





ABSTRACT
Total syntheses of borolithochromes H1, H2, I1, and I2, the red pigments isolated from fossils of Jurassic putative red alga Solenopora jurassica, have been achieved. The naphthoquinone possessing a chiral sec-butyl side chain has been synthesized from (S)-2-methylbutanol. The Diels-Alder reaction of the chiral naphthoquinone and the previously reported diene was followed by one pot S-methylation/intramolecular Corey-Chaykovsky reaction/epoxide rearrangement to provide the benzo[gh]tetraphene skeleton. Complexation of the resulting ligand with trimethyl borate and the following O-demethylation furnished a 1:1 mixture of borolithochromes I1 and I2, which were separated by HPLC using CHIRALPAK IC® to afford optically pure borolithochromes I1 (6) and I2 (7). On the other hand, borolithochromes H1 and H2 were not separated by HPLC in our laboratory. Fortunately, the mixture of the methyl ethers of borolithochromes H1 and H2 were separated and O-demethylation with magnesium iodide furnished optically pure borolithochromes H1 (4) and H2 (5).
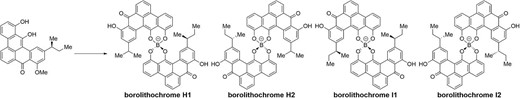
Total syntheses of borolithochromes H1, H2, I1, and I2 have been achieved.
Borolithochromes (Figure 1) have been isolated as red pigments from fossils of Jurassic putative red alga Solenopora jurassica (Wolkenstein, Gross and Falk 2010). Structures of them were proved to be the first boronated aromatic polyketides (Wolkenstein et al. 2015). The aromatic ligands possessed benzo[gh]tetraphene skeletons, which have been rare in natural products (Only clostrubins A and B are known as natural products possessing benzo[gh]tetraphene skeletons. Pidot et al. 2014; Shabuer et al. 2015). Thirteen borolithochromes have been isolated so far, and the diversity of this family is due to the combination of two of four types of ligands (Scheme 1) and the configuration of boron. Borolithochromes A (1), D (2), and G (3) were isolated as racemic forms, whereas other borolithochromes, possessing ligand(s) with a chiral carbon, were isolated as optically pure forms (Wolkenstein et al. 2015). Although borolithochromes have unique structures, the availability of only very small quantities of them has prevented assessment of their properties. Therefore, synthesis of borolithochromes are required to promote further studies.

Structures of borolithochromes.
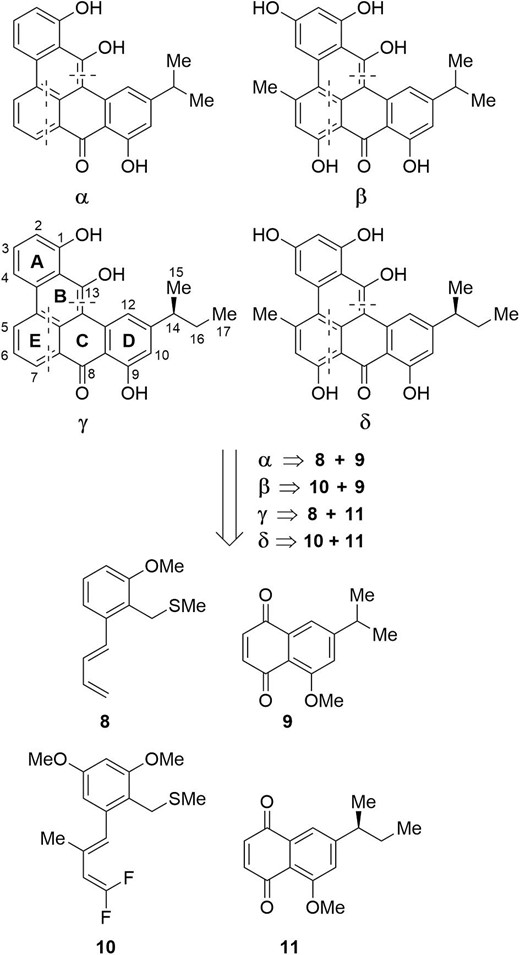
Ligands of borolithochromes and a strategy of the divergent synthesis.
Recently, we achieved the total syntheses of borolithochromes A (1), D (2), and G (3) as racemic forms (Kirita et al. 2024). The strategy for the divergent synthesis of ligands and a method to synthesize the hetero-complex were developed. However, efforts to synthesize optically pure borolithochromes have never been reported. Herein, we report the total syntheses and optical resolution of borolithochromes H1 (4), H2 (5), I1 (6), and I2 (7).
The diversity of 13 borolithochromes is due to the combination of two of the four types of ligands (Scheme 1). Recently, we reported the synthesis of α and β type ligands, which constituted borolithochromes A (1), D (2), and G (3), by Diels-Alder reaction between diene 8 and dienophile 9 and between 10 and 9, respectively. On the other hand, borolithochromes I1 (6) and I2 (7) are consisted of γ type ligand, whereas H1 (4) and H2 (5) are of α and γ type ligands. Therefore, in order to access these borolithochromes, synthesis of γ type ligand is required. Based on the diversity-oriented synthesis shown in Scheme 1, which might allow to synthesize all ligands by combination of two dienes (8 and 10) and two quinones (9 and 11), γ type ligand would be synthesized by combination of diene 8 and naphthoquinone 11. Since the synthesis of diene 8 has already reported previously (Kirita et al. 2024), chiral naphthoquinone 11 (CD rings of γ ligand) is required to be synthesized.
Results and discussion
Synthesis of naphthoquinone 11 started from TEMPO oxidation (Anelli et al. 1987) of commercially available (S)-2-methylbutanol (12) (Scheme 2). The resulting aldehyde was immediately subjected to the Grignard reaction to give alcohol 14. AZADO oxidation (Shibuya et al. 2006) of 14 was followed by the Honer-Wadsworth-Emmons reaction to give α,β-unsaturated ester 16. Deprotonation at the β-methyl group of 16 and O-silylation of the resulting dienolate afforded the corresponding vinylketene silyl acetal, which was subjected to the Diels-Alder reaction with benzoquinone to give naphthoquinone 11. In this reaction, benzoquinone played dual roles, including dienophile and oxidant for naphthohydroquinone derived in situ from the Diels-Alder adducts (Xu et al. 2014). Thus, naphthoquinone 11 was synthesized in six steps.
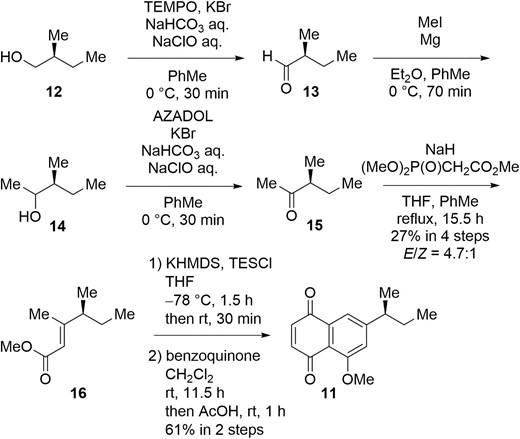
Synthesis of naphthoquinone 11.
The γ-type ligand (Scheme 1) was synthesized based on the previously reported synthesis of the α-type ligand (Scheme 3) (Kirita et al. 2024). The Diels-Alder reaction between diene 8 and dienophile 11 proceeded at 130 °C and the sequential air oxidation was promoted under the basic conditions to give anthraquinone 17. 17 was subjected to the one-pot sequential transformation including S-methylation, intramolecular Corey-Chaykovsky reaction (Lupattelli et al. 2009) and rearrangement of the resulting epoxide (19) (Rao et al. 1993) to produce benzo[gh]tetraphene 20 in excellent yield (98% from 17). Exposure of pentacyclic 20 with lithium chloride in N,N-dimethylformamide (DMF) at 80 °C promoted C1-selective O-demethylation to give ligand 21 (Bernard et al. 1989). The regioselectivity of this reaction might be due to the reagent delivery of C13 hydroxy group to react with C1-OMe group C. Similarity of nuclear magnetic resonance (NMR) spectral data to the corresponding intermediate in α type ligand showed that dimethyl ether 20 took C8 keto and C13 hydroxy form, whereas monomethyl ether 21 existed as C8 hydroxy and C13 keto form (Kirita et al. 2024).
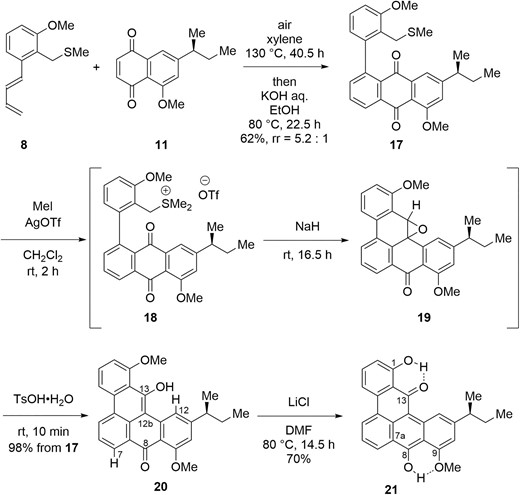
Synthesis of pentacyclic ligand 21.
Completion of the total synthesis of borolithochromes I1 (6) and I2 (7) is shown in Scheme 4. Treatment of monomethyl ether 21 with trimethyl borate in the presence of potassium carbonate promoted complexation to afford bis(aryl methyl ether) 22 in excellent yield as a 1:1 diastereomeric mixture. O-Demethylation of 22 proceeded with lithium iodide in DMF at 150 °C to furnish a 1:1 diastereomeric mixture of borolithochromes I1 (6) and I2 (7) in 98% yield (Harrison 1969). Finally, Separation of the diastereomers by HPLC using CHIRALPAK IC® gave optically pure borolithochrome I1 (6) and borolithochrome I2 (7). 1H NMR spectral data of synthetic 6 and 7 were identical with those of the natural products (Wolkenstein et al. 2015). Therefore, the first total syntheses of borolithochromes I1 (6) and I2 (7) have been achieved. It is noteworthy that borolithochromes I1 (6) and I2 (7) showed very large absolute value of optical rotation (6: [α]D20 ‒ 4322, 7: [α]D20 +3964).
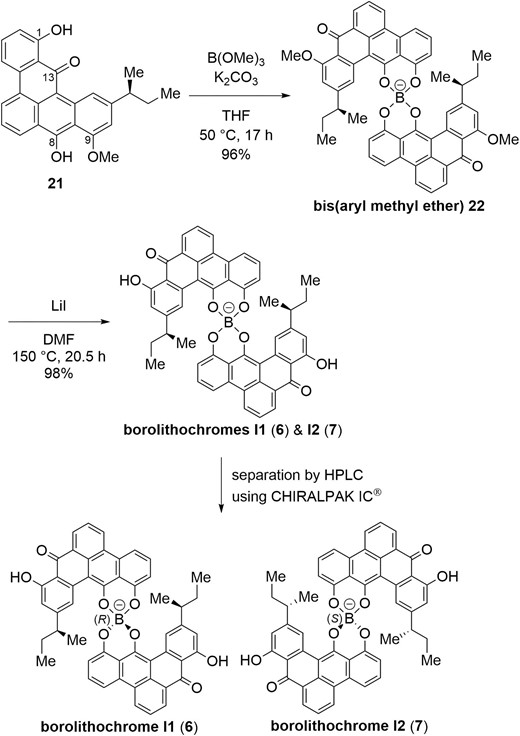
Synthesis and separation of borolithochromes I1 (6) and I2 (7).
Next, syntheses of borolithochromes H1 (4) and H2 (5), hetero-complexes possessing different ligands, were investigated (Scheme 5). Based on the previously reported method to synthesize borolithochrome D (2) (Kirita et al. 2024), a spiroborate of hetero-complex, 2-(dimethylamino)ethyl dimethyl borate (23) and α type ligand 24 were reacted at room temperature to obtain 1:1 ligand-borate complex 25. Subsequent reaction of 25 with γ type ligand 21 proceeded at 40 °C to afford bis(methyl ether) 26. Exposure of 26 with lithium iodide in DMF at 150 °C promoted O-demethylation to give a 1:1 mixture of borolithochromes H1 (4) and H2 (5). Unfortunately, the mixture was not separated by HPLC using chiral columns in our laboratory, including CHRAKPAK AD-H®, AS®, IB®, IC®, ID® IF®, and OD® (Wolkenstein group isolated borolithochromes H1 and H2 as single isomers by HPLC using a Phenomenex Gemini C18 column. Wolkenstein et al. 2015).
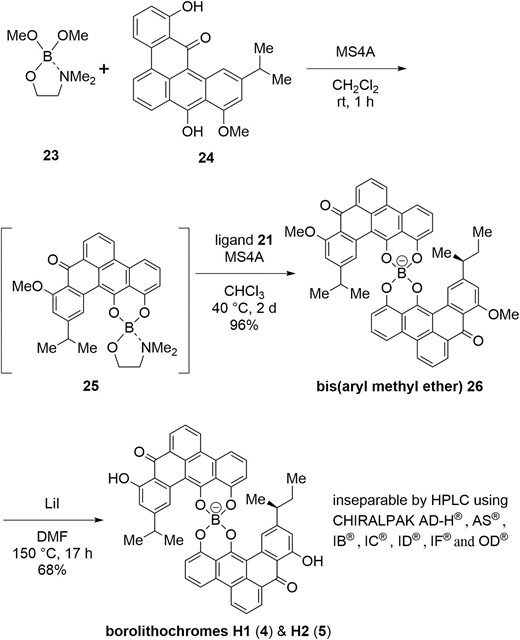
Synthesis of borolithochromes H1 (4) and H2 (5) as a mixture.
Although borolithochromes H1 (4) and H2 (5) were inseparable in our laboratory, we found that bis(aryl methyl ether) 26 was separable by HPLC using CHIRALPAK IB® (Scheme 6). Exposure of borolithochromes H1-type bis(aryl methyl ether) 27 with lithium iodide in DMF at 150 °C promoted O-demethylation, however, partial epimerization at boron was observed. Conditions including aluminum chloride in dimethyl sulfide (Node et al. 1981; Fuji and Node 1984; Reddy, Gudup and Ghorai 2016), used in the total synthesis of racemic borolithochromes A and D, also caused partial epimerization. Finally, we found that magnesium iodide promoted O-demethylation of 27 in THF at 50 °C without epimerization to furnish borolithochromes H1 (4) in high yield. Similarly, O-demethylation of 28 employing magnesium iodide (Yamaguchi et al. 1999) proceeded without epimerization to provide borolithochromes H2 (5) in 90% yield. 1H NMR spectral data of both synthetic 4 and 5 were identical with those of natural products (Wolkenstein et al. 2015). Therefore, the total syntheses of borolithochromes H1 (4) and H2 (5) were achieved.
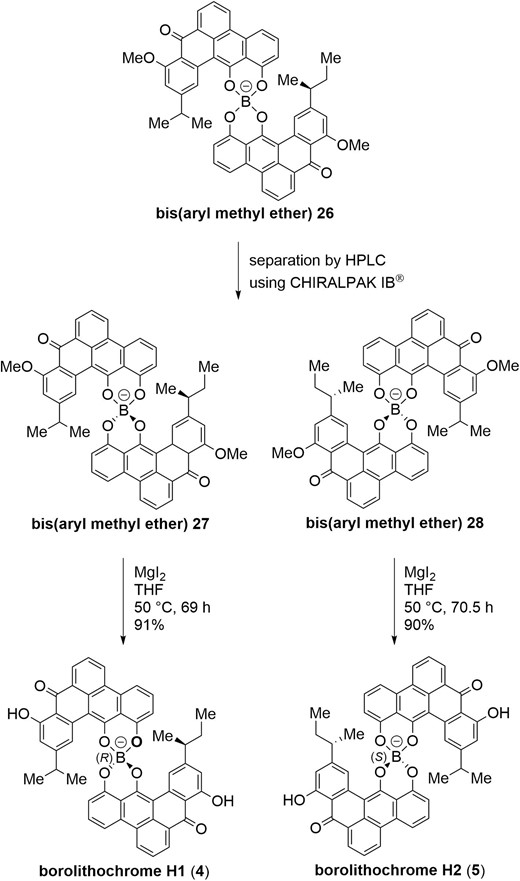
Separation of borolithochrome H dimethyl ether and O-demethylation to afford optically pure borolithochromes H1 (4) and H2 (5).
Conclusion
In conclusion, the first total syntheses of borolithochromes H1 (4), H2 (5), I1 (6), and I2 (7) have been achieved.
The common pentacyclic ligand (γ type ligand in Scheme 1) was synthesized by the strategy including the Diels-Alder reaction of diene 8 and chiral naphthoquinone 11 followed by the intramolecular Corey-Chaykovsky reaction and rearrangement of the resulting epoxide. Complexation of the common ligand 21 with trimethyl borate and the following O-demethylation furnished a 1:1 mixture of borolithochromes I1 (6) and I2 (7), which were separated by HPLC using CHIRALPAK IB® to afford optically pure borolithochromes I1 (6) and I2 (7). On the other hand, stepwise complexation employing borate 23 with ligand 24 and 21 provided borolithochrome H-type bis(aryl methyl ether) 26, which was subjected to O-demethylation afforded a 1:1 mixture of borolithochromes H1 (4) and H2 (5). Although the mixture was inseparable by HPLC in our laboratory, bis(aryl methyl ether) 26, the mixture of bis(aryl methyl ether) 27, and 28, was found to be separable. The resulting optically pure borolithochromes H1-type bis(aryl methyl ether) 27 and H2-type bis(aryl methyl ether) 28 was exposed to magnesium iodide to promote O-demethylation to give borolithochromes H1 (4) and H2 (5), respectively, without epimerization. These results show the methods to synthesize optically pure borolithochromes and related spiroborates possessing two polycyclic aromatic ligands.
Experimental
General procedure
1H NMR spectra were recorded at 400 MHz with JEOL ECS400 instruments or 500 MHz with JEOL JNM-ECZ500R instruments. 13C NMR spectra were recorded at 101 MHz with JEOL ECS400 instruments or 126 MHz with JEOL JNM-ECZ500R instruments. 11B NMR were recorded at 160 MHz with JEOL JNM-ECZ500R instruments. Chemical shits (δ) of 1H and 13C NMR spectra are quoted in parts per million (ppm) and referenced to the residual solvent peak. Chemical shifts (δ) of 11B NMR spectra were referenced to the chemical shifts of boron trifluoride etherate in chloroform-d as an external reference (0 ppm). Attenuated total reflectance-infrared spectroscopy (ATR-IR) spectra were recorded with Thermo Fisher Scientific Nicolet 6700. High resolution mass spectrometry (HR-MS) were obtained with Bruker Compact(ESI) ESI-TOF MS. UV-Vis spectra were recorded at JASCO V-630. Fluorescence spectra were recorded with JASCO FP-8200. Dry dichloromethane and dry chloroform were distilled form Phosphorus pentoxide. Dry tetrahydrofuran was distilled from lithium aluminum hydride immediately before use. Chlorotriethylsilane and dry N, N-dimethylformamide were distilled calcium hydride immediately before use. All reactions except those with individual instructions were carried out in a static nitrogen atmosphere in glass with magnetic stirring.
12 → 13
To the mixture of 12 (5.0 mL, 46.2 mmol) in toluene (125 mL) were added potassium bromide (550 mg, 4.62 mmol), a saturated aqueous solution of sodium hydrogen carbonate (47.0 mL), and 2,2,6,6-tetramethylpiperizine N-oxyl (75.9 mg, 0.486 mmol) at 0 ºC. Then, a mixture of a saturated aqueous solution of sodium hydrogen carbonate (78.6 mL) and an aqueous solution of sodium hypochlorite (>5% effective chlorine concentration, 103 mL) was added in several portions over 10 min. After stirring 30 min at 0 ºC, brine was added to the mixture. The organic layer was separated, and the aqueous layer was extracted with diethyl ether (100 mL × 2). The combined organic layer containing 13 was dried over sodium sulfate and was used for the next reaction without purification.
13 → 14
To the mixture of magnesium (2.34 g, 96.3 mmol) and a catalytic amount of iodine in dry diethyl ether (100 mL) was dropwise added methyl iodide (5.76 mL, 92.5 mmol) at 0 ºC. After stirring for 1.5 h, a crude solution 13 in toluene (125 mL) and diethyl ether (200 mL) was dropwise added over 1 h via canula. After stirring for additional 10 min, a saturated aqueous solution of sodium hydrogen carbonate (350 mL) was added to the mixture. The organic layer was separated, and the aqueous layer was extracted with diethyl ether (100 mL × 2). The combined organic solvent was concentrated by distillation of diethyl ether (oil bath temperature : 80 ºC-100 ºC) with Vigreux column φ = 1.9 cm, h = 20 cm. After 10 mL of toluene was added to wash the Vigreux column, the solution containing 14 was used for the next reaction without further purification.
14 → 15
To the mixture of a crude solution of 14 in toluene (135 mL) were added potassium bromide (550 mg, 4.62 mmol), a saturated aqueous solution of sodium hydrogen carbonate (47.0 mL), and 2-hydroxy-2-azaadamantane (38.9 mg, 0.254 mmol) at 0 ºC. Then, a mixture of a saturated aqueous solution of sodium hydrogen carbonate (78.6 mL) and an aqueous solution of sodium hypochlorite (effective chlorine concentration >5%, 103 mL) was added in several portions over 10 min. After stirring 30 min, brine was added to the mixture. The organic layer was separated and dried over sodium sulfate. The solution containing 15 was used for the next reaction without further purification.
15 → 16
To a solution of trimethyl phosphonoacetate (8.92 g, 49.0 mmol) in dry tetrahydrofuran (100 mL) was carefully added sodium hydride (1.94 g, 48.5 mmol, 60% dispersion in oil) at 0 ºC. After stirring for 15 min, a crude solution 15 in toluene (135 mL) was dropwise added to the mixture. The resulting mixture was stirred in reflux for 15.5 h. After cooling to room temperature, a saturated aqueous solution of sodium hydrogen carbonate (250 mL) was added to the mixture. The organic layer was separated, and the aqueous layer was extracted with toluene (50 mL × 2). The combined organic layer was concentrated by distillation (oil bath temperature : 100 ºC-140 ºC) with Vigreux column φ = 1.9 cm, h = 20 cm. The residue was purified by vacuum distillation (1.5 kPa, oil bath temperature : 140 ºC) with Vigreux column to give 16 (1.93 g, 12.4 mmol, 27% in four steps, E : Z = 4.7 : 1) as a colorless liquid.: Rf value 0.67 (n-hexane : ethyl acetate) = 5 : 1; [α]D20 + 16.6 (c 1.09, chloroform); 1H NMR (chloroform-d, 400 MHz) (data in brackets are those of Z-isomer) : δ(ppm) 5.67 (1H, s), [5.65 (1H, q, J = 1.0 Hz)], [3.80 (1H, ddq, J = 7.0, 7.0, 7.0 Hz)], 3.69 (3H, s), [3.67 (3H, s)], 2.13 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 2.09 (3H, d, J = 1.0 Hz), [1.77 (3H, d, J = 1.0 Hz)], 1.45 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.38 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.04 (3H, d, J = 7.0 Hz), [1.02 (3H, d, J = 7.0 Hz)], [0.84 (3H, dd, J = 7.0, 7.0 Hz)], 0.83 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (chloroform-d, 126 MHz) : δ(ppm) (only peaks of E-isomer are listed) 167.4, 164.7, 114.6, 50.8, 45.7, 27.5, 18.8, 15.4, 11.9; HRMS (ESI) m/z calcd for C9H16O2Na [M + Na]+ 179.1043, found 179.1051; IR (ATR) : 2964, 2877, 1717, 1645, 1434, 1378, 1222, 1158, 1032, and 868 (cm−1).
(S)-7-(but-2-yl)-5-methoxy-1,4-naphthoquinone 11
To a solution of potassium bis(trimethylsilyl)azide (0.5 m, 1.53 mL, 0.765 mmol) in freshly distilled tetrahydrofuran (2.0 mL) was dropwise added 16 (100.5 μL 0.640 mmol) at −78 ºC. After stirring for 1 h, freshly distilled chlorotriethylsilane (139.4 μL, 0.823 mmol) was dropwise added. The resulting mixture was stirred at −78 ºC for 30 min and at room temperature for 30 min. After the solvent was removed in vacuo, the residue was filtered through a pad of celite® with n-hexane, and the filtrate was concentrated in vacuo. This operation was repeated until the residue got colorless. The residue was dissolved in dichloromethane (2.0 mL), and benzoquinone (417.9 mg, 3.87 mmol) was added at 0 ºC. The mixture was stirred at 0 ºC for 1 h and at room temperature for 11.5 h. Then, acetic acid (142.5 μL, 2.49 mmol) was added to the reaction mixture. After stirring for 1 h, the resulting mixture was concentrated in vacuo. (Before concentration, toluene (2.0 mL) was added to remove acetic acid by azeotrope) The residue was purified by column chromatography on silica gel (toluene : ethyl acetate = 20 : 1) to give 11 (94.9 mg, 0.388 mmol, 61%) as a dark brown syrup.: Rf value 0.18 (n-hexane : ethyl acetate = 5 : 1); [α]D20 + 27.5 (c 0.23, chloroform); 1H NMR (chloroform-d, 500 MHz) : δ(ppm) 7.58 (1H, d, J = 1.0 Hz), 7.09 (1H, d, J = 1.0 Hz), 6.85 (1H, d, J = 10.0 Hz), 6.83 (1H, d, J = 10.0 Hz), 4.01 (3H, s), 2.72 (1H, tq, J = 6.8, 6.8 Hz), 1.66 (2H, dq, J = 7.2, 6.8 Hz), 1.29 (3H, d, J = 6.8 Hz), 0.85 (3H, t, J = 7.2 Hz); 13C NMR (chloroform-d, 126 MHz) : δ(ppm) 185.6, 184.1, 159.9, 156.2, 141.0, 136.1, 134.0, 117.9, 117.9, 116.6, 56.4, 42.5, 30.6, 21.3, 12.1; HRMS (ESI) m/z calcd for C15H16O3Na [M + Na]+ 267.0992, found 267.0993; IR (ATR) : 2960, 2930, 2873, 1654, 1593, 1458, 1305, 1272, 1237, 1170, 1033, and 845 (cm−1).
(S)-1-methoxy-5-[3-methoxy-2{(methylsulfanyl)methyl}phenyl]-3-(but-2-yl)anthraquinone 17
The mixture of 8 (1.39 g, 6.31 mmol) and 11 (557.2 g, 2.26 mmol) in xylene (10.0 mL) was stirred at 130 ºC for 40.5 h under air in dark. After cooling to room temperature, ethanol (10.0 mL) and an aqueous solution of potassium hydroxide (0.85 M, 1.0 mL) were added to the mixture. The resulting mixture was stirred at 80 ºC for 22.5 h. After cooling to room temperature, hydrochloric acid (1.0 m, 15 mL) and ethyl acetate (15 mL) were added. After the organic layer was separated, the aqueous layer was extracted with ethyl acetate (15 mL × 2). The combined organic layer was concentrated in vacuo. The residue was purified by column chromatography on silica gel (toluene : ethyl acetate = 10 : 1) to give the inseparable mixture of 17 and 17′ (641.6 mg, 1.39 mmol, 62%, rr = 5.2 :1) as a brown syrup.: Rf value 0.46 (toluene : ethyl acetate = 10 : 1); [α]D20 + 27.5 (c 0.18, chloroform, mixture of 17 and 17′); 1H NMR (chloroform-d, 500 MHz) (Data of 17′ are indicated in bracket) : δ(ppm) 8.37 (1H, dd, J = 7.5, 0.5 Hz), [8.31 (1H, dd, J = 8.0, 1.0 Hz)], 7.75 (1H, dd, J = 7.8, 7.8 Hz), [7.69 (1H, dd, J = 7.8, 7.8 Hz)], [7.59 (1H, d, J = 1.0 Hz)], 7.58 (1H, d, J = 0.8 Hz), 7.56 (1H, dd, J = 7.8, 1.0 Hz), 7.31 (1H, dd, J = 8.0, 8.0 Hz), 7.07 (1H, d, J = 0.5 Hz), [7.04 (1H, s)], 6.96 (1H, d, J = 8.0 Hz), [6.91 (1H, d, J = 8.0 Hz)], 6.75 (1H, d, J = 7.5 Hz), 4.05 (3H, s), 3.92 (3H, s), [3.90 (3H, s)], [3.87 (3H, s)], 3.58 (1H, d, J = 12.5 Hz), [3.58 (1H, d, J = 12.5 Hz)], [3.39 (1H, d, J = 12.5 Hz)], 3.35 (1H, d, J = 12.5 Hz), [2.72 (1H, tq, J = 7.0, 7.0 Hz)], 2.65 (1H, tq, J = 7.0, 7.0 Hz), [2.36 (3H, s)], 1.86 (3H, s), [1.67 (2H, dq, J = 7.5, 7.5 Hz)], 1.60 (2H, dq, J = 7.5, 7.0 Hz), [1.30 (3H, d, J = 7.0 Hz)], 1.23 (3H, d, J = 7.0 Hz), [0.86 (3H, t, J = 7.5 Hz)], 0.80 (3H, t, J = 7.5 Hz); 13C NMR (chloroform-d, 126 MHz) (Data of 17 are listed) : δ(ppm) 183.8, 182.5, 160.2, 157.5, 156.1, 143.3, 141.0, 136.9, 136.6, 136.0, 132.6, 129.0, 127.6, 127.3, 123.7, 120.9, 118.5, 117.6, 116.2, 109.4, 56.5, 55.5, 42.6, 30.6, 30.0, 21.3, 15.9, 12.1; HRMS (ESI) m/z calcd for C28H28O4SNa [M + Na]+ 483.1601, found 483.1597; IR (ART) : 2959, 2918, 1668, 1596, 1578, 1458, 1314, 1256, 1048, 990, and 716 (cm−1).
(S)-13-hydroxy-1,9-dimethoxy-11-(but-2-yl)-8H-benzo[gh]tetraphene-8-one 20
To a solution of 17 (618.2 mg, 1.34 mmol, mixture with 17′) in dry dichloromethane (12.5 mL) were added iodomethane (125.3 μL, 2.01 mmol) and silver trifluoromethanesulfonate (413.8 mg, 1.61 mmol) at room temperature. After stirring for 2 h, sodium hydride (135.0 mg, 3.38 mmol, 60% dispersion in oil) was added to the reaction mixture. The resulting mixture was stirred for 16.5 h at room temperature and the solution became slightly viscous. After adding dry dichloromethane (6.5 mL) and stirring for 30 min to ensure sufficient mixing, p-toluenesulfonic acid hydrate (968.5 mg, 4.94 mmol) was added to the mixture. The resulting mixture was stirred for 10 min, and hydrochloric acid (0.5 m, 18.0 mL) was added to the mixture. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (18.0 mL × 5). The combined organic layer was concentrated in vacuo. The residue was purified by column chromatography on silica gel (chloroform → methanol) to give 20 (455.0 mg, 1.10 mmol, 98% from 17) as a brown solid.: Rf value 0.17 (toluene : ethyl acetate = 10 : 1); [α]D20 + 2.23 (c 0.11, chloroform); m.p. 185.5 ºC-186.5 ºC; 1H NMR (chloroform-d, 500 MHz) : δ(ppm) 11.46 (1H, s), 8.86 (1H, s), 8.67 (2H, d, J = 7.8 Hz), 8.18 (1H, d, J = 8.0 Hz), 7.63 (1H, dd, J = 7.8, 7.8 Hz), 7.47 (1H, dd, J = 8.0, 8.0 Hz), 6.98 (1H, d, J = 7.8 Hz), 6.87 (1H, s), 4.11 (3H, s), 4.07 (3H, s), 2.77 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 1.77 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.72 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.38 (3H, d, J = 7.0 Hz), 0.93 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (chloroform-d, 126 MHz) : δ(ppm) 183.7, 161.0, 156.8, 155.5, 153.6, 139.0, 133.7, 130.1, 129.4, 128.9, 128.4, 127.6, 124.3, 124.0, 120.6, 119.2, 117.0, 115.7, 109.0, 108.1, 107.5, 56.9, 56.3, 42.9, 31.0, 21.6, 12.4; HRMS (ESI) m/z calcd for C27H24O4Na [M + Na]+ 435.1567, found 435.1573; IR (ATR) : 3216, 2922, 1634, 1579, 1441, 1282, 1254, 1039, 782, and 741 (cm−1); UV-vis (methanol, 0.010 m m) : {λmax nm (log ε)} 451 (4.46); Fluorescence (methanol, λexc 451 nm) λem 523 nm.
(S)-1,8-dihydroxy-9-methoxy-11-(but-2-yl)-13H-benzo[gh]tetraphene-13-one 21
To a solution of 20 (406.1 mg, 0.985 mmol) in dry dimethylformamide (8.1 mL) was added lithium chloride (414.3 mg, 9.77 mmol). The reaction mixture was stirred at 80 ºC for 14.5 h. After cooling to room temperature, hydrochloric acid (3.0 m, 10.0 mL) and chloroform (10.0 mL) was added to the reaction mixture. The organic layer was separated, and the aqueous layer was extracted with chloroform (10.0 mL × 3). The combined organic layer was concentrated in vacuo. The residue was purified by column chromatography on silica gel (chloroform) to give 21 (272.9 mg, 0.685 mg, 70%) as a dark purple powder.: Rf value 0.90 (ethyl acetate : acetone = 5 : 1); m.p. 215.0 ºC-216.0 ºC; [α]D20 + 26.5 (c 0.16, chloroform); 1H NMR (chloroform-d, 500 MHz) : δ(ppm) 14.96 (1H, s), 11.61 (1H, s), 9.84 (1H, s), 8.62 (1H, d, J = 8.0 Hz), 8.60 (1H, d, J = 8.0 Hz), 7.81 (1H, d, J = 8.0 Hz), 7.55 (2H, dd, J = 8.0, 8.0 Hz), 7.02 (1H, d, J = 8.0 Hz), 6.78 (1H, s), 4.20 (3H, s), 2.92 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 1.81 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.75 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 1.42 (3H, d, J = 7.0 Hz), 0.95 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (chloroform-d, 126 MHz) : δ(ppm) 188.4, 163.5, 162.4, 157.5, 152.9, 138.1, 135.7, 133.4, 130.7, 128.1, 126.0, 125.9, 123.3, 119.6, 118.9, 117.7, 115.8, 112.4, 112.1, 110.1, 103.7, 56.5, 43.1, 30.8, 21.5, 12.4; HRMS (ESI) m/z calcd for C26H22O4Na [M + Na]+ 421.1410, found 421.1415; IR (ATR) : 3187, 2923, 1619, 1556, 1424, 1400, 1235, 1179, 1152, 1052, 758, and 658 (cm−1); UV-vis (methanol, 0.011 m m) : {λmax nm (log ε)} 528 (3.98); Fluorescence (methanol, λexc 491 nm) λem 573 nm.
Bis(aryl methyl ether) (22)
To a solution of 21 (31.4 mg, 78.8 μmol) in dry tetrahydrofuran (630 μL) were added trimethyl borate (4.50 μL, 39.4 μmol) and potassium carbonate (16.9 mg, 0.122 mmol). The mixture was stirred at 50 ºC for 17 h. After cooling to room temperature, the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate → acetone → methanol) to give 22 (30.3 mg, 37.9 μmol, 96%) as an orange solid.: Rf value 0.13 (ethyl acetate : acetone = 5 : 1); m.p. >300 ºC; 1H NMR (dimethyl sulfoxide-d6, 500 MHz) (Data in brackets are those of stereoisomers at the boron. Data in and out of brackets are interchangeable) : δ(ppm) 9.00 (2H, d, J = 7.0 Hz), [8.98 (2H, d, J = 7.0 Hz)], 8.72 (2H, s), [8.68 (2H, s)], 8.37 (2H, d, J = 7.0 Hz), [8.36 (2H, d, J = 7.0 Hz)], 8.29 (2H, d, J = 7.0 Hz), [8.27 (2H, d, J = 7.0 Hz)], 7.70-7.63 (8H, m), 7.09 (2H, d, J = 7.5 Hz), [7.06 (2H, d, J = 7.5 Hz)], 6.57 (2H, s), [6.51 (2H, s)], 3.73 (6H, s), [3.70 (6H, s)], 1.62 (2H, ddq, J = 7.0, 7.0, 7.0 Hz), [1.45 (2H, ddq, J = 7.0, 7.0, 7.0 Hz)], 1.04 (2H, ddq, J = 14.0, 7.0, 7.0 Hz), [0.87 (2H, ddq, J = 14.0, 7.0, 7.0 Hz)], 0.83 (2H, ddq, J = 14.0, 7.0, 7.0 Hz), [0.73 (2H, ddq, J = 14.0, 7.0, 7.0 Hz)], 0.48 (6H, d, J = 7.0 Hz), [0.29 (6H, d, J = 7.0 Hz)], 0.13 (6H, dd, J = 7.0 Hz), [0.09 (6H, dd, J = 7.0, 7.0 Hz)]; 13C NMR (dimethyl sulfoxide-d6, 126 MHz) (some peaks are overlapped) (data in brackets are those of stereoisomers at the boron. Data in and out of brackets are interchangeable.) : δ(ppm) 181.6, [181.6], 160.3, [160.2], 156.4, [156.2], 155.7, 152.8, [152.6], 139.0, 132.5, [132.4], 130.6, [130.5], 129.7, [129.6], 129.5, [129.4], 127.7, 127.3, [127.2], 124.9, [124.8], 123.1, 119.5, [119.2], 117.6, [117.6], 115.8, [115.7], 113.6, 113.0, 112.9, 107.6, [107.5], 104.0, [103.7], 55.6, [55.6], 41.5, [41.5], 29.5, [29.2], 21.2, [20.8], 11.7, [11.5]; 11B NMR (dimethyl sulfoxide-d6, 160 MHz) : δ(ppm) 2.0; HRMS (ESI) m/z calcd for C52H40O8B [M]− 803.2822, found 803.2793; IR (ATR) : 2955, 1602, 1560, 1509, 1468, 1337, 1305, 1213, 1128, 931, 868, 778, and 709 (cm−1); UV-vis (methanol, 6.0 μm) : {λmax nm (log ε)} 481 (4.95); Fluorescence (methanol, λexc 482 nm) λem 552 nm.
Borolithochrome I1 (6) and borolithochrome I2 (7)
To a solution of 22 (15.7 mg, 19.0 μmol) in dry dimethylformamide (320 μL) was added lithium iodide (56.4 mg, 0.421 μmol). The mixture was stirred at 150 ºC for 20.5 h. After cooling to room temperature, the solvent was removed in vacuo. Tetrahydrofuran (5.0 mL) and pH 7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate buffer (10.0 mL) were added, and the mixture was stirred well. After brine was added, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10.0 mL × 2). The combined organic layer was concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 5 : 1) to give the mixture of 6 and 7 (14.9 mg, 18.7 μmol, 98%) as a red solid. The NMR spectrum and the data of physical properties of borolithochrome I were measured after separation into borolithochrome I1 (6) and borolithochrome I2 (7) (described later).
Separation of borolithochrome I1 (6) and I2 (7)
The mixture of 6 and 7 (9.7 mg) in n-hexane : 2-propanol = 5 : 4 (9.0 mL) was loaded into HPLC column (CHIRALPAK IC®, n-hexane : 2-propanol : water : diethyl amine = 5 : 4 : 0.56 : 0.022) and separated into 6 and 7. Each solvent was concentrated in vacuo. Tetrahydrofuran (5.0 mL) and pH 7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate buffer (10.0 mL) were added, and the mixture was stirred well. Then after brine was added, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10.0 mL × 2). The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 5 : 1) to give 6 (1.9 mg, 20%) and 7 (2.1 mg, 22%).
6 : Rf value 0.54 (ethyl acetate : acetone = 5 : 1); [α]D20 −4321.6 [c 0.0018, methanol (the solution is highly diluted due to strong light absorption)]; m.p. >300 ºC; 1H NMR (acetone-d6, 500 MHz) : δ(ppm) 14.45 (2H, s), 9.11 (2H, d, J = 7.8 Hz), 8.80 (2H, s), 8.69 (2H, d, J = 7.5 Hz), 8.32 (2H, d, J = 8.0 Hz), 7.73 (2H, dd, J = 8.0, 7.5 Hz), 7.70 (2H, dd, J = 7.8, 7.5 Hz), 7.16 (2H, d, J = 7.5 Hz), 6.36 (2H, s), 1.66 (2H, ddq, J = 7.0, 7.0, 7.0 Hz), 0.94 (2H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.90 (2H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.51 (6H, d, J = 7.0 Hz), 0.29 (6H, dd, J = 7.0, 7.0 Hz); 13C NMR (acetone-d6, 126 MHz) : δ(ppm) 188.0, 164.9, 160.5, 157.8, 157.3, 139.1, 134.1, 132.1, 131.9, 130.2, 128.6, 127.7, 126.7, 123.7, 119.3, 119.2, 115.0, 114.4, 113.7, 111.9, 105.2, 43.3, 32.0, 21.8, 12.4; 11B NMR (acetone-d6, 160 MHz) : δ(ppm) 2.5; HRMS(ESI) m/z calcd for C50H36O8B [M]− 775.2509 found 775.2492; IR (ATR) : 3448, 2956, 2928, 1701, 1618, 1561, 1474, 1321, 1284, 1256, 1218, 1125, 1081, 934, and 859 (cm−1); UV-vis (methanol, 4.9 μm) : {λmax nm (log ε)} 504 (4.27).
7 : Rf value 0.54 (ethyl acetate : acetone = 5 : 1); [α]D20 + 3963.7 [c 0.0026, methanol (the solution is highly diluted due to strong light absorption)]; m.p. >300 ºC; 1H NMR (acetone-d6, 500 MHz) : δ(ppm) 14.48 (2H, s), 9.10 (2H, d, J = 8.0 Hz), 8.85 (2H, s), 8.70 (2H, d, J = 7.5 Hz), 8.30 (2H, d, J = 8.0 Hz), 7.71 (2H, dd, J = 8.0, 7.5 Hz), 7.70 (2H, dd, J = 8.0, 7.5 Hz), 7.13 (2H, d, J = 7.5 Hz), 6.41 (2H, s), 1.77 (2H, ddq, J = 7.0, 7.0, 7.0 Hz), 1.13 (2H, ddq, J = 13.5, 7.5, 7.0), 1.02 (2H, ddq, J = 13.5, 7.5, 7.0), 0.61 (6H, d, J = 7.0 Hz), 0.24 (6H, dd, J = 7.5, 7.5 Hz); 13C NMR (acetone-d6, 126 MHz) : δ(ppm) 188.0, 165.0, 160.3, 157.7, 157.1, 139.1, 134.1, 132.2, 131.9, 130.2, 128.7, 127.7, 126.6, 123.7, 119.5, 119.4, 115.0, 114.4, 113.6, 111.8, 105.0, 43.2, 32.0, 21.9, 12.2; 11B NMR (acetone-d6, 160 MHz) : δ(ppm) 2.4; HRMS(ESI) m/z calcd for C50H36O8B [M]− 775.2509 found 775.2503; IR (ATR) : 3354, 2958, 2925, 1702, 1619, 1560, 1472, 1319, 1217, 1124, 935, and 871 (cm−1); UV-vis (methanol, 5.1 μm) : {λmax nm (log ε.)} 505 (4.18).
Bis(aryl methyl ether) 26
To a solution of trimethyl borate (3.40 μL, 29.8 μmol) in dry dichloromethane (1.0 mL) with molecular sieves 4A was added 2-(dimethylamino)ethanol (3.29 μL, 32.5 μmol). After stirring at room temperature for 1 h, 24 (10.2 mg, 26.5 μmol) was added to the mixture. The reddish-purple mixture was stirred for another 1 h, and the color of the solution turned orange. The mixture was filtered through a glass filter with dry dichloromethane, and the filtrate was concentrated in vacuo. The residue was purified by reprecipitation with a mixture of n-hexane—dichloromethane (8 : 1). The precipitate was dissolved in dry chloroform (1.0 mL) with molecular sieves 4A. Then, 21 (10.8 mg, 27.1 μmol) was added to the mixture. The resulting mixture was stirred at 40 ºC for 2 d. After cooling to room temperature, the solvenent was removed in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 1 : 1 → acetone) to give 26 (20.6 mg, 25.3 μmol, 96%) as an orange solid. The NMR spectrum and the data of physical properties of borolithochrome H dimethyl ether were measured after separation into bis(aryl methyl ether) (27) and bis(aryl methyl ether) (28) (described later).
Borolithochrome H1 (4) and H2 (5)
To a solution of 26 (3.2 mg, 3.94 μmol) in dry dimethylformamide (200 μL) was added lithium iodide (10.5 mg, 78.4 μmol). The mixture was stirred at 150 ºC for 17 h. After cooling to room temperature, the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 5 : 1) to give an inseparable 1 : 1 mixture of 4 and 5 (2.1 mg, 2.68 μmol, 68%) as a red solid.
Separation of bis(aryl methyl ether) (27) and bis(aryl methyl ether) (28)
The mixture of crude 26 (25.3 mg) in 2-propanol (30 mL) was loaded into HPLC column (CHIRALPAK IB®, n-hexane : 2-propanol : water : diethyl amine = 5 : 4 : 0.56 : 0.022) and separated into 27 and 28. Each solvent was concentrated in vacuo. Tetrahydrofuran (5.0 mL) and pH 7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate buffer (10.0 mL) were added, and the mixture was stirred well. Then after brine was added, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10.0 mL × 2). The residue was purified by column chromatography on silica gel (ethyl acetate → acetone → methanol) to give 27 (9.3 mg, 38%) and 28 (7.8 mg, 31%).
27 : Rf value 0.14 (ethyl acetate : acetone = 5 : 1); [α]D20 −722.8 (c 0.10, methanol); m.p. >300 ºC; 1H NMR (N, N-dimethylformamide-d7, 400 MHz) : δ(ppm) 9.04 (2H, d, J = 7.5 Hz), 8.99 (1H, s), 8.93 (1H, s), 8.47 (2H, d, J = 7.5 Hz), 8.34 (2H, d, J = 8.0 Hz), 7.74-7.64 (4H, m), 7.12 (2H, d, J = 7.5 Hz), 6.68 (1H, s), 6.63 (1H, s), 3.81 (3H, s), 3.80 (3H, s), 1.96 (1H, qq, J = 7.0, 7.0 Hz), 1.63 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 1.02 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.88 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.62 (3H, d, J = 7.0 Hz), 0.54 (3H, d, J = 7.0 Hz), 0.44 (3H, d, J = 7.0 Hz), 0.25 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (N, N-dimethylformamide-d7, 101 MHz) (some peaks were overlapped) : δ(ppm) 182.3, 182.3, 161.2, 161.1, 157.5, 157.4, 156.9, 156.9, 154.4, 153.4, 140.0, 140.0, 133.3, 133.3, 130.9, 130.8, 130.7, 130.6, 130.4, 128.0, 127.9, 127.6, 125.7, 125.7, 123.3, 120.3, 119.7, 118.6, 118.5, 116.8, 114.1, 114.0, 113.2, 108.0, 107.5, 104.7, 56.0, 55.7, 42.5, 23.0, 23.0, 21.2, 21.1, 11.9; 11B NMR (N, N-dimethylformamide-d7, 160 MHz) : δ(ppm) 2.3; HRMS(ESI) m/z calcd for C51H38O8B [M]− 789.2665 found 789.2656; IR (ATR) : 3363, 2924, 1603, 1561, 1512, 1468, 1337, 1310, 1214, 1128, 1073, 987, 940, and 763 (cm−1).
28 : Rf value 0.14 (ethyl acetate : acetone = 5 : 1); [α]D20 + 1194.3 (c 0.12, methanol); m.p. >300 ºC; 1H NMR (acetone-d6, 400 MHz) (the signal of H14 is overlapped with the solvent peak) : δ(ppm) 9.09 (1H, d, J = 1.0 Hz), 8.97 (1H, d, J = 1.0 Hz), 8.94 (2H, d, J = 8.0 Hz), 8.48 (1H, dd, J = 7.5, 1.0 Hz), 8.47 (1H, dd, J = 7.5, 1.0 Hz), 8.23 (1H, d, J = 8.0 Hz), 8.22 (1H, d, J = 8.0 Hz), 7.65 (1H, dd, J = 8.0, 8.0 Hz), 7.65 (1H, dd, J = 7.5, 7.5 Hz), 7.65 (1H, dd, J = 7.5, 7.5 Hz), 7.64 (1H, dd, J = 8.0 Hz), 7.08 (1H, d, J = 7.5 Hz), 7.05 (1H, d, J = 7.5 Hz), 6.64 (1H, d, J = 1.0 Hz), 6.58 (1H, d, J = 1.0 Hz), 3.80 (3H, s), 3.79 (3H, s), 1.68 (1H, ddq, J = 7.0, 6.8, 6.8 Hz), 1.08 (1H, ddq, J = 13.0, 7.0, 6.8 Hz), 0.90 (1H, ddq, J = 13.0, 7.0, 6.8 Hz), 0.68 (3H, d, J = 6.8 Hz), 0.61 (3H, d, J = 6.8 Hz), 0.55 (3H, d, J = 6.8 Hz), 0.15 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (acetone-d6, 126 MHz) (some peaks were overlapped) : δ(ppm) 183.2, 161.7, 161.6, 158.3, 157.8, 157.8, 155.0, 153.8, 140.9, 140.8, 133.9, 131.2, 131.0, 128.3, 128.2, 126.3, 126.2, 123.5, 121.5, 120.8, 119.3, 117.5, 114.7, 114.6, 113.3, 108.6, 108.2, 105.2, 56.4, 43.0, 35.6, 32.3, 23.6, 23.6, 23.3, 22.0, 14.3, 12.2; 11B NMR (acetone-d6, 160 MHz) : δ(ppm) 3.4; HRMS(ESI) m/z calcd for C51H38O8B [M]− 789.2665 found 789.2656; IR (ATR) : 3358, 2830, 1603, 1564, 1517, 1468, 1339, 1306, 1235, 1214, 1126, 1048, 978, 938, and 761 (cm−1).
Borolithochrome H1 (4)
To a solution of 27 (7.3 mg, 8.98 μmol) in dry tetrahydrofuran (200 μL) was added magnesium iodide (28.6 mg, 0.103 mmol). The mixture was stirred at 50 ºC for 69 h. After cooling to room temperature, the solvent was removed in vacuo. Tetrahydrofuran (5.0 mL) and pH 7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate buffer (10.0 mL) were added, and the mixture was stirred well. Then after brine was added, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10.0 mL × 2). The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 5 : 1) to give 4 (6.4 mg, 8.14 μmol, 91%) as a red solid.: Rf value 0.19 (ethyl acetate : acetone = 5 : 1); [α]D20 −1493.9 (c 0.1, methanol); m.p. >300 ºC; 1H NMR (acetone-d6, 500 MHz) (the signal of H14 is overlapped with the solvent peak) : δ(ppm) 14.45 (2H, s), 9.11 (1H, d, J = 7.0 Hz), 9.10 (1H, d, J = 7.0 Hz), 8.85 (1H, s), 8.79 (1H, s), 8.69 (2H, d, J = 7.5 Hz), 8.32 (1H, d, J = 7.5 Hz), 8.31 (1H, d, J = 7.0 Hz), 7.76-7.66 (4H, m), 7.16 (1H, d, J = 7.5 Hz), 7.15 (1H, d, J = 7.5 Hz), 6.41 (1H, s), 6.37 (1H, s), 1.67 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 0.94 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.91-0.84 (1H, m), 0.65 (3H, d, J = 7.0 Hz), 0.58 (3H, d, J = 7.0 Hz), 0.52 (3H, d, J = 7.0 Hz), 0.30 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (acetone-d6, 126 MHz) (some peaks are overlapped): δ(ppm) 188.0, 165.0, 164.9, 160.4, 158.3, 157.7, 157.3, 139.1, 139.1, 134.1, 134.1, 132.1, 132.0, 131.9, 131.8, 130.2, 130.2, 128.6, 127.6, 126.7, 126.6, 123.7, 119.2, 118.8, 117.3, 117.3, 115.1, 114.3, 113.7, 113.7, 111.9, 111.2, 105.1, 43.3, 35.7, 32.0, 23.6, 23.4, 21.8, 12.4; 11B NMR (acetone-d6, 160 MHz) : δ(ppm) 2.4; HRMS(ESI) m/z calcd for C49H34O8B [M−] 761.2352 found 761.2347; IR (ATR) : 3386, 2958, 2926, 1702, 1560, 1473, 1320, 1217, 1125, 1090, 939, 872, and 742 (cm−1).
Borolithochrome H2 (5)
To a solution of 28 (5.9 mg, 7.26 μmol) in dry tetrahydrofuran (200 μL) was added magnesium iodide (23.1 mg, 83.1 μmol). The mixture was stirred at 50 ºC for 70.5 h. After cooling to room temperature, the solvent was removed in vacuo. Tetrahydrofuran (5.0 mL) and pH 7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate buffer (10.0 mL) were added, and the mixture was stirred well. Then after brine was added, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10.0 mL × 2). The residue was purified by column chromatography on silica gel (ethyl acetate : acetone = 5 : 1) to give 5 (5.1 mg, 6.50 μmol, 90%) as a red solid.: Rf value 0.19 (ethyl acetate : acetone = 5 : 1); [α]D20 + 2903.9 [c 0.001, methanol (The solution is highly diluted due to strong light absorption.)]; m.p. >300 ºC; 1H NMR (acetone-d6, 500 MHz) (the signal of H14 is overlapped with the solvent peak) : δ(ppm) 14.48 (1H, s), 14.47 (1H, s), 9.10 (2H, d, J = 7.0 Hz), 8.91 (1H, d, J = 1.5 Hz), 8.80 (1H, d, J = 1.5 Hz), 8.70 (1H, d, J = 7.0 Hz), 8.69 (1H, dd, J = 7.0, 1.5 Hz), 8.30 (1H, d, J = 8.0 Hz), 8.30 (1H, d, J = 7.5 Hz), 7.75-7.67 (4H, m), 7.15 (1H, d, J = 7.5 Hz), 7.12 (1H, d, J = 7.5 Hz), 6.45 (1H, s), 6.39 (1H, s), 1.71 (1H, ddq, J = 7.0, 7.0, 7.0 Hz), 0.96 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.90 (1H, ddq, J = 14.0, 7.0, 7.0 Hz), 0.71 (3H, d, J = 7.0 Hz), 0.65 (3H, d, J = 7.0 Hz), 0.58 (3H, d, J = 7.0 Hz), 0.20 (3H, dd, J = 7.0, 7.0 Hz); 13C NMR (acetone-d6, 126 MHz) (some peaks are overlapped) : δ(ppm) 188.0, 165.0, 164.9, 160.4, 158.3, 158.2, 157.7, 157.1, 139.2, 139.1, 134.1, 132.1, 132.1, 131.9, 130.2, 128.7, 127.6, 126.7, 123.7, 119.5, 119.4, 118.8, 117.2, 115.1, 115.0, 114.3, 113.7, 111.8, 111.7, 111.2, 105.0, 43.2, 35.8, 32.0, 23.6, 23.5, 21.9, 12.2; 11B NMR (acetone-d6, 160 MHz) : δ(ppm) 2.4; HRMS(ESI) m/z calcd for C49H34O8B [M−] 761.2352 found 761.2351; IR (ATR) : 3401, 2958, 2928, 1697, 1619, 1588, 1471, 1282, 1216, 1124, 1088, 939, 872, and 742 (cm−1).
Data availability
The authors confirm that the data underlying this article are available in the article and in its online supplementary material.
Author contribution
S.H. designed this study and wrote the manuscript. S.H. and K.K. designed the synthetic route, and K.K., H, M., G.E., and K.I. conducted the synthetic experiments.
Funding
This work was financially supported by Kosé Cosmetology Research Foundation and JSPS KAKENHI Grant [numbers JP21J20095, JP22KJ2878]. This work was the result of using research equipments in Material Characterization Central Laboratory at Waseda University shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant [number JPMXS0440500024].
Disclosure statement
No potential conflict of interest was reported by the authors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}