





Abstract
Swine harbor genetically diverse influenza A viruses (IAVs) with the capacity to host-switch to humans, causing global pandemics. Spain is the largest swine producer in Europe and has a mixed production system that includes ‘white coat’ pigs raised intensively in modern buildings and free-range Iberian pigs that interface differently with humans, wildlife, and other swine. Through active longitudinal IAV surveillance in nine Spanish provinces during 2015–9, we generated forty-seven complete or near-complete genome sequences from IAVs collected from swine in both systems. Genetically diverse IAVs were identified in intensively raised white pigs and free-range Iberian pigs, including new H3N1 reassortants. Both systems are dynamic environments for IAV evolution, but driven by different processes. IAVs in white pigs were genetically related to viruses found in swine raised intensively in other European countries, reflecting high rates of viral introduction following European trade routes. In contrast, IAVs in Iberian pigs have a genetic makeup shaped by frequent introductions of human IAVs, reflecting rearing practices with high rates of human contact. Transmission between white and Iberian pigs also occurred. In conclusion, Iberian swine with high rates of human contact harbor genetically diverse IAVs and potentially serve as intermediary hosts between white pigs and humans, presenting an understudied zoonotic risk that requires further investigation.
1. Introduction
The expanding genetic diversity of influenza A viruses (IAVs) circulating in swine globally presents an ongoing pandemic risk for humans (Lewis et al. 2016; Sun et al. 2020). The 2009 H1N1 pandemic is an example of how swine serve as reservoirs for diverse IAVs of human and avian origin that can exchange genome segments via reassortment and rapidly generate novel viruses capable of host-switching to humans (Smith et al. 2009; Mena et al. 2016). A large genetically diverse reservoir of H1N1, H1N2, and H3N2 viruses circulates in swine herds in Europe, including viruses from the Eurasian avian-like H1N1 (EA-H1N1) lineage with high antigenic divergence from human viruses (Lewis et al. 2016; Henritzi et al. 2020), documented zoonotic infection of humans (Zhu et al. 2016; Parys et al. 2021), and a capacity to host-switch to other non-human mammalian species, including canines (Chen et al. 2018). Alarmingly, the genetic diversity of IAV diversity in European swine has further expanded since the 2009 pandemic, driven by frequent reverse zoonosis of 2009 H1N1 (pdmH1N1) viruses from humans to swine. Novel genotypes emerge following genomic reassortment with the existing pool of avian-origin and human-origin viruses circulating in European swine, including EA-H1N1 (Moreno et al. 2011; Starick et al. 2011; Lange et al. 2013; Watson et al. 2015; Beato et al. 2016; Krog et al. 2017; Chastagner et al. 2018; Zell et al. 2020). However, our understanding of IAV evolution in Europe is obscured by major gaps in surveillance, including in Spain, which recently surpassed Germany as the European Union’s largest swine producer. With more than 31 million swine, Spain is the world’s fourth-largest swine producer after China, the USA, and Brazil.
Spain is a country where modern indoor and traditional free-ranging practices of swine production both sustainably coexist. Approximately 90 per cent of Spain’s domestic swine are ‘white pigs’ of the Landrace and Large White breeds that are bred across Europe for high-efficiency production and housed in large modern buildings. Spain’s rapidly growing commercial sector is concentrated in Spain’s northeast region, where the Catalonia and Aragon regions account for more than half of the country’s total swine census (Fig. 1A). Influenza is endemic and widespread in Spain’s intensively raised white pigs (Sosa Portugal et al. 2021). All four major IAV lineages found in European swine also circulate in Spain: (1) pdmH1N1, (2) human seasonal-like H3N2 (huH3N2), (3) human seasonal-like H1 (huH1), and (4) EA-H1N1, which is dominant across Europe (Meritxell et al. 2012; Martín-Valls Gerard et al. 2014).
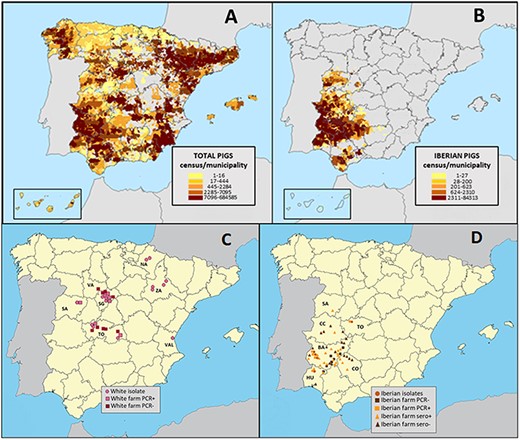
Map of Spanish swine and sample collection. Distribution and density of (A) all domestic pigs and (B) Iberian pigs in Spain in 2020. Each colored rectangle represents the number of pigs per municipality. The locations (Global Positioning System coordinate) of the farms for (C) white pigs and (D) Iberian pigs where respiratory specimens (squares) and serum samples (triangles) were collected for this study including positive (light color) and negative (dark color) farms. Samples were collected from white pigs in seven Spanish provinces (NA, Navarra; SA, Salamanca; SG, Segovia, TO, Toledo; VAL, Valencia; VA, Valladolid; ZA, Zaragoza) and from Iberian pigs in six provinces (BA, Badajoz; CC, Cáceres; CO, Córdoba; HU, Huelva; SA, Salamanca; TO, Toledo).
In contrast, there is no information about IAVs circulating in Iberian pigs, a dark-pigmented autochthonous breed that constitutes approximately 10 per cent of Spain’s swine population. Iberian pigs originate from Sus scrofa mediterraneous and have been reared for centuries on a section of the Iberian Peninsula spanning Portugal and Spain (Rodríguez-Estévez et al. 2010; Fig. 1B). Iberian pigs are raised primarily in a Mediterranean oak forest ecosystem called dehesa where pigs feed on naturally fallen acorns and herbaceous plants during a seasonal fattening phase called montanera that runs from October to February (Garrido-Fernández and Manuel 2019). There are strict criteria for the diet, range, and seasonal environment of a certified Iberian pig product, the most expensive pork in the world. Iberian pigs share habitat with ruminant livestock and wildlife, creating opportunities for transmission of respiratory pathogens such as tuberculosis (Morris, Pfeiffer, and Jackson 1994). High contact rates between Iberian swine and human handlers also facilitate bidirectional transmission of other pathogens (Wiethoelter et al. 2015; Roxana et al. 2020). However, IAV has not been reported in Iberian swine, and it is possible that influenza epidemiology differs due to a combination of farming practices, genetic susceptibility, different circulating IAVs, or other coinfecting pathogens.
Iberian farms tend to be small, no larger than a few hundred animals, whereas white pigs in Spain are increasingly raised in large modern facilities housing thousands of animals. The number of large swine farms intensively raising thousands of white pigs steadily increased in Spain between 2007 and 2020 while the number of small farms with fewer than twenty-five pigs nearly halved (Supplementary Table S1), representing a shift in the industry toward larger operations that has been observed in many countries globally. Given these trends, it is possible that larger Iberian farms will emerge in the future. Intensive farming of white pigs also tends to be a horizontal industry, where farrowing (sows giving birth), weaning, and fattening occur on different premises, sometimes in different countries. Many of Spain’s piglets are imported from the Netherlands (Supplementary Fig. S1), providing opportunities for IAV migration following the movement of pigs. In contrast, the production system in most Iberian farms is ‘ciclo cerrado’ (‘closed cycle’), meaning that the entire production process, including breeding, rearing and fattening, takes place on the same farm, without outsourcing production steps.
To understand IAV evolution in different swine farming systems, we conducted active longitudinal surveillance in free-ranging Iberian swine during 2015–9. In parallel, we also conducted active and passive surveillance for IAV in Spanish white pigs raised in enclosed high-density modern farms in order to compare IAV evolutionary trajectories in two distinct ecosystems. Iberian swine had lower rates of IAV infection compared to white pigs, but the viruses isolated from Iberian herds were genetically diverse, reflecting a distinct and dynamic ecosystem with zoonotic risk that merits further study.
2. Materials and methods
2.1 Sample collection
Active and passive surveillance was carried out throughout the year from 2015 to 2019 in the main swine-producing regions of Spain, including free-range and intensive farms (Fig. 1C, D). In total (positive and negative farms), we collected 2,149 nasal swabs for this study, including 821 nasal swabs from white pigs and 1,328 nasal swabs from Iberian pigs. We also collected 2,853 serum samples from Iberian pigs. Sampling in free-range farms of Iberian pigs was conducted on sixty-nine farms from forty-one different towns in the six southwestern provinces of Badajoz, Cáceres, Huelva, Segovia, Salamanca, Toledo and Córdoba. Sampling in intensive farms of white pigs was conducted on thirty farms from fourteen towns spanning the central provinces of Toledo, Valladolid, Salamanca, and Segovia; the eastern province of Valencia; and the northeastern provinces of Zaragoza and Navarra. Passive collection of samples was predominantly lung or tracheal tissues from euthanized white pigs with respiratory disease taken by the official farm veterinarian and submitted to a specialized veterinary diagnostic laboratory covering the main intensive swine producing regions of Spain. Active collection of nasal swabs and sera was performed in random batches of approximately twenty to thirty pigs per farm of Iberian and white swine with or without clinical signs of respiratory disease. Sampling was carried out in weaning pigs (3–9 weeks old) in intensive farms with Landrace and Large White breeds (white pigs) and in free-range Iberian pig farms. Nasal swabs and sera from fattening Iberian pigs (10–12 months old) were also collected. Seroconversion, as a correlate of IAV circulation, was a criterion for Iberian farm selection for sampling. All farms were visited at least twice and tested by serology (see Section 2.3 ), for a total of 137 Iberian farm visits during the study.
In total, over the study period, 821 nasal swabs and lung samples were collected from white pigs and 1,328 nasal swabs and 2,853 serum samples from Iberian pigs. Sterile cotton swabs were inserted deep into the ventral part of the nose and gently rotated for a few seconds around the conduct. The moisten swabs were then inserted into viral transport medium (VTM): phosphate buffer solution containing 0.5 per cent bovine serum albumin (BSA), 200 U/ml penicillin, 200 mg/ml streptomycin, 100 U/ml polymyxin B sulfate, and 250 mg/ml gentamicin. Swabs were chilled on ice and sent to the laboratory within 24–36 h after collection. Lung tissues from pneumonic lesions from sick animals sacrificed in the farms were sent refrigerated to the laboratory for analysis.
Since our surveillance protocol differed somewhat for white pigs compared to Iberian pigs to accommodate the lower detection rates of virus on Iberian farms, we did not compare the overall rates of polymerase chain reaction (PCR) positivity, as these numbers are biased by different denominators. Rates would be inflated in Iberian pigs because serologically negative farms were not tested by PCR. We instead report the rate of PCR(+) pigs on farms with at least one PCR(+) animal (Fig. 2). The seropositive farms were primarily fattening farms with adult pigs (twenty-eight of forty), whereas the other twelve seropositive were farrow-to-finish. Five of the twelve farrow-to-finish farms had piglets with PCR(+) samples. Of the 109 piglets tested at the five PCR(+) farms, eighty-nine piglets were PCR(−) and twenty were PCR(+). In the twenty PCR(+) pigs, eleven IAVs were successfully isolated and sequenced. At least one virus was isolated and sequenced from each of the five Iberian farms that was PCR(+) during this study, including one farm in Huelva, one farm in Toledo, and three farms in Badajoz (Fig. 1C).
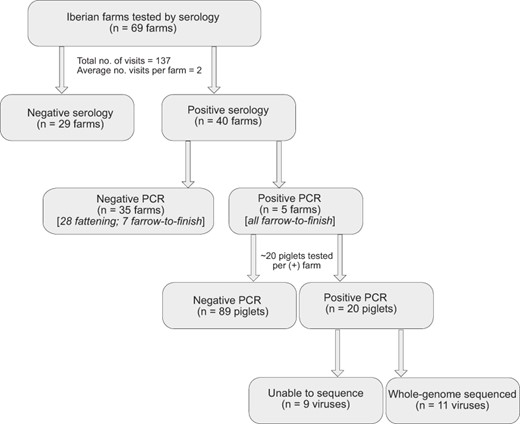
Flowchart of sampling protocol in Iberian farms during 2015–9.
2.2 Virus isolation and RNA extraction
The swab and tissue samples, previously minced in viral transport media, were centrifuged for 15 minutes and the supernatants were used for RNA extraction and for inoculation of cell cultures and chicken embryos for virus isolation. Total RNA from samples was extracted using the QIAamp Viral RNA Mini Kit (Qiagen). IAV detection was made by real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR) of Matrix (M) gene with the primers forward: GAC CRA TCC TGT CAC CTC TGA C, reverse: AGG GCA TTY TGG ACA AAK CGT CTA, and probe: TGC AGT CCT CGC TCA CTG GGC ACG (labeled with 6-FAM, MGBNFQ; Invitrogen). The reaction conditions were in accordance with CDC (SOP REF#1-007-05): RT (45°C, 30 min), activation Taq (95°C, 10 min) followed by 40 cycles: denaturation (95°C, 15 sec) and annealing/extension (60°C, 60 sec). The RT-qPCR was performed with the High ScripTools-Quantimix Easy Probes kit (Biotools) in an Applied BiosystemsTM Fast 7500 thermal cycler. IAV isolation from RT-PCR-positive samples was made in 10-day-old chicken embryos or Madin–Darby canine kidney cells cultures grown in advanced Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher) supplemented with glutamine (4 µM), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (20 µM), BSA fraction V, 100 U/ml streptomycin, 100 U/ml penicillin and TPCK (6-(1-tosylamido-2-phenyl)-ethyl-chloromethyl-ketone)-treated trypsin (2 mg/ml) from bovine pancreas (Merck Life Science) at 37°C and 5 per cent CO2 according to the Manual on Animal Influenza Diagnosis and Surveillance (World Health Organization 2002). Before inoculation in embryos and cells, swab fluids and lung extracts in VTM were spinning 4000 rpm, 10 min, and filtered through 0.22-mm Millex syringe filters (Millipore). Inoculated cell cultures were cultured at 37°C, 5 per cent CO2, and examined daily for evidence of cytopathic effects (CPEs). If CPE is positive, or after 4 days, supernatants were tested by RT-PCR. Allantoic fluid was tested for the presence of virus by hemagglutination assay (HA) or RT-PCR. Negative allantoic fluid and cell supernatants were passaged up to three times. Virus isolates were submitted to antigenic and genetic analyses to determine the subtype by conventional one-step RT-PCR with Superscript III one-step RT-PCR Kit with Platinum Taq polymerase (Invitrogen; Zhang and Harmon 2014).
2.3 Serological assays
Serological analysis was performed in two successive steps: first an influenza type A (against IAV nucleocapsid) indirect Enzyme-Linked Immunosorbent Assay (ELISA) (INGEZIM Influenza A, Eurofins-Ingenasa, Spain) was used to screen large numbers of serum samples from pigs and then a hemagglutination inhibition (HI) test to assay the specificity of the IAV-positive sera against the different swine virus subtypes, H1N1, H1N2, and H3N2. The ELISA was performed according to the manufacturer’s instructions, and samples were positive when the percentage of inhibition was ≥45 per cent. HI test was performed according to standard procedures (World Health Organization 2002). Previously to the HI test, the ELISA-positive sera were treated overnight with receptor-destroying enzyme from Vibrio cholerae (Sigma, Spain) to remove nonspecific inhibitors, were then inactivated at 56°C for 60 min, and finally were adsorbed with a 50 per cent chicken red blood cell (RBC) suspension at 4°C and to avoid nonspecific binding to RBC. The cutoff of HI was set to dilution ≥1:40. The swine influenza A strains used in HI analysis were as follows: A/sw/Spain/54008/2004 (reassortant human-like H3N2), A/sw/Spain/53207/2004 (EA-H1N1), A/sw/Spain/40564/2002 (reassortant human-like H1N2), and A/California/07/09 (pdmH1N1). To study the potential exposure of Iberian pigs to avian or human influenza viruses, especially in farms located near wetlands and water reservoirs, we performed ELISA tests with recombinant HAs from representative avian and human subtypes. In case of doubtful ELISA results, suspicious serum samples were also tested with selected avian IAVs by HI. The recombinant HAs were provided by Dr Florian Krammer (Icahn School of Medicine at Mount Sinai, New York, USA; Meade et al. 2017).
2.4 Genome sequencing
The complete genomes of swine IAV isolates were sequenced at the Icahn School of Medicine at Mount Sinai using methods previously described (Venkatesh et al. 2018). The entire genome was amplified from 3 µl of RNA template using a multisegment RT-PCR (M-RTPCR) strategy (Zhou et al. 2009). RNA was extracted using a QIAamp viral RNA minikit (Qiagen). M-RTPCR amplification was performed with a SuperScript III high-fidelity RT-PCR kit (Invitrogen) according to the manufacturer’s instructions using the Opti1 primer set, consisting of primers Opti1-F1 (5´-GTTACGCGCCAGCAAAAGCAGG), Opti1-F2 (5´-GTTACGCGCCAGCGAAAGCAGG), and Opti1-R1 (5´-GTTACGCGCCAGTAGAAACAAGG). DNA amplicons were purified using an Agencourt AMPure XP 5 ml kit (Beckman Coulter). Sequencing libraries were prepared, and sequencing was performed on a MiSeq instrument (Illumina, Cambridge, UK) with 2 × 150-base paired end reads. Handling of the data for the raw sequence reads and extraction of consensus sequences were performed at Icahn School of Medicine at Mount Sinai (ISMMS) as described previously (Mena et al. 2016). All sequences were deposited in the National Center for Biotechnology Information’s (NCBI) GenBank, with accession numbers MZ945737-MZ373166 (Supplementary Table S2).
2.5 Phylogenetic analysis
All available whole-genome sequence data for IAVs collected from swine in Europe during 2009–9 were downloaded from the NCBI Influenza Virus Resource, which curates data submitted to GenBank (Bao et al. 2008). Sequence alignments were generated separately for each genome segment (and separately for H1, N1, H3, and N2) and aligned using MUSCLE v3.8.3 (Edgar 2004), with manual correction in Seqotron (Fourment and Holmes 2016). Phylogenetic relationships were inferred using the maximum likelihood (ML) method available in the program RAxML v7.2.6 (Stamatakis 2006), incorporating a general time-reversible (GTR) model of nucleotide substitution with a gamma-distributed rate variation among sites. Due to the size of the dataset, we used the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (http://biowulf.nih.gov). To assess the robustness of each node, a bootstrap resampling process was performed (500 replicates), again using the ML method available in RAxML and the Biowulf Linux cluster. Lineage assignment was performed using reference sequences as well as the H1 swine classification tool (Anderson et al. 2016), available at the Influenza Research Database (https://www.fludb.org/; Zhang et al. 2017), and octoFlu (Chang et al. 2019). For clarification, Supplementary Table S3 in the Supplementary Materials lists the reference strains and names for swine lineages used in this study.
2.6 Bayesian analysis
To investigate the evolutionary relationships between human-like H3 viruses in swine and humans in finer detail, an additional dataset was downloaded from the Influenza Virus Resource (Bao et al. 2008) that included 40 H3 sequences available from humans in Europe. TempEst v1.5.3 (Rambaut et al. 2016) was used to identify potential sequencing errors that substantially deviated from the linear regression of root-to-tip genetic distance against time. A time-scaled Bayesian analysis used the Markov chain Monte Carlo (MCMC) method available in the BEAST v1.10.4 package (Suchard et al. 2018), again using the Biowulf Linux cluster. A relaxed uncorrelated lognormal molecular clock was used with a constant population size and a GTR model of nucleotide substitution with gamma-distributed rate variation among sites. The MCMC chain was run separately three times for each of the datasets for at least 100 million iterations with subsampling every 10,000 iterations, using the BEAGLE 3 library, to improve computational performance (Ayres et al. 2019). All parameters reached convergence, as assessed visually using Tracer v.1.7.1, with statistical uncertainty reflected in values of the 95 per cent highest posterior density. At least 10 per cent of the chain was removed as burn-in, and runs were combined using LogCombiner v1.10.4, and a maximum clade credibility (MCC) tree was summarized using TreeAnnotator v1.10.4 and visualized in FigTree v1.4.4. The number of live swine traded between Spain and other European countries during 2001–9 was obtained from the United Nations Comtrade Database (https://comtrade.un.org/data/; Supplementary Table S4). The number of swine per European country was obtained from the Food and Agriculture Organization of the United Nations FAOSTAT (http://www.fao.org/faostat/en/#data; Supplementary Table S5).
2.7 Pandemic H1N1 viruses
To investigate the evolutionary relationships between pdmH1N1 viruses in swine and humans in finer detail, an additional dataset was downloaded from the Influenza Virus Resource (Bao et al. 2008) that included all available pdmH1N1 sequences from humans in Europe during 2009–9. ML trees were inferred using methods described above for all eight segments and are available at the GitHub repository (https://github.com/mostmarmot/Iberian). Epidemiological data on the number of pdmH1N1 viruses collected from humans in Spain during 2009–9 were obtained from the World Health Organization’s FluNet (Supplementary Table S6).
3. Results
3.1 IAV identified in Iberian swine by serology, PCR, and whole-genome sequencing
From 2015 to 2019, IAV was detected every year in Iberian pigs by serology (Supplementary Table S7). Of the sixty-nine Iberian farms tested multiple times for this study, forty were positive for IAV by serology (58.0 per cent; Fig. 2). In total, 953 of 2853 total Iberian pigs tested were seropositive (33.4 per cent), a fraction that is lower than previously reported for white pigs in Spain (Meritxell et al. 2012). Five Iberian farms also were positive for IAV by RT-PCR: three farms in Badajoz, one farm in Huelva, and one farm in Toledo (Fig. 1C). All PCR-positive swabs were collected from weaning piglets (3–9 weeks old), with no positive swabs from fattening pigs (10 or 12 months old). Serum collected from older Iberian swine provided evidence of prior infection with viruses from the EA-H1, huH3, and pdmH1 lineages, but only viruses with HA segments from the EA-H1 lineage were successfully isolated and sequenced from piglets. Viruses were isolated and whole-genome sequenced (n = 11 viruses) from all five Iberian farms that were PCR positive (Fig. 3).
3.2 Genetic diversity of IAV in Iberian swine
Complete or near-complete genome sequences were obtained for eleven viruses collected for this study from Iberian swine (Fig. 3) and thirty-six viruses from white pigs. Of the twelve genotypes identified in all pigs in this study (G1–G12, Fig. 4), two were identified in both Iberian and white pigs (G1 and G2), two genotypes were observed in Iberian pigs only (G6 and G7), and eight genotypes were observed in white pigs only. Of the seven IAV lineages detected in Spanish swine in this study, three were observed in both Iberian and white pigs (EA-H1N1, 1970s-like huH3N2, and pdmH1N1), one was observed in Iberian swine only (1990s-like huH3N2, N2 segment only), and three were observed in white pigs only (a second 1970s-like huH3N2, huH1N1, and 2000s-like huH3N2). The vast majority of viruses had six internal genes belong to a single lineage (EA-H1N1 or pdmH1N1), with reassortment primarily involving the HA and NA antigens. Nine different HA–neuraminidase (NA) pairings were observed. Notably, three reassortant genotypes were identified in this study that were not previously observed in European swine: G4 and G12 in white pigs and G7 in Iberian swine.

IAV genotypes in Iberian pigs. The farm (#1–#5, see map) and genotype is listed for the eleven viruses sequenced for this study from Iberian pigs. Each circle represents a segment of the IAV genome, shaded by IAV lineages. HA and NA surface proteins are listed first, followed by the six internal gene segments.

Twelve genotypes in Spanish swine. Each box represents a segment of the IAV genome, shaded by lineage, similar to Fig. 3. The Spanish provinces where each genotype was identified are listed for Iberian pigs and white pigs.
3.3 Rare H3N1 subtype with a human-origin H3 identified in Spanish white pigs
Five reassortant H3N1 viruses were found between 6 November 2018 and 25 September 2019 in white pigs in three Spanish provinces (Fig. 5). The viruses have seven segments from the EA-H1N1 lineage and one H3 segment that was introduced from humans into swine in Europe in the 2000s. To date, this human-origin H3 has only been detected in swine in the Netherlands and Denmark (e.g. A/sw/Denmark/10115-1/2013(H3N2); Fig. 5). In contrast to the Spanish H3N1 viruses, all other viruses positioned in this clade from Denmark and the Netherlands still have the H3N2 subtype. Spain imports more live swine from the Netherlands than another other country (Supplementary Table S3, Supplementary Fig. S1), providing opportunities for a new human-origin H3 to enter Spain along these trade routes. The estimated time of viral entry into Spain is 2013–7, based on the tMRCAs on the branch leading up to the Spanish clade (indicated with a star, Fig. 5). Along this branch, eight amino acid changes in the H3 also occurred (Fig. 5).
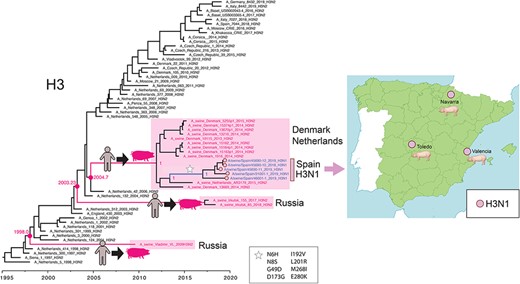
H3N1 viruses in Spanish white pigs. A time-scale MCC tree of the H3 segment reconstructs the evolutionary relationships of 5 H3N1 viruses collected in Spanish white pigs, 12 closely related H3N2 viruses from swine in Denmark and the Netherlands, 40 human seasonal H3N2 viruses collected during 1997–2019 in Europe, and 3 human-like H3N2 viruses collected from swine in Russia. Posterior probabilities and estimated tMRCAs are provided for key nodes. Cartoons and pink boxes indicate the three introductions of human seasonal H3N2 viruses to swine in Europe since 1997. A white star indicates amino acid substitutions observed along the branch leading to the H3N1 Spanish clade (H3 numbering). A map shows the three locations where H3N1 viruses were identified in Spanish white pigs.
3.4 Reassortant H1N2 virus with Spanish human-origin N2 identified in Iberian swine
A novel reassortant virus was also identified in Iberian swine in Badajoz. The G7 (H1N2) viruses have segments derived from three IAV lineages: N2 segments estimated to have transmitted from humans to swine in the mid-1990s (Fig. 6), HA from EA-H1N1, and six pdmH1N1 internal genes (Fig. 3). Based on the phylogeny, the human-like N2 has been circulating undetected in swine for decades but to date has only been detected in swine in Spain, with no evidence of spread to other countries. Similar N2-1990s segments were found in viruses collected previously in white pigs in northeastern Spain (Martín-Valls Gerard et al. 2014), suggesting that the virus may have originally transmitted from humans to white pigs before infecting Iberian swine. However, the direction of transmission is difficult to resolve because so much time elapsed between the reverse zoonosis event in the mid-1990s and first detection in swine in 2010, resulting in a very long branch on the tree. Overall, the N2 phylogeny indicates that there have been at least six introductions of human seasonal H3N2 viruses into swine in Europe since 1990 (Fig. 6). These clades also tend to be small and limited to one country (Italy, Germany, Russia (twice), France, and Spain), with little evidence of spread to additional countries.
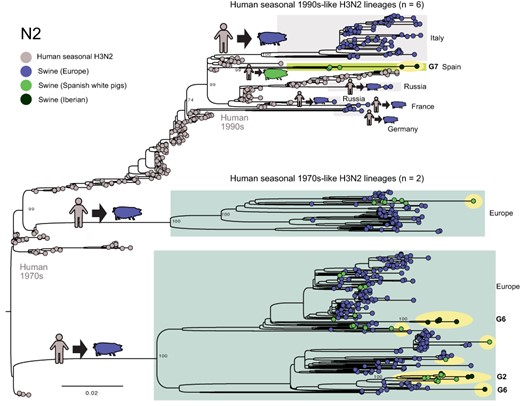
Human seasonal 1990s-like N2 lineage identified in Iberian pigs. A ML tree reconstructs the evolutionary relationships of the N2 segment from H3N2 and H1N2 viruses collected in European swine and humans. Each virus is represented by a circle, shaded by host and location. Viruses sequenced for this study are highlighted in yellow. Bootstrap values are provided for key nodes. The genotype (Fig. 3) is listed for all viruses from Iberian pigs.
The human seasonal 1970s-like N2 lineages that are widespread in European swine were introduced seven times into our study population from swine in other European countries. In most cases, long branch lengths make it difficult to infer which European country was the source of the introduction. The only direct evidence of gene flow between Spanish white pigs and Iberian swine involved a clade of viruses with the G2 genotype identified in Toledo province, where both white pigs and Iberian pigs are raised (Fig. 7).
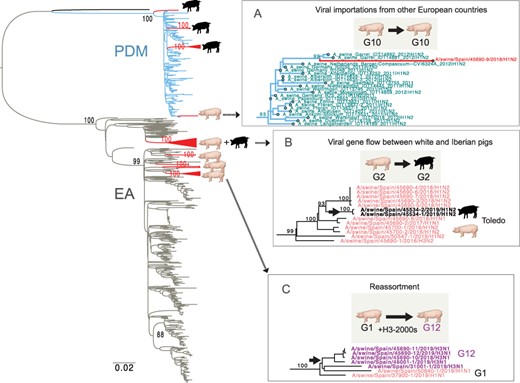
Three mechanisms of IAV evolution in Spanish swine. A ML tree of viruses collected in European swine from 2009 to 2019 (PB2 segment). Red triangles indicate clades of viruses collected from Spanish swine for this study, with cartoons indicating host (Iberian or white pig). Three clades of interest have been expanded (A–C). Complete trees for all segments, including pdmH1N1 trees that include human and swine viruses, are available in the GitHub repository (https://github.com/mostmarmot/Iberian).
3.5 Reverse zoonotic transmission of pdmH1N1 viruses into Iberian swine
Reverse zoonosis of pdmH1N1 viruses from humans to swine has occurred at least sixty-six times in Europe since the 2009 pandemic, repeatedly seeding new human-origin pdmH1N1 lineages in most European countries that conduct IAV surveillance in swine (Fig. 7A). The vast majority (83 per cent, 55/66) of independent human-to-swine transmission events occurred during the first years of the pandemic (2009–11; Fig. 8A) when the virus was circulating at its highest levels in humans (Fig. 8B). The vast majority of pdmH1N1 viruses are phylogenetically positioned within clades that are monophyletic by country, with no evidence of spread between countries. Reassortant viruses with pdmH1N1 segments were identified in both Iberian and white pigs from our study (Figs 3 and 4). The pdmH1N1 segments observed in Iberian pigs derived from viruses that appear to be introduced directly from humans into Iberian pigs on at least three occasions during 2009–11, as evidenced by three monophyletic Iberian clades (Figs 7–9).
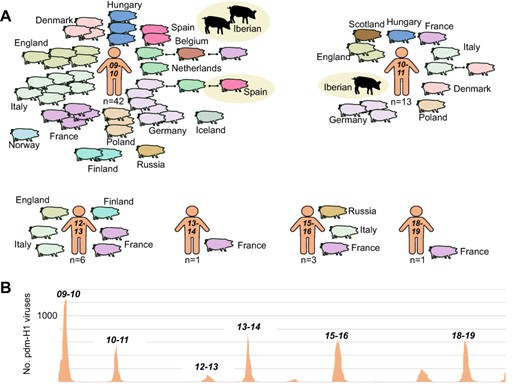
Transmission of pdmH1N1 viruses from humans to swine in Europe, 2009–19. (A) Each pig represents a discrete introduction of a pdm-H1 virus from humans into swine, shaded by European country. Instances of onward transmission to swine in another European country are indicated by black lines. Viruses identified in Spanish swine in this study are highlighted by beige circles. Introductions are ordered chronologically by the seven winter epidemics in humans from 2009–10 to 2018–9 in which reverse zoonosis was observed. The total number of introductions from humans into European swine per winter season is provided. (B) The number of pdm-H1 viruses collected in humans in Spain during 2009–19. Data available from WHO FluNet.
Thereafter, the pdmH1N1 viruses in Iberian pigs acquired new HA and NA segments through reassortment with EA-H1N1, huH3N2, and huN2-1990s lineages, retaining only the pdmH1N1 internal genes (Fig. 9). In contrast, pdmH1N1 viruses observed in Spanish white pigs (e.g. G10, Fig. 4) were positioned in clades containing viruses from swine in Germany and the Netherlands (Figs 8–9) and were introduced not by humans but by white pigs entering Spain from other countries via high-volume EU live swine trade (Supplementary Fig. S1).

Sources of pdmH1N1 reassortants in Spanish swine. Arrows indicate introductions of two reassortant viruses with pdmH1N1 segments into Spanish white pigs from swine in other European countries and three pdmH1N1 viruses from humans into Iberian pigs, followed by reassortment with other swine viruses. Genotypes G4 and G7 (red font) have only been observed in Spain.
4. Discussion
By conducting active genomic surveillance for IAVs in Spain’s Iberian swine, our study uncovered IAV in a free-range ecosystem in southern Spain that is rarely studied. Although influenza is not considered to be a clinical problem in Iberian swine, the pigs were found to be infected with a range of genetically diverse IAVs introduced from both humans and white pigs. At least three independent reverse zoonosis transmission events of pdmH1N1 occurred since 2009. The introduction of pdmH1N1 viruses into Iberian pigs from humans provided new opportunities for reassortment, creating new genotypes with pdmH1N1 internal genes and HA and NA segments from other H3N2 and H1N1 viruses circulating in swine, similar to observations in other European swine-producing countries (Bányai et al. 2012; Grøntvedt et al. 2013; Nokireki et al. 2013). Prior to 2009, reverse zoonosis contributed human-origin surface proteins, including a novel N2 segment observed in Iberian swine that is related to human H3N2 viruses from the 1990s and has only been detected in Spain to date. A H3 segment found in Spanish white pigs in 2018–9 was introduced from humans to European swine (possibly Denmark) in the early 2000s and has reassorted with EAswH1N1 viruses in Spain to create rare H3N1 reassortants. The H3N1 viruses were circulating in Spanish herds as recently as September 2019; ongoing surveillance will monitor whether the genotype will persist.
The observation of multiple diverse H1N1 and H3N2 viruses transmitting from humans to swine in Spanish herds is not entirely unexpected, given the frequency of reverse zoonosis of IAV to swine in other countries (Cappuccio et al. 2011; Wong et al. 2018). Iberian swine have particularly high contact rates with their farmers, providing opportunities for transmission. Whether the viruses circulating in Iberian swine transmit back to their human handlers is unknown, as no IAV surveillance has been conducted in Iberian swine farmers at this time. It would be of great interest to study how frequently Iberian swine farmers are exposed to swine IAVs, given that Iberian swine harbor genetically diverse IAVs, including reassortants with EA-H1N1 surface proteins that have caused prior zoonotic infections and to which humans have little preexisting immunity.
Further study also is needed to understand how IAVs transmit between white pigs and Iberian pigs. Although Iberian and white pig farms are geographically segregated and no farms are known that raise both Iberian and white pigs on their premises, Toledo is an example of a province where white pig farms are located within areas rich in Iberian farms. Being separate systems, viruses would have to travel considerable distance via wind, environmentally, or via an intermediate host such as a free-ranging wild boar to spread between the two populations. The low amount of viral gene flow between white and Iberian pigs reflects such barriers, although the fact that IAVs are periodically transmitted from white to Iberian pigs is an indication of how difficult it is to keep even a relatively isolated Iberian swine population free of a pathogen as transmissible as influenza.
Going into this study, one hypothesis was that free-range Iberian swine would have higher rates of exposure to wildlife viruses, including IAVs carried by wild boars that roam the European countryside and wild birds that are a natural reservoir for sixteen HA subtypes and nine NA subtypes. However, we found no evidence of IAV transmission between wildlife and Iberian pigs in serological or virological data. This does not necessarily mean wildlife-to-pig transmission does not occur, only not at a frequency high enough to be detected at our level of sampling. Such findings support prior conclusions that swine are far more susceptible to human viruses than avian viruses, which is consistent with their sialic acid receptor distributions being more similar to humans than birds (Nicholls et al. 2008; Wasik, Barnard, and Parrish 2016). These findings have implications for biosecurity for IAV on swine farms going forward. For decades, traditional backyard swine farms have been systematically replaced by large enclosed facilities designed to produce swine with greater efficiency as well as reduce transmission of pathogens between swine and wildlife. Our findings suggest that Iberian swine raised in a free-range outdoors system appear to have a low risk of acquiring IAVs from wild birds and IAV control strategies should instead be focused on interfaces between Iberian pigs and their human handers, as well as with intensively raised white pigs. Uptake of seasonal influenza vaccines in humans indirectly protects animals with human contact, preventing reassortment between animal and human strains. Systematic surveillance of influenza viruses and other relevant infectious agents, in domestic or wild pigs, is highly recommended due to the rapid reorganization of ecosystems caused by human intervention.
Supplementary data
Supplementary data is available at Virus Evolution online.
Acknowledgements
We acknowledge the veterinarians and swine holders who submitted samples to our laboratory, especially Lourdes Muñoz and Juan Pablo Rosas from Inga Food S.A. (Tres Cantos, Madrid, Spain) and Gema Chacón and Rafael Baselga from Exopol S.L. (San Mateo del Gállego, Zaragoza, Spain). We also thank our collaborators Jaime Maldonado from Hipra S.A. (Amer, Gerona, Spain) who provided influenza strains for the HI assays, Florian Krammer from the Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, USA who provided the recombinant hemagglutinins for the HA ELISA assays, Zhain Khalil of the Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, USA for her assistance in submitting virus genomic sequences to GenBank, and Jaime Bosch from Centro de Vigilancia Sanitaria Veterinaria of the Universidad Complutense, Madrid who provided us the Spanish swine density maps in Fig. 1.
Funding
This work was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID) Intramural Research Program and by the Center for Research on Influenza Pathogenesis (CRIP), an NIAID Center of Excellence for Influenza Research and Surveillance (CEIRS, contract # HHSN272201400008C) and the Center for Research on Influenza Pathogenesis and Transmission (CRIPT), a NIAID Center of Excellence for Influenza Research and Response (CEIRR, contract # 75N93019R00028).
Conflict of interest:
None declared.
Disclaimer
The content does not necessarily reflect the views or policies of the U.S. Government or imply endorsement of any products. The A.G.-S. Laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-fold LLC, Model Medicines, and Merck, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, and Pfizer, outside of the reported work. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work.
References
World Health Organization. (
Author notes
Current address: National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, 8600 Rockville Pike, Bethesda, MD 20894 USA.
Paloma Encinas and Gustavo del Real contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}