





Abstract
Cancer cells are often exposed to cell intrinsic stresses and environmental perturbations that may lead to accumulation of unfolded and/or misfolded proteins in the lumen of endoplasmic reticulum (ER), a cellular condition known as ER stress. In response to ER stress, the cells elicit an adaptive process called unfolded protein response (UPR) to cope with the stress, supporting cellular homeostasis and survival. The ER stress sensors inositol requiring protein 1α (IRE1α), eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3, also called PERK), and activating transcription factor 6 (ATF6) constitute the three branches of UPR to resolve ER stress. IRE1α, PERK, and ATF6 play an important role in tumor cell growth and survival. They are also involved in chemotherapy resistance of cancers. These have generated widespread interest in targeting these UPR branches for cancer treatment. In this review, we provide an overview of the role of IRE1α, PERK, and ATF6 in cancer progression and drug resistance and we summarize the research advances in targeting these UPR branches to enhance the efficacy of chemotherapy of cancers.
Introduction
The endoplasmic reticulum (ER) is the central intracellular organelle in the secretory pathway. It is responsible for protein translocation, protein folding, and protein post-translational modification that allows further transport of proteins to the Golgi apparatus and ultimately to vesicles for secretion or display on the plasma surface. The accumulation of unfolded and/or misfolded proteins in the ER, a process named ‘ER stress’, triggers the unfolded protein response (UPR). The UPR is a defense mechanism activated by cells during stressful conditions in response to an accumulation of unfolded/misfolded proteins in ER [1–4]. The UPR is a tightly orchestrated collection of intracellular signal transduction reactions. It functions to induce the transcription of genes encoding ER chaperones and ER-associated degradation (ERAD) components to enhance the protein folding and clearance capacity and restore ER homeostasis.
UPR initiates survival signals to restore ER homeostasis. The ER membrane-localized ER stress sensors inositol requiring enzyme 1α (IRE1α), eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2A3K, also called PERK), and activating transcription factor 6 (ATF6) constitute the three branches of UPR to resolve ER stress (Fig. 1). Under normal condition, the immunoglobulin-binding protein (BiP, also called GRP78) binds the ER-luminal domains of IRE1α, PERK, and ATF6 and sequesters them in an inactive form [5]. The unfolded and/or misfolded proteins in the ER bind to BiP and cause BiP to be released from IRE1α, PERK, and ATF6, which triggers their signaling pathways. BiP functions as the gatekeeper that binds and limits activation of these three transmembrane proteins. The UPR activates adaptive and pro-survival signals in response to ER stress, and the UPR’s pro-survival effect can be converted to a pro-death signal when the stress stimuli are acute or persistent that the cells cannot restore ER homeostasis [1,2]. Thus, the UPR has a dual role in cell fate decisions, which depends on the intensity and duration of the stress.
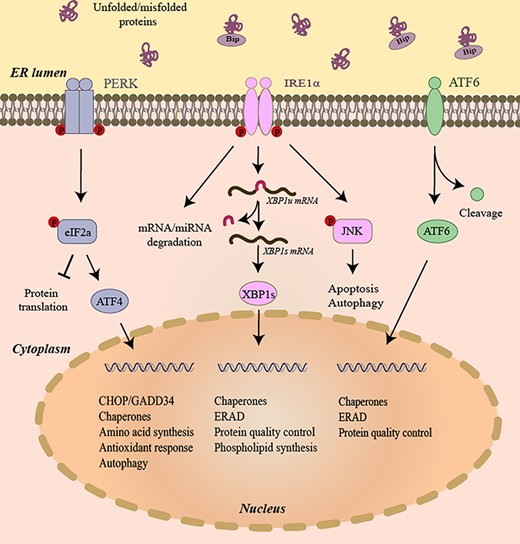
The three branches of UPR In the presence of unfolded and/or misfolded proteins, the IRE1α’s kinase domain trans-autophosphorylates. IRE1α phosphorylation leads to allosteric activation of the adjacent RNase. In response to low level of ER stress, IRE1α’s RNase excises a 26-nucleotide intron from the mRNA encoding the XBP1 transcription factor to produce transcription factor XBP1s. XBP1s then translocates to the nucleus and induces transcription of many genes that attempt to restore ER homeostasis. In the presence of unfolded/misfolded proteins, PERK dimerizes and phosphorylates eIF2α. Phosphorylation inhibits eIF2α activity and hence slows down global protein translation. In contrast, translation of the transcription factor ATF4 is selectively upregulated when the amount of active eIF2α is limited. ATF4 expression transcriptionally upregulates CHOP, which tips the ER toward homeostasis through the induction of a number of corrective genes. When activated, ATF6 translocates to the Golgi where it is cleaved by S1P and S2P protease to generate active transcription factor ATF6. The active ATF6 induces transcription of genes that restore ER homeostasis. XBP1u mRNA, unspliced XBP1 mRNA. XBP1s mRNA, spliced XBP1 mRNA.
IRE1α is the most evolutionally conserved among the three UPR branches. It is ubiquitously expressed and possesses endoribonuclease and protein kinase activities. In ER stress, IRE1α dissociates from BiP, and dimerizes and/or oligomerizes, followed by autophosphorylation and activation of IRE1α cytosolic RNase domain [5]. Upon activation, IRE1α removes a 26-nt intron from the mRNA encoding X-box-binding protein 1 (XBP1) to generate a spliced active form of this transcription factor XBP1s. XBP1s controls the expressions of genes involved in protein chaperones, ERAD, protein quality control, and phospholipid synthesis [5]. Activation of IRE1α also regulates the IRE1α-dependent decay (RIDD) of mRNAs [6], which may reduce the protein-folding load on the ER. When activated, IRE1α can directly cleave selected microRNAs that normally repress proapoptotic targets [7]. In addition to endoribonuclease activity, IRE1α also has kinase activity. Upon severe ER stress, IRE1α is overactivated and it recruits TRAF2 to facilitate phosphorylation of c-Jun N-terminal kinase (JNK) [8], which may trigger cell apoptosis.
Upon activation, PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α) and results in attenuation of protein translation, thus restoring ER protein-folding homeostasis [5]. The phosphorylated eIF2α causes inhibition of a global protein biosynthesis that is concurrent with the increased translation of activating transcription factor 4 (ATF4). ATF4 enters the nucleus and activates the transcription of genes that are necessary for the antioxidant response and amino acid biosynthesis and transport [9]. ATF4 also induces the expression of CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP). CHOP and ATF4 form heterodimers to upregulate transcription of genes that function in UPR, autophagy, and mRNA translation [10]. CHOP induces cell cycle arrest and apoptosis in response to ER stress. Once ER protein-folding homeostasis is restored, ATF4 and CHOP induce the transcription of growth arrest and DNA-damage-inducible protein 34 (GADD34) to direct dephosphorylation of p-eIF2α and restart global mRNA translation, which is essential for cells that are to survive an acute insult [11].
Under ER stress, ATF6 moves to Golgi where it is cleaved by S1P and S2P proteases to form a cytosolic fragment [12,13]. The ATF6 cytosolic fragment then enters the nucleus and initiates transcription of ER chaperones and proteins involved in protein quality control and ERAD [14].
UPR Branches in Cancer Progression
Accumulating evidence suggests that disrupted ER proteostasis is a hallmark of cancer [15]. Cancer cells are usually exposed to many stressful conditions, such as hypoxia, nutrient starvation, oxidative stress, and metabolic dysregulation, which result in continued ER stress [15–17] (Fig. 2). Oncogenes can also induce ER stress [15–17]. Oncogene activation causes an increased burden on cells to augment biosynthetic pathways and rewire metabolism to meet the demand of rapid proliferation, inducing replicative and metabolic stresses in cells. ER stress has a profound effect on cancer cell proliferation and survival [18]. In general, tumors are prone to ER stress and have elevated activation of UPR compared with normal tissues. Sustained activation of UPR has been found in different human cancers and the activation of UPR helps cancer cells escape from ER-stress-induced cell death [19,20]. Bunch of evidence has shown that the activated UPR branches IRE1α, PERK, and ATF6 contribute to oncogenic processes and play important roles in cancer development [19–22].
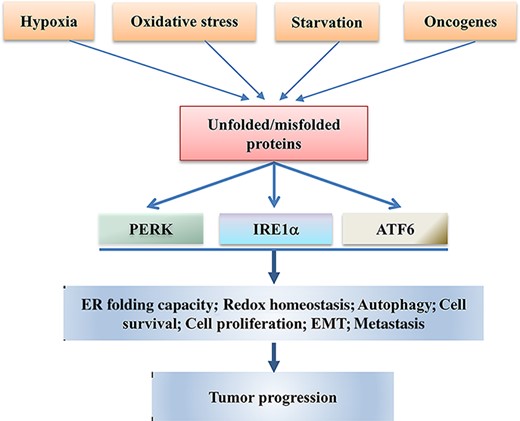
The IRE1α, PERK, and ATF6 UPR branches are involved in tumor progression Different extrinsic (hypoxia, oxidative stress, and nutrient deprivation) and intrinsic (oncogene activation) factors cause formation and accumulation of unfolded and/or misfolded proteins that trigger the activation of IRE1α, PERK, and ATF6, the three branches of UPR. The activated IRE1α, PERK, and ATF6 regulate the expressions of genes that induce adaptation, survival, proliferation, EMT, and autophagy of tumor cells, ultimately promoting tumor progression.
IRE1α–XBP1 signaling in progression of cancer
IRE1α is one of the proteins that are liable to become cancer drivers when mutated [23]. The IRE1α–XBP1 signaling is implicated in tumor progression. This signaling was found to be overactive in triple-negative breast cancer (TNBC), conferring on these tumor cells a highly aggressive phenotype [24]. XBP1 assembles a transcriptional complex with hypoxia-inducing factor 1α (HIF1α), which regulates the expressions of HIF1α target genes, thus driving TNBC tumorigenicity [24]. In TNBC MDA-MB-231 cells, the IRE1α–XBP1 signaling is activated to enhance the productions of interleukin (IL)-6, IL-8, C-X-C motif chemokine 1 (CXCL1), granulocyte-macrophage colony-stimulating factor (GM-CSF), and TGFβ2, the pro-tumorigenic factors that promote cell proliferation [25]. Inhibition of IRE1α RNase activity with MKC8866 attenuates the proliferation of the breast cancer cell [25]. The IRE1α–XBP1 signaling plays an important role in pathogenesis of multiple myeloma (MM) [26] and it is emerging as a pathway involved in MM development [27]. XBP1s overactivation has been suggested to be part of the etiology of MM. Enhanced proliferative potential of cells overexpressing XBP1 was observed in MM [26]. High expression of XBP1s correlates with poor prognosis in glioblastoma [28] and pre-B acute lymphoblastic leukemia [29].
It has been found that IRE1α is crucial in the initiation and development of colon cancer [30]. Genetic ablation of IRE1α in intestinal epithelial cells prevented the colitis-associated colonic tumorigenesis in mice. Inhibition of IRE1α RNase activity with 4μ8C suppressed colon cancer cell xenograft growth. Mechanistically, depletion of IRE1α repressed the expression of β-catenin, a key factor that drives colonic tumorigenesis. Similarly, in hepatocytes, deletion of IRE1α was found to reduce the occurrence of diethylnitrosamine-induced hepatocellular carcinoma (HCC) in liver-specific IRE1α knockout mice [31]. Loss of IRE1α in hepatocytes resulted in a decrease in activation of STAT3. IRE1α–XBP1 signaling was shown to promote prostate cancer cell proliferation by activating c-MYC signaling [32]. The IRE1α branch plays a role in cell survival upon ER stress. It has been demonstrated that activation of IRE1α–XBP1 signaling is important for the survival of chronic lymphocytic leukemia (CLL) cells and inhibition of IRE1α RNase activity with B-I09 induces CLL cell apoptosis through compromising the B cell receptor (BCR) signaling [33]. Luan et al. [34] demonstrated in melanoma cells that, upon ER stress, IRE1α–XBP1 was activated to induce the expression of RIPK1, which promoted cell survival through autophagy.
The IRE1α–XBP1 signaling is associated with the metastatic potential of cancers. The TNBC cells expressing XBP1s metastasized more efficiently to the lungs [24]. It was shown that high expression of XBP1s in lung tumors was associated with cancer aggressiveness and epithelial-to-mesenchymal transition (EMT) [35]. Expression of XBP1 was found to be upregulated in metastatic and poorly differentiated colon cancer tissue samples, and loss of XBP1 was shown to severely inhibit tumor metastasis in vitro and in vivo [36]. Recently, IRE1α was found to promote the metastasis of colon cancer cells through the fibronectin 1 (FN1)-proto-oncogene tyrosine-protein kinase Src (Src)/focal adhesion kinase (FAK)/guanosine triphosphatase (GTPase) signaling [37]. XBP1 promoted tumor invasion through upregulating the expression of matrix metalloproteinase (MMP) 9 in esophageal squamous cell carcinoma [38] and triggering EMT through inducing snail in breast cancer cells [39] and HCC cells [40]. These results suggest that the IRE1α–XBP1 signaling promotes migration and invasion of cancer cells.
PERK signaling in cancer progression
Similar to IRE1α, the PERK branch also has pro-survival effects in response to ER stress [41]. The survival mechanism is linked to the upregulation of genes in a phospho-eIF2α-dependent fashion [21,22] and to the direct activation of Nrf2 [42,43], a transcription factor that plays a key role in the response to oxidative stress. In Burkitt’s lymphoma, activation of PERK induced miR-211, a microRNA that suppresses core circadian regulators Bmal1 and Clock. Suppression of Bmal1 and Clock impacted the expression of circadian genes, and Bmal1 repression was essential for UPR-dependent inhibition of protein synthesis and cell adaptation to stresses [44]. These results suggest that the PERK–miR-211 axis suppresses circadian regulators and protein synthesis to promote cancer cell survival. The PERK branch has been implicated in initiation and progression of both solid and hematological cancers [45–50]. Activation of PERK was required for tumor cell adaptation to hypoxic condition and PERK-deficient cells gave rise to smaller tumors compared to wild-type cells [45]. Activation of PERK was found in human breast cancer, and the activated PERK prevented mammary epithelial cells from anoikis through activating antioxidant response and autophagy [46]. It is known that autophagy protects cells from stress conditions [47]. UPR was shown to mediate autophagy in ER stress [48], with the PERK branch as an inducer [49]. Blais et al. [50] demonstrated that PERK plays a role in tumor cell adaptation to hypoxic stress by regulating the translation of angiogenic factors that are necessary for the development of functional microvessels. Bobrovnikova-Marjon et al. [51] demonstrated in a mouse breast cancer model that PERK promoted cancer cell proliferation and tumor growth by limiting oxidative DNA damage through regeneration of intracellular antioxidants. In an orthotopic mouse model for HCC, Vandewynckel et al. [52] found that the PERK pathway was activated during tumor progression. The authors showed that upon ER stress the PERK inhibitor GSK2656157 reduced cell viability and proliferation via escalating proteotoxic stress in vitro. Notably, the PERK inhibitor decreased tumor burden in the mouse model [52]. Inhibition of PERK significantly decreased Myc-induced colony formation and tumor formation, and the activation of PERK signaling was required in Myc-induced lymphomas [49]. In a transgenic mouse model of insulinoma, Gupta et al. [53] showed that PERK deficiency reduced tumor growth and vascularity. A recent study demonstrated in prostate cancer that activation of PERK–eIF2α signaling reset global protein synthesis to a level that fosters aggressive tumor development [54].
The PERK branch also plays a role in promoting the migration and invasion of cancer cells. Feng et al. [55] demonstrated that EMT cells had a constitutive activated PERK-eIF2α axis and PERK activation was required for EMT cells to invade and metastasize. The authors found that, in human breast, colon, gastric, and lung tumor tissues, EMT gene expression correlated strongly with PERK–eIF2α signaling. It was reported that the PERK–eIF2α signaling promoted the metastasis of hypoxic cervical, breast, and head and neck cancer cells through activating the pro-metastatic factor lysosome-associated membrane protein 3 (LAMP3) [56–58]. ATF4, the downstream target of PERK, was upregulated in human esophageal squamous cell carcinoma (ESCC), and ATF4 expression was correlated with multiple malignant characteristics and indicated poor prognosis in ESCC patients [59]. ATF4 promoted cell invasion and metastasis through upregulating the expression of MMP2 and 7 [59].
ATF6 in progression of cancer
Elevated expression of active ATF6 was seen in patient tissues of HCC [60] as well as in Hodgkin lymphoma [61]. In HCC cells, activation of ATF6 promoted the expression of cell cycle/proliferation-associated genes [62]. The activation of ATF6 was important for survival of quiescent squamous carcinoma cells, and knockdown of ATF6 prolonged the survival of nude mice bearing the dormant tumor cells through the ATF6–Ras homolog enriched in brain (Rheb)–Mammalian target of rapamycin (mTOR) pathway [63]. In patients with colon cancer, ATF6 was associated with reduced time of disease-free survival [64]. In a mice model, the activation of ATF6 in colon epithelial cells induced intestinal dysbiosis and innate immune response, which promoted tumorigenesis [64]. In a TNBC cell model with missense TP53 mutation, Sicari et al. [65] showed that mutant p53-enhanced activation of ATF6 was necessary for viability and invasion phenotypes. Together, these studies implicated a positive role of ATF6 in tumor progression.
UPR branches in chemotherapy resistance of cancer
Cancer cells are capable of developing resistance to chemotherapy. The development of multidrug resistance (MDR) is the primary obstacle to cancer treatment. MDR is a phenomenon by which, after exposure to a chemotherapeutic agent, cancer cells develop resistance, and simultaneous cross-resistance, to a wide range of functionally and structurally unrelated chemotherapeutic drugs [66]. Intrinsic or acquired MDR is one of the main causes for chemotherapy failure, leading to the recurrence of malignant tumors and ultimately patient relapse or death. A variety of mechanisms have been attributed to MDR, such as enhanced drug efflux, increased DNA damage repair, reduced apoptosis, elevated autophagy, and altered drug metabolism.
Chemotherapeutic agents can trigger ER stress [16,17,67]. UPR is activated in response to drugs, and activated UPR plays an important role in chemotherapy resistance of tumors [16,19,68]. For example, there is a correlation between UPR activation and resistance of cancer cells to a topoisomerase II-targeting drug [69]. The UPR was found to be activated and confer resistance to chemotherapy in multiple cancers such as liver [70], breast [71], glioma [72], and colon [73] cancers. Activation of UPR in hepatoma cells is related to the upregulation of P-glycoprotein (also known as ABCB1) [74], which increases the efflux of drugs and induces MDR. The UPR is one mechanism exploited by cancer cells to ensure survival upon exposure to chemotherapeutic agents, with all three branches of UPR being implicated. Tumor cells might co-opt the ER stress response to adapt to and survive various types of stress including chemotherapy. Thus, the UPR represents an adaptive mechanism that supports not only survival but also chemoresistance of tumor cells.
IRE1α–XBP1 signaling in cancer drug resistance
IRE1α–XBP1 signaling is linked to resistance in cancer chemotherapy. The elevated level of IRE1α predicts a poor response to drugs in glioblastoma cells [75]. In cancer cells, overexpression of XBP1 confers drug resistance by preventing drug-induced cell cycle arrest and mitochondrial permeability and apoptosis [76] (Fig. 3). UPR activation and XBP1 expression have been described in both ERα+ and ERα− breast cancer cells [77]. XBP1 is involved in resistance to endocrine therapy, molecular-targeted agents, and traditional chemotherapeutics of breast cancers. In vitro estrogen stimulates UPR, and UPR activation induces resistance to both doxorubicin and 5-fluorouracil (5-FU) of breast cancer cells [78]. Expression of XBP1 was upregulated in tamoxifen-insensitive breast cancer MCF-7 cells and inhibition of XBP1 splicing re-established tamoxifen sensitivity to tamoxifen-resistant MCF-7 cells [79]. Upregulation of XBP1 is identified as a predictive marker of tamoxifen resistance in ER-positive breast cancer cells and is associated with reduced time to recurrence and poor survival [80].
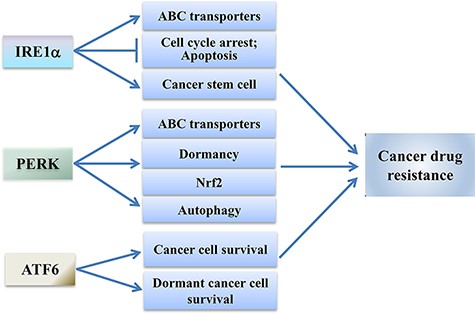
The IRE1α, PERK, and ATF6 branches induce drug resistance of cancer cells When activated, IRE1α can enhance the expression of ABC transporters, inhibit cell cycle arrest and apoptosis, and promote cancer stem cells, which contribute to the drug resistance of cancer cells. PERK signaling may confer drug resistance of cancer cells through enhancing the expression of ABC transporters, promoting cancer cell dormancy, inducing Nrf2, and activating autophagy. The activated ATF6 may promote chemotherapy resistance of cancer cells through promoting survival of cancer cells.
Cancer stem cells (CSCs) have been implicated in the development of resistance to chemotherapy, and therapy-driven expansion of CSCs provokes therapy resistance and tumor relapse [81]. In breast cancer MDA-MB-231 cells, knockdown of XBP1 decreased the proportion of CD44high/CD24low cells post-chemotherapy and reduced the ability of these cells to form tumor spheres [24]. Likewise, inhibiting IRE1α RNase activity reduced the ability of MDA-MB-231 cells to form tumor spheres post-paclitaxel treatment [25]. These results suggest that the IRE1–XBP1 signaling promotes breast CSC expansion to overcome chemotherapy. Fujimoto et al. [82] isolated sphere-forming (cancer stem-like) cells from the cervical cancer cell line SiHa, and they found that cisplatin at a low concentration failed to shift sphere-forming cells to apoptosis. Apoptosis occurred in sphere-forming cells by treatment with cisplatin plus the IRE1α inhibitor 4μ8C. The results indicate that a combination of IRE1α inhibitor and chemotherapeutic drug may shift cancer stem-like cells to apoptosis.
Overexpression of the ATP-binding cassette (ABC) transporters, particularly ABCB1, ABCC1, and ABCG2, is one of the most prominent mechanisms underlying drug resistance of cancers [83]. In colon cancer cells, the chemotherapeutic drug 5-FU activated the IRE1α–XBP1 signaling, which contributed to 5-FU resistance of colon cancer cells through upregulating the expression of ABCB1, ABCC1, and ABCG2 [84]. Inhibition of IRE1α RNase activity with 4μ8C repressed 5-FU-induced expression of these ABC transporters and sensitized 5-FU-resistant colon cancer cells to 5-FU [84].
PERK branch in cancer drug resistance
The PERK branch is involved in drug resistance of cancer cells. The activation of PERK is associated with upregulation of P-glycoprotein and resistance to adriamycin in hepatocarcinoma cells [85]. PERK is also involved in the upregulation of an MDR-related protein, multidrug resistance-associated protein 1 (MRP1; also known as ABCC1), through nuclear factor erythroid 2-related factor 2 (Nrf2) in the chemotherapy-resistant human colon adenocarcinoma HT29 cells [86]. Activation of ATF4 also leads to drug resistance of cancer. Tanabe et al. [87] showed that ATF4 was upregulated in cisplatin-resistant lung cancer cells and the expression of ATF4 was correlated with cisplatin sensitivity of cancer cells. The cells overexpressing ATF4 had less sensitivity to cisplatin treatment. ATF4 was also found to confer drug resistance of ESCC and gastric cancer cells through signal transducer and activator of transcription 3 (STAT3) [88] and NAD-dependent protein deacetylase sirtuin-1 (SIRT1) [89], respectively.
The activation of PERK also contributes to the drug resistance of dormant carcinoma cells. In dormant human epidermoid carcinoma HEp3 (D-HEp3) cells, PERK signaling was activated and the activated PERK contributed cell resistance to etoposide and doxorubicin, probably due to dormancy-related survival mechanisms [90]. The activated PERK induced the Nrf2 detoxifying pathway and promoted cell survival [43]. Del Vecchio et al. [91] demonstrated that the constitutive PERK–Nrf2 signaling protected dedifferentiated cells from chemotherapy by reducing reactive oxygen species (ROS) and increasing drug efflux. Additionally, analysis of patient tumor data sets showed that the PERK pathway signature correlates strongly with chemotherapy resistance. In addition, there is a report demonstrating that activated PERK is the major contributor to UPR-induced etoposide resistance [92].
Autophagy, a process that involves the enzymatic breakdown of cytoplasmic components within the lysosome, is proposed to play a key role in relieving cells from the burden of accumulating aberrant proteins. There is a bunch of evidence indicating that autophagy promotes cell survival. ER stress induces autophagy, and autophagy serves as a pro-survival mechanism [93]. The PERK and IRE1α branches are implicated in transcriptional upregulation of numerous autophagy-related genes [16]. And crosstalk between these two branches may account for chemotherapy resistance in many cancers because the chemoresistant phenotypes are usually concomitant with increasing autophagy when drugs are administrated [94]. Activation of ATF4 was suggested to promote drug resistance of cancer cells [95]. Ma et al. [96] showed that, in BRAFV600E melanoma cells, the inhibitor of BRAF induced cytoprotective autophagy, and inhibition of autophagy enhanced the efficacy of BRAF inhibitor to induce cell death. The authors found that PERK was activated and activated PERK was responsible for the activation of cytoprotective autophagy. Blockade of PERK limited the BRAF inhibition-induced autophagy and enhanced cell death. Thus, inhibition of autophagy may be a strategy to improve the efficacy of chemotherapeutic drugs.
ATF6 in cancer drug resistance
ATF6 also plays a role in chemoresistance of cancer cells. Kim et al. [73] found that the expression of ATF6 was enhanced in 5-FU-resistant SNUC5 colon cancer cells. Knockdown of ATF6 made the 5-FU-resistant SNUC5 cells more sensitive to 5-FU than control cells. These results suggest that upregulated ATF6 is associated with 5-FU resistance. Higa et al. [97] found that ATF6 was activated in imatinib-treated leukemia K562R cells and inhibition of ATF6 expression partially restored sensitivity of the cells to imatinib. Yarapureddy et al. [98] demonstrated that knockdown of ATF6 enhanced sensitivity of osteosarcoma cells against chemotherapy-induced cell death. The authors showed that activation of ATF6 protected osteosarcoma cells from drug-induced apoptosis via ATF6 downstream effectors BiP, protein disulfide isomerase (PDI), and endoplasmic reticulum oxidoreductase 1-dependent pro-survival mechanisms. The authors also analyzed the primary tumors from osteosarcoma patients and found that the patients with higher level of nuclear ATF6 had poorer response to chemotherapy. Tumor dormancy has been implicated in several tumor types as the culprit of therapy resistance [99]. Schewe et al. [63] showed that ATF6 activated mTOR through inducing the expression of mTOR-activator Rheb, thus promoting drug resistance of dormant squamous carcinoma. Together, these studies implied that ATF6 plays a role in chemotherapy resistance of cancer cells.
Targeting UPR Branches to Enhance Efficacy of Cancer Chemotherapy
Due to the critical role of IRE1α, PERK, and ATF6 in cancer cell survival and chemotherapy resistance, targeting these UPR branches has been considered a promising approach to treat cancers [5,100,101]. A few inhibitors targeting these branches have been developed and these inhibitors exert inhibitory effects on cancer cells [102]. Combination of these inhibitors and traditional anticancer drugs may be a promising strategy for improving the efficacy of cancer chemotherapy. These inhibitors are summarized in Table 1.
Inhibitors for IRE1αPERK,and ATF6 signaling
| Inhibitor | Target | Mechanism | Pharmacological effects | References |
|---|---|---|---|---|
| 4μ8C | IRE1α | Inhibits RNase | Inhibits tumor growth; enhances efficacy of cisplatin and 5-FU to tumor cells | [29,79,81] |
| MKC-3946 | IRE1α | Inhibits RNase | Induces apoptosis; enhances efficacy of bortezomib or 17-AAG to tumor cells | [100] |
| A-I06 | IRE1α | Inhibits RNase | Cytotoxic to tumor cells; enhances cytotoxicity of bortezomib to tumor cells | [102] |
| Toyocamycin | IRE1α | Inhibits RNase | Inhibits tumor cells; has synergistic effect with bortezomib on tumor cells | [104] |
| MKC8866 | IRE1α | Inhibits RNase | Tumor growth inhibition; increases efficacy of paclitaxel and docetaxel | [24,31,104] |
| STF-083010 | IRE1α | Inhibits RNase | Tumor growth inhibition; induces tumor cell apoptosis | [76] |
| B-I09 | IRE1α | Inhibits RNase | Reduces tumor burden | [32,103] |
| Kira8 | IRE1α | Inhibits kinase | Inhibits of tumor growth; enhances efficacy of bortezomib and lenalidomide | [101] |
| GSK2656157 | PERK | Inhibits PERK | Inhibition of tumor growth and angiogenesis | [49,107,108] |
| GSK2606414 | PERK | Inhibits PERK | Tumor growth inhibition | [83,110] |
| ISRIB | eIF2α | Inhibits p-eIF2α | Inhibits tumor aggression; enhances efficacy of gemcitabine | [110] |
| Ceapin-A7 | ATF6 | Blocks ATF6 activity | Sensitizes tumor cells to ER stressor | [116] |
| Inhibitor | Target | Mechanism | Pharmacological effects | References |
|---|---|---|---|---|
| 4μ8C | IRE1α | Inhibits RNase | Inhibits tumor growth; enhances efficacy of cisplatin and 5-FU to tumor cells | [29,79,81] |
| MKC-3946 | IRE1α | Inhibits RNase | Induces apoptosis; enhances efficacy of bortezomib or 17-AAG to tumor cells | [100] |
| A-I06 | IRE1α | Inhibits RNase | Cytotoxic to tumor cells; enhances cytotoxicity of bortezomib to tumor cells | [102] |
| Toyocamycin | IRE1α | Inhibits RNase | Inhibits tumor cells; has synergistic effect with bortezomib on tumor cells | [104] |
| MKC8866 | IRE1α | Inhibits RNase | Tumor growth inhibition; increases efficacy of paclitaxel and docetaxel | [24,31,104] |
| STF-083010 | IRE1α | Inhibits RNase | Tumor growth inhibition; induces tumor cell apoptosis | [76] |
| B-I09 | IRE1α | Inhibits RNase | Reduces tumor burden | [32,103] |
| Kira8 | IRE1α | Inhibits kinase | Inhibits of tumor growth; enhances efficacy of bortezomib and lenalidomide | [101] |
| GSK2656157 | PERK | Inhibits PERK | Inhibition of tumor growth and angiogenesis | [49,107,108] |
| GSK2606414 | PERK | Inhibits PERK | Tumor growth inhibition | [83,110] |
| ISRIB | eIF2α | Inhibits p-eIF2α | Inhibits tumor aggression; enhances efficacy of gemcitabine | [110] |
| Ceapin-A7 | ATF6 | Blocks ATF6 activity | Sensitizes tumor cells to ER stressor | [116] |
Inhibitors for IRE1αPERK,and ATF6 signaling
| Inhibitor | Target | Mechanism | Pharmacological effects | References |
|---|---|---|---|---|
| 4μ8C | IRE1α | Inhibits RNase | Inhibits tumor growth; enhances efficacy of cisplatin and 5-FU to tumor cells | [29,79,81] |
| MKC-3946 | IRE1α | Inhibits RNase | Induces apoptosis; enhances efficacy of bortezomib or 17-AAG to tumor cells | [100] |
| A-I06 | IRE1α | Inhibits RNase | Cytotoxic to tumor cells; enhances cytotoxicity of bortezomib to tumor cells | [102] |
| Toyocamycin | IRE1α | Inhibits RNase | Inhibits tumor cells; has synergistic effect with bortezomib on tumor cells | [104] |
| MKC8866 | IRE1α | Inhibits RNase | Tumor growth inhibition; increases efficacy of paclitaxel and docetaxel | [24,31,104] |
| STF-083010 | IRE1α | Inhibits RNase | Tumor growth inhibition; induces tumor cell apoptosis | [76] |
| B-I09 | IRE1α | Inhibits RNase | Reduces tumor burden | [32,103] |
| Kira8 | IRE1α | Inhibits kinase | Inhibits of tumor growth; enhances efficacy of bortezomib and lenalidomide | [101] |
| GSK2656157 | PERK | Inhibits PERK | Inhibition of tumor growth and angiogenesis | [49,107,108] |
| GSK2606414 | PERK | Inhibits PERK | Tumor growth inhibition | [83,110] |
| ISRIB | eIF2α | Inhibits p-eIF2α | Inhibits tumor aggression; enhances efficacy of gemcitabine | [110] |
| Ceapin-A7 | ATF6 | Blocks ATF6 activity | Sensitizes tumor cells to ER stressor | [116] |
| Inhibitor | Target | Mechanism | Pharmacological effects | References |
|---|---|---|---|---|
| 4μ8C | IRE1α | Inhibits RNase | Inhibits tumor growth; enhances efficacy of cisplatin and 5-FU to tumor cells | [29,79,81] |
| MKC-3946 | IRE1α | Inhibits RNase | Induces apoptosis; enhances efficacy of bortezomib or 17-AAG to tumor cells | [100] |
| A-I06 | IRE1α | Inhibits RNase | Cytotoxic to tumor cells; enhances cytotoxicity of bortezomib to tumor cells | [102] |
| Toyocamycin | IRE1α | Inhibits RNase | Inhibits tumor cells; has synergistic effect with bortezomib on tumor cells | [104] |
| MKC8866 | IRE1α | Inhibits RNase | Tumor growth inhibition; increases efficacy of paclitaxel and docetaxel | [24,31,104] |
| STF-083010 | IRE1α | Inhibits RNase | Tumor growth inhibition; induces tumor cell apoptosis | [76] |
| B-I09 | IRE1α | Inhibits RNase | Reduces tumor burden | [32,103] |
| Kira8 | IRE1α | Inhibits kinase | Inhibits of tumor growth; enhances efficacy of bortezomib and lenalidomide | [101] |
| GSK2656157 | PERK | Inhibits PERK | Inhibition of tumor growth and angiogenesis | [49,107,108] |
| GSK2606414 | PERK | Inhibits PERK | Tumor growth inhibition | [83,110] |
| ISRIB | eIF2α | Inhibits p-eIF2α | Inhibits tumor aggression; enhances efficacy of gemcitabine | [110] |
| Ceapin-A7 | ATF6 | Blocks ATF6 activity | Sensitizes tumor cells to ER stressor | [116] |
Targeting IRE1α to enhance chemotherapeutic efficacy of cancers
The IRE1α–XBP1 signaling has been suggested as a potential target for chemotherapy. It was found that inhibition of IRE1α RNase activity with a specific inhibitor STF-083010 was toxic to MM cells [103]. The IRE1α RNase inhibitor MKC-3946 enhanced cytotoxic effects of bortezomib or 17-AAG on the MM cells, although it alone triggered modest growth inhibition in MM cell lines with little toxicity in normal mononuclear cells [104]. Inhibition of IRE1α with Kira8, a compound inhibiting IRE1α kinase and inactivating IRE1α RNase, suppressed subcutaneous or orthometastatic growth of MM tumors in mice and enhanced efficacy of bortezomib and lenalidomide, two antimyeloma agents [105]. The expression of XBP1 is upregulated in acute myeloid leukemia (AML) cell lines and AML patient samples. The IRE1α inhibitor A-I06 exhibited cytotoxic effect on AML cells, and a combination of A-I06 with bortezomib was synergistic in AML cytotoxicity [106]. The IRE1α RNase inhibitor B-I09 synergized with ibrutinib, a Food and Drug Administration-approved bruton tyrosine kinase (BTK) inhibitor, to induce apoptosis in B cell leukemia, lymphoma, and multiple myeloma [33]. B-I09 improved cytotoxicity of doxorubicin or vincristine in Burkitt’s lymphoma cells [107]. Toyocamycin, an inhibitor of IRE1α RNase, not only inhibited human MM xenograft growth but also showed synergistic effects with bortezomib [108].
The use of specific inhibitors of the IRE1α RNase is also beneficial in breast cancers. In MDA-MB-231 cells the anticancer drug paclitaxel activated IRE1α, which contributed to the production of pro-tumorigenic factors such as IL-6 and IL-8 [25]. Inhibition of IRE1α RNase activity with MKC8866 suppressed the formation of these pro-tumorigenic factors, increased the paclitaxel-mediated tumor suppression and delayed tumor relapse post-therapy. In preclinical mouse models of prostate cancer, MKC8866 synergized with enzalutamide, abiraterone acetate, and cabazitaxel, the central prostate cancer drug regimens that are currently used in the clinic [32]. STF-083010 re-established tamoxifen sensitivity to resistant MCF-7 cells [79], and in a xenograft mammary tumor model, administration of mice with STF-083010 and tamoxifen significantly delayed breast cancer progression. A recent report demonstrated that MKC8866 substantially enhanced the efficacy of docetaxel chemotherapy, resulting in rapid regression of MYC-overexpressing breast tumors in a PDX model in mice [109]. Inhibition of the IRE1–XBP1 axis is also a potential strategy for overcoming endocrine resistance in breast cancer [110].
Taken together, these studies suggest that inclusion of inhibition of IRE1α in therapeutic strategies may enhance the efficacy of chemotherapeutics.
Targeting PERK to enhance efficacy of cancer chemotherapy
Reduced tumor growth and restored chemosensitivity in resistant tumors were observed after PERK silencing. Treatment with PERK-specific inhibitor GSK2656157 resulted in a dose-dependent inhibition of growth of multiple human tumor xenografts (such as pancreatic cancer cell BxPC3 xenograft and multiple myeloma cell NCI-H929 xenograft) in mice [111]. Shi et al. [112] discovered that activation of the PERK pathway was required for colon cancer cells to survive treatment of 5-FU, and the PERK inhibitor GSK2656157 greatly sensitized colon cancer cells to 5-FU treatment. Most importantly, this inhibitor synergized with 5-FU in suppressing the growth of colon cancer cells in mouse models. Ranganathan et al. [90] showed that PERK activation protected dormant tumor cells from chemotherapy and inhibition of PERK rendered the dormant tumor cells susceptible to drug-induced apoptosis. ISRIB, a drug targeting PERK–eIF2α signaling, increased death of pancreatic cancer cells induced by gemcitabine [113]. The drug-resistant breast cancer cells are also sensitive to PERK inhibition. PERK inhibitor-titration and -time course experiments showed that the drug-resistant MCF-7 breast cancer cells were more sensitive to PERK inhibitor GSK2606414 than the parental MCF-7 cells [114]. Kusio-Kobialka et al. [115] demonstrated that inactivation of the PERK–eIF2α branch decreased the clonogenic and proliferative capacities of chronic myelogenous leukemia (CML) cells and sensitized the cells to imatinib, a protein tyrosine kinase inhibitor that is usually used for the treatment of CML.
Malignant carcinomas that recur following therapy are typically dedifferentiated and multidrug resistant. Dedifferentiated cancer cells acquire MDR through upregulating ROS-scavenging enzymes and drug efflux pumps. Del Vecchio et al. [89] showed that the constitutive PERK–Nrf2 signaling protects dedifferentiated cells from chemotherapy and inhibition of this signaling reversed the therapy resistance of breast cancer cells. In dual ER stress–chemotherapy resistant colon cancer cells that had enhanced activation of the PERK–Nrf2 signaling and expression of ABCC1, silencing of PERK reduced tumor growth and restored chemosensitivity in resistant tumor xenografts [86]. Chemotherapy may induce cytoprotective autophagy through UPR. The BRAFV600E melanoma cells often become resistant to BRAF inhibition, probably through the PERK-activated autophagy [86]. And inhibition of PERK with its specific inhibitor GSK2606414 enhanced the sensitivity of the BRAF inhibition-resistant cells to BRAF inhibitor PLX4720. Together, these results suggest that PERK can serve as a target in the treatment of drug-resistant cancers.
Blocking ATF6 to enhance the effectiveness of anticancer drugs
ATF6 targeting can also enhance the efficacy of anticancer drugs. Kim et al. [74] reported that knockdown of ATF6 increased the sensitivity of 5-FU-resistant SNUC5 colon cancer cells to 5-FU treatment. A role for ATF6 in cancer chemoresistance through the PDIA5 upon ER stress has been identified [97]. Inhibition of PDIA5–ATF6 axis could restore the sensitivity of the imatinib-resistant leukemia K562R cells to imatinib, a drug that is usually used to treat certain types of leukemia [97]. Ceapin-A7, an inhibitor that selectively inhibits the ATF6 branch, has been developed [116]. It was shown that administration of Ceapin-A7 sensitized human bone osteosarcoma U2-OS cells to the treatment of ER stressor Tunicamycin [116]. These results suggest that ATF6 targeting is a potential strategy for chemotherapy of cancers.
Conclusions and Perspectives
Chemotherapy resistance is the tolerance of cancer cells to chemotherapeutic drugs, which is a significant issue in the management of cancers. And it is a major barrier for physician in the treatment of cancers. While the development of new chemotherapies has proceeded quickly, the drugs that are effective against the advanced stages of cancers have not been developed due to a poor understanding of drug resistance. The UPR plays a critical role in the adaptive survival signaling that promotes resistance to chemotherapy in cancer cells. And the drug resistance resulted from UPR is more or less regulated by IRE1α, PERK, and ATF6. Thus, these UPR branches have become potential and effective targets for developing strategies against chemotherapy resistance of cancers.
The involvement of the UPR in cancer drug resistance is complex, and the detailed mechanisms of the relationship between these UPR branches and chemotherapy resistance remain poorly understood. Therefore, a global understanding of the molecular mechanisms controlling UPR-mediated drug resistance is highly needed. Complete understanding of the molecular mechanisms underlying chemotherapy resistance can allow the development of combined therapeutic approaches with inhibitors that specifically target the UPR branches to bypass anticancer drug resistance and enhance efficacy of treatment.
The UPR represents an adaptive mechanism that supports survival and chemoresistance of tumor cells. In such a scenario, a possible pharmaceutical strategy to counteract tumor progression and drug resistance is to target UPR pro-survival components and the complex interplay between UPR and apoptosis, thus favoring a shift toward cell death instead of cell survival. Future challenges will certainly lead to the development of combined therapeutic approaches with new drugs that specifically target the UPR branches to bypass chemotherapy resistance of cancers. It should be noted that UPR manages ER stress through restoring and maintaining proteostasis, and it is essential to protect cells from stress. Therefore, any inhibition of UPR activity might impair this critical cellular defense and carry the risk of severe immediate damage. To avoid the risk, extensive studies are warranted and long-term side effects need to be defined and tested.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (Nos. 31670785 and 82073061 to J.F., and No. 32000434 to M.Y.), the Ministry of Science and Technology of China (No. 2020YFA0803300 to J.F.), and the Shandong Natural Science Foundation (No. ZR2020MH206 to J.F. and No. ZR2020QH193 to M.Y.).
Conflict of Interest
The authors declare that they have no conflict of interest.
References
Author notes
Mengchao Yu and Jie Lun contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}