale is an interesting challenge. Heating of multi-metallic clusters under H2 gas is a promising approach to promote alloying since the metal atoms become effusive at lower temperature due to the dissolved hydrogen.90,91
(2) XAS is a powerful tool to gain insights into the local geometrical structure and electronic state of specific elements of polymer-stabilized MC,92 under the restriction that single-crystal X-ray diffraction analysis cannot be applied. Nevertheless, even when the MCs are synthesized with atomic precision, their inherent properties make it difficult to clarify the correlation between the structures and catalytic properties: the individual MCs may take isomeric structures and may undergo isomerization under ambient as well as catalytic conditions. Therefore, establishment of the energy landscape of MCs is one of the ultimate goals to clarify the origin of the catalysis. This issue can be partly addressed by time-lapse HRTEM imaging93–96 of structural fluctuation combined with the help of theoretical simulation. Operando characterization is a more straightforward but nevertheless challenging approach.
(3) For the practical applications of polymer-stabilized MC catalysts, the robustness and reusability of MCs should be improved. Aoshima and co-workers successfully prepared a stable and recyclable Au NP (4 nm) catalyst by using vinyl ether star polymer, star(EOEOVE), as a scaffold for suppressing the particle coalescence during catalysis.97 The Au:star(EOEOVE) could be easily recovered from the reaction solution by raising the temperature above 40 °C: star(EOEOVE) showed lower critical solution temperature type phase separation behavior around 40 °C. Such studies may pave for new polymers that provide better platforms to study the catalysis of MCs.
Acknowledgment
The authors thank the following for fruitful collaboration: Prof. Miho Yamauchi; Prof. Seiji Yamazoe; Dr. Shinjiro Takano; Dr. M. J. Sharif; Dr. P. Maity; Dr. Shun Hayashi; Dr. Tatsuya Higaki; Ms. Setsuka Arii; Mr. Ryo Ishida; Mr. Atsushi Matsuo; Ms. Koto Hirano. This research was financially supported by the Elements Strategy Initiative for Catalysts & Batteries (ESICB) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
References
T. Tsukuda, H. Häkkinen, ed., Protected Metal Clusters: From Fundamentals to Applications (Elsevier, Amsterdam, 2015).

Shingo Hasegawa
Shingo Hasegawa received his M.S. degree in 2019 from The University of Tokyo. He is currently a Ph.D. candidate at The University of Tokyo under the supervision of Prof. Tatsuya Tsukuda. His Ph.D. project is the atomically-precise synthesis and catalytic application of polymer-stabilized Au-based clusters.

Tatsuya Tsukuda
Tatsuya Tsukuda received a Ph.D. degree in 1994 from The University of Tokyo under the supervision of Prof. Tamotsu Kondow. After postdoctoral research at RIKEN, he was appointed Assistant Professor at The University of Tokyo in 1994, and in 2000 he became an Associate Professor at Institute for Molecular Science. He was promoted to Professor at Catalysis Research Center of Hokkaido University in 2007, before moving to The University of Tokyo in 2011. His research interests cover the atomically-precise synthesis and characterization of gold clusters protected by ligands and their catalytic applications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}