




Abstract
Tyrosine kinase inhibitors such as imatinib mesylate have changed the clinical course of chronic myeloid leukemia; however, the observation that these inhibitors do not target the leukemia stem cell implies that patients need to maintain lifelong therapy. The mechanism of this phenomenon is unclear: the question of whether tyrosine kinase inhibitors are inactive inside leukemia stem cells or whether leukemia stem cells do not require breakpoint cluster region (Bcr)-Abl signaling is currently under debate. Herein, I propose an alternative model: perhaps the leukemia stem cell requires Bcr-Abl, but is dependent on its kinase-independent functions. Kinases such as epidermal growth factor receptor and Janus kinase 2 possess kinase-independent roles in regulation of gene expression; it is worth investigating whether Bcr-Abl has similar functions. Mechanistically, Bcr-Abl is able to activate the Ras, phosphatidylinositol 3-kinase/Akt, and/or the Src-kinase Hck/Stat5 pathways in a scaffolding-dependent manner. Whereas the scaffolding activity of Bcr-Abl with Grb2 is dependent on autophosphorylation, kinases such as Hck can use Bcr-Abl as substrate, inducing phosphorylation of Y177 to enable scaffolding ability in the absence of Bcr-Abl catalytic activity. It is worth investigating whether leukemia stem cells exclusively express kinases that are able to use Bcr-Abl as substrate. A kinase-independent role for Bcr-Abl in leukemia stem cells would imply that drugs that target Bcr-Abl's scaffolding ability or its DNA-binding ability should be used in conjunction with current therapeutic regimens to increase their efficacy and eradicate the stem cells of chronic myeloid leukemia
Introduction
Tyrosine kinase inhibitors (TKIs) have dramatically changed the clinical course of chronic myeloid leukemia (CML), improving the 5-year survival from 35% to greater than 90% with imatinib mesylate (Gleevec, Novartis International, Basel, Switzerland, http://www.gleevec.com/index.jsp) [1, 2]. Imatinib mesylate is a TKI that binds to the Abl kinase domain of the breakpoint cluster region (Bcr)-Abl chimerical protein and inhibits the protein's catalytic activity. The development of this drug provided the proof of principle for rational drug design [3], yielding dramatically increased survival of chronic phase CML patients with limited side effects [1, 2]. Nevertheless, TKI therapy is not curative: relapse remains a significant problem for patients with CML, particularly patients in the blast crisis phase of the disease. Early in the course of therapy, relapse is caused by acquired drug resistance [4, 5], but the failure of TKIs to cure CML may also be due to the fact that imatinib mesylate and other TKIs do not eradicate the leukemia stem cells (LSCs) [6, 7]; thus, most patients are required to continue TKI treatment for life given that discontinuation of therapy often results in relapse [8, 9].
Imatinib is the first approved TKI specifically designed to inhibit the catalytic activity of the Bcr-Abl oncogene of CML [10]. Unfortunately, in spite of impressive response rates and tolerability of this drug, a substantial minority of patients eventually develops resistance to imatinib. Mechanisms of resistance include the acquisition of point mutations in the catalytic domain of Bcr-Abl that render imatinib ineffective and restore kinase activity to the oncoprotein in the presence of imatinib [4, 5, 11], amplification of the Bcr-Abl gene resulting in overexpression of the Bcr-Abl protein [4, 12, 13], and clonal evolution [14, 15]. Hence, using knowledge of the topology of the kinase domain in wild-type and mutant Bcr-Abl, second-generation TKIs—dasatinib and nilotinib—were developed that showed efficacy in many imatinib-resistant patients [16–18], although neither imatinib nor the second-generation inhibitors are effective in patients with the common T315I mutation. The third-generation tyrosine kinase inhibitor ponatinib is able to inhibit most Bcr-Abl mutations and is effective in patients with T315I [19, 20]. However, it is not known whether CML stem cells are susceptible to ponatinib treatment.
Acquired Versus Inherent Resistance
Relapse of the disease following discontinuation of a drug is not synonymous with the acquisition of resistance. Resistance can be subdivided into acquired and inherent, where acquired resistance is defined as the acquisition of mutations that allow the cell to become refractory to treatment, and inherent resistance is defined as the presence of a population (or subpopulation) of cells that are intrinsically refractory to treatment. Acquired resistance may be further categorized as Bcr-Abl-dependent or Bcr-Abl-independent. Most patients who are initially sensitive to treatment with TKIs but later become unresponsive develop acquired resistance that is associated with mutations in the Bcr-Abl oncogene [21]. In fact, the T315I mutation can be detected in some patients even prior to treatment [17]. Other forms of acquired resistance have been described that are independent of mutation in Bcr-Abl but can be attributed to increased expression of efflux and influx proteins [22–24], deregulation of apoptosis/survival pathways [25–30], or other acquired mutations including amplification of Bcr-Abl [31]. Although this is an interesting and extremely important topic, acquired resistance is not the scope of this article.
Inherent (primary) resistance, on the other hand, is a state in which drugs lack efficacy from the outset of treatment. One may envision a situation in which the entire CML cell population is homogeneously refractory to treatment or another in which a subpopulation of a patient's CML cells is resistant to treatment: in the latter case, treatment creates a selective pressure that accelerates the outgrowth of the pre-existing resistant clone. Indeed, the presence and outgrowth of pre-existing mutations in the Bcr-Abl oncogene have been described in patients [32, 33]. The scope of this article is not to discuss inherent resistance per se, but rather to discuss a specific instance of this phenomenon: the inherent resistance of CML stem cells to TKIs. This differs from the usual notion of inherent resistance because the overall population of leukemia cells, predominantly composed of leukemia progenitor cells (LPCs), remains sensitive to drug, whereas the LSCs are refractory and serve as a reservoir of cells that can subsequently re-establish the disease. It is unlikely that the phenomenon of resistance of LSCs to TKIs is merely the result of the outgrowth of a pre-existing resistant clone, because in this scenario the entire population of clonal progeny would be refractory to treatment, whereas, in fact, immunophenotypically defined CML progenitor cells are sensitive, and the inherently resistant CML cells express stem cell markers and are a distinct subpopulation. In order for stem cells' resistance to TKIs to be the result of acquired mutation, one would need to envision a scenario in which an acquired mutation not only confers resistance to TKIs, but also confers expression of stem cell markers.
Relevance of LSC Resistance to TKIs to Clinical Outcome
It is worth emphasizing that the phenomenon of acquired drug resistance is independent of the phenomenon that is the subject of this article: that LSCs in CML are refractory to TKIs that target Bcr-Abl. Most cases of acquired resistance develop early in the disease from the outgrowth of clones that contain mutations in Bcr-Abl that impair its ability to bind the TKIs [4, 5]. On the other hand, it has been observed that patients who develop a complete cytogenetic response retain Bcr-Abl positive clones [2] and that these clones contain LSCs that can cause relapse upon discontinuation of imatinib [8, 9, 34]. More directly, CML cells with an LSC immunophenotype are able to survive in the presence of TKIs [6, 35]. Interestingly, it must be conceded that not all patients relapse upon discontinuation of TKIs: in the study of Rousselot et al. [9] only 6 of 12 patients who stopped TKI therapy experienced relapse of CML. Other studies have since corroborated these data, showing that although the majority of patients relapse upon drug discontinuation, 40%–50% do not [8, 36]. How can this be explained? If imatinib does not act on the leukemia stem cell, how could these patients achieve control of their disease and eradication of the Bcr-Abl transcript at the molecular level? This apparent paradox may be explained by the induction of an antileukemia immune response that targets the LSCs [37]. In the study by Rousselot et al. [9], the patients who did not have a molecular relapse upon discontinuation of imatinib had previously been treated with interferon (IFN)-α. Likewise, Takahashi et al. showed that prior treatment with IFN-α was predictive of relapse-free survival upon drug discontinuation [36]. In contrast to imatinib, IFN-α preferentially targets primitive leukemia cells including the LSC [38]. Although the precise mechanism of IFN-α action is not known, it is thought to act by facilitating an antileukemia immune response. Clinical trials testing the ability of IFN-α to maintain remission upon TKI drug discontinuation are currently in progress [37].
Nevertheless, another interpretation of the above data is that not every single leukemia stem cell needs to be eradicated to attain long-term remissions. In the above studies, patients who did not relapse upon drug discontinuation retained LSCs that were positive at the genomic level for the Philadelphia chromosome [39–41]. Although it is not certain whether this minimal residual disease comprises quiescent stem cells that have entered a state of deep dormancy, or whether the stem cells do in fact cycle at low rates and contribute to leukemia progenitor cells that are cleared by the immune system, it is really exciting that at least in some patients clearance of the entire leukemia clone is not a prerequisite for long-term remission. Notwithstanding, many patients with CML are expected to remain on TKIs for life in an effort to prevent relapse of their disease.
Cancer Stem Cells in General Are Difficult Therapeutic Targets
The notion that cancer stem cells are more resistant to chemotherapy is not specific to CML. Stem cells in general have several properties, shared with cancer stem cells, that make them poor targets of chemotherapy. Stem cells are quiescent, a property that is able to make some types of cancer stem cells refractory to drugs that selectively kill cycling cells [42–44]. Second, stem cells have enhanced capacity for DNA repair, which has been reported to make some types of cancer stem cells refractory to drugs that function by induction of DNA damage [45–48]. Finally, stem cells have increased expression of drug efflux molecules; it is possible to enrich for normal tissue-specific stem cells in a variety of normal tissues including bone marrow [49], testis [50], and heart [51], as well as cancer stem cells in a variety of tumors including those of the brain [52], gastrointestinal tract [53], and kidney [54] based on efflux of the Hoechst 33342 dye using flow cytometry due to expression of the Bcrp1/ABCG2 drug transporter [55]. Although it is not valid to extrapolate that cancer stem cells possess the same properties as noncancerous tissue-specific stem cells, and although the presence of cancer stem cells in solid tumors needs to be evaluated based on the strength of the experimental evidence for each cancer type, nevertheless, in some cancers, putative populations of cancer-initiating cells are more chemoresistant.
Defining the Leukemia Stem Cells of CML
The very definition of LSCs in CML is worth discussing as it is not sufficient to simply assume that the putative existence of leukemia stem cells in acute leukemia [56] is a concept that can be seamlessly transferred to CML, nor is it correct to assume that the immunophenotype of normal hematopoietic stem cells is the immunophenotype of the CML stem cell. In CML, this is especially relevant given that CML is a triphasic disease in which an indolent chronic phase characterized by a hyperproliferative hematopoietic stem cell biased toward myeloid differentiation and excessive production of myeloid progenitor cells [57] is followed by an accelerated phase that ultimately transitions to an aggressive terminal phase termed blast crisis that resembles acute leukemia and is characterized by additional acquired genetic mutations, abrogated differentiation, and poor response to treatment. Whereas it is thought that the initial Bcr-Abl transformation occurs in a CD34+CD38− hematopoietic stem cell, the leukemia stem cell that drives blast crisis is a granulocyte-macrophage progenitor cell that has acquired the ability to self-renew [58]. For the purposes of this discussion, the resistance of chronic phase CD34+CD38−CD90+Lin− leukemia stem cells is discussed. The resistance of LSCs in blast crisis CML to TKIs is a distinct phenomenon.
Even after restricting the discussion to chronic phase LSCs, caveats remain. For example, whereas it is feasible technically to fractionate leukemia cells according to immunophenotype in such a way as to achieve significant enrichment for LSCs versus LPCs, it remains impossible to purify these populations to homogeneity. Although it is tempting, in the name of simplicity, to use a blanket definition of LSCs and LPCs based on immunophenotype—for example, to define the population of CD34+CD38− versus CD34+CD38+ as human LSCs versus LPCs, respectively—it is crucial to acknowledge that these populations are not exclusively LSC versus LPC, and also that this classification is not equally valid in all patients [59–63]. The “best” definition of the LSC is functional, reflecting the unique capacity of this cell to reconstitute leukemia in a healthy recipient. However, this definition is not experimentally tractable in most circumstances: in this article, the definitions of these populations have been deliberately left vague to allow room for investigator innovation. There are multiple ways to define leukemia stem cells, none of which is without limitations; one must be cognizant of the limitations of each model system in the interpretation of the data and to incorporate appropriate controls and methodology to overcome the caveats.
Models Explaining Resistance of CML Stem Cells to TKIs
Two contrasting mechanisms have been proposed to explain the refractoriness of LSCs to TKIs. The first suggests that TKIs are unable to inhibit kinase activity in LSCs [64, 65], and the second suggests that TKIs are able to inhibit kinase activity in LSCs but that LSCs do not require Bcr-Abl signaling for survival [7, 35]. I propose an alternative model, predicated on the notion that fundamental differences exist in the signaling cascades orchestrated by Bcr-Abl in LSCs versus nonstem leukemia cells. In this model, LSCs would indeed require Bcr-Abl signaling for the clinical manifestation of CML, but in contrast to nonstem leukemia cells, the signaling that is crucial to the LSC would be independent of Bcr-Abl's catalytic activity. If indeed Bcr-Abl is active in LSCs in a kinase-independent fashion, then to achieve cure, we need to devise methods of targeting Bcr-Abl that do not rely on inhibition of its catalytic activity.
Can Resistance of LSCs to TKIs Be Explained by Increased Drug Efflux?
Some of the properties that make stem cells resistant to chemotherapy have been described for hematopoietic stem cells (HSCs) and in CML. HSCs express high levels of drug efflux molecules [66], and evidence suggests that LSCs in CML also express these molecules [67]. Furthermore, both HSCs and LSCs share the property of quiescence [68]. Nevertheless, it is improbable that the TKI resistance of LSCs is simply a function of their stem cell properties.
It is true that TKIs are substrates for drug efflux proteins such as ABCB1 and ABCG2 [69, 70], but it is not likely that drug efflux is the reason LSCs are refractory to TKIs. First, mouse models of CML that are created on the multidrug resistance (Mdr)1a/1b null backgrounds and hence lack the P-glycoprotein (P-gp) had similar responses to imatinib as wild-type animals [71]. Second, overexpression of P-gp failed to confer resistance to TKIs in the CML cell line K562 [71, 72], although other studies have reported the opposite result [73–75]. It has been argued that the difference between the studies that showed that overexpression of P-gp was sufficient to confer resistance and the ones that showed no effect were that the former were conducted on cell lines that were previously selected for antibiotic resistance clones [71]. Finally, several compelling studies demonstrate that there is no significant difference in the ability of TKIs to inhibit the kinase activity of Bcr-Abl in stem cells (CD34+CD38−, CD133+), which express drug efflux molecules, versus progenitor (CD34+CD38+) cells [35], which do not, and that the TKI dasatinib is not able to eradicate LSCs even though it abrogated the catalytic activity of Bcr-Abl [76].
The hypothesis that LSCs might be resistant to TKIs because of quiescence is also not likely because imatinib can effectively kill both dividing and nondividing cells [35, 77]. Nevertheless, it has been observed that quiescent CML stem cells are in fact more resistant to TKIs [6, 78–81]; however, whether this is by virtue of their cell cycle status or because quiescent stem cells inherently possess different Bcr-Abl-signaling mechanisms than nonstem cell cancers remains an open question. Finally, enhanced DNA repair mechanisms are not likely to be the reason that LSCs are resistant to TKIs as resistant clones in CML actually have increased genomic instability [82, 83].
Overview of Bcr-Abl Signaling
CML is a clonal myeloproliferative disorder with an incidence of 1 or 2 per 100,000 per year [84]. It is associated with expression of the specific chromosomal abnormality, the Philadelphia chromosome [85], characterized by the reciprocal translocation t(9;22)(q34;q11) [86]. This translocation gives rise to the Bcr-Abl chimeric protein, in which the first exon of Abl is replaced with the N-terminal exon(s) of the Bcr gene. Depending on where the breakpoint occurs, the chimeric Bcr-Abl gene may contain one, two, three, or even more exons of the Bcr gene, giving rise to fusion proteins of various sizes [87]. The p210BCR-ABL protein containing the first two or three N-terminal exons of Bcr is the most common variant of Bcr-Abl occurring in 90% of CML patients. This chimeric gene encodes the Bcr-Abl fusion protein that is characterized by a high level of tyrosine kinase activity derived from the kinase domain of Abl.
The Bcr-Abl protein aberrantly activates a number of signaling pathways that result in activation of proliferation, promotion of genetic instability, suppression of apoptosis, and weakening of cell adhesion [88–90]. Bcr-Abl when phosphorylated at Y177 induces activation of the Janus kinase (Jak)-signal transducer and activator of transcription (Stat) pathway in the absence of cytokine activation [91–95] by directly stimulating Jak2 [96, 97] and Stat [92, 98]. Bcr-Abl induces activation of the Ras pathway by binding to the adaptor proteins Grb2, Shc, Crk, and Cbl, which form a complex and recruit Sos [99–105], which then activates the Ras/Raf/Mek/Erk pathway ultimately activating c-Myc [106–108]. In addition, the Bcr-Abl-Grb2 complex recruits Gab2 [89], which in turn activates phosphatidylinositol 3-kinase (PI3K)/Akt [109]. The Src family kinase Hck is activated by Bcr-Abl in a kinase-independent manner [110] (Fig. 1A). Bcr-Abl kinase activity is not required either for binding or activation of Hck catalytic activity. The physical interaction between Bcr-Abl and Hck involves multiple domains of both proteins [111]. Bcr-Abl preferentially binds inactive conformations of Hck (either dephosphorylated Hck or the Y390F and the K269R mutant Hck proteins whose autophosphorylation function is inhibited and hence possesses the inactive conformation) [110]. Activation of Hck protein occurs subsequent to binding to Bcr-Abl, putatively by undergoing a conformational change that alters the orientation of amino acid residues necessary for catalytic activity. Activated Hck is able to phosphorylate Stat5 [112, 113], whose activation is critical for the transforming ability of Bcr-Abl [114]. Finally, Bcr-Abl promotes resistance to cell death by increasing the expression of Bcl-XL [115, 116] and Bcl-2 [117, 118].
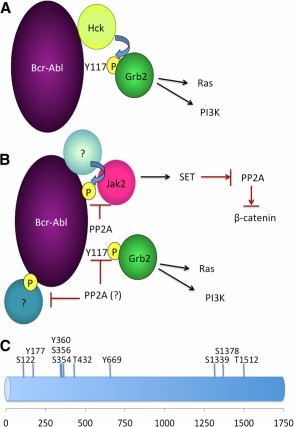
A model of how Bcr-Abl can activate downstream signaling targets by acting as a molecular scaffold. (A): Grb2 physically interacts with residue Y117 from Bcr-Abl, a site that is typically autophosphorylated. Nevertheless, other kinases (e.g., Hck) are able to phosphorylate Y177 on Bcr-Abl even in the absence of Bcr-Abl catalytic activity. (B): Persistence of quiescent chronic myeloid leukemia (CML) stem cells requires inhibition of PP2A. In these cells, Jak2 activation requires the expression but not kinase activity of Bcr-Abl. Although the mechanism of how Jak2 activation occurs is not completely understood—for example, we do not know which kinase phosphorylates Bcr-Abl, enabling its scaffolding activities—the observation that PP2A can prevent the recruitment and activation of Jak2 suggests that Jak2 uses Bcr-Abl as a molecular scaffold. Activated Jak2 then activates β-catenin through a mechanism that involves increasing levels of SET, the endogenous PP2A inhibitor, which decreases levels of PP2A that subsequently increases β-catenin activity. It will be interesting to determine which are the critical targets of PP2A in CML. Whether PP2A specifically dephosphorylates Bcr-Abl at the site where Jak2 interacts or whether PP2A acts to dephosphorylate other residues on Bcr-Abl that may potentially serve as sites for signaling molecules is not known at this time. (C): Putative phosphorylation sites of the Bcr-Abl protein that can serve as docking sites of recruitment of signaling proteins to Bcr-Abl. Phosphorylation sites were determined by PhosphoSitePlus. Abbreviations: Jak2, Janus kinase 2; PI3K, phosphatidylinositol 3-kinase; PP2A, protein phosphatase 2A.
Scaffolding Functions of Bcr-Abl
The activation of the Ras pathway and PI3K/Akt appears to be dependent on the scaffolding ability of Bcr-Abl, as the Y177F mutation that disrupts binding of Grb2 prevents activation of these pathways [89, 103]. The Bcr-Abl-Grb2 interaction involves binding of the SH2 domain of the Grb2 protein with a phosphorylated tyrosine 177 in the Bcr portion of Bcr-Abl. Tyrosine 177 is autophosphorylated by the Abl tyrosine kinase domain in Bcr-Abl; hence, it is dependent on the catalytic activity of Bcr-Abl. Mutation of this residue in Bcr-Abl (Y177F) abrogates binding of Grb2 and also inhibits activation of Ras in fibroblasts [103], suggesting that activation of Ras is dependent on both the scaffolding and catalytic ability of Bcr-Abl. Similarly, activation of PI3K is dependent on the ability of the p85 subunit of PI3K to bind to the SH2 domain of Bcr-Abl; abrogation of this interaction inhibits activation of Akt, which is essential for induction of growth-factor-independent growth, leukemogenesis in SCID mice, and upregulation of c-Myc and Bcl-2 [119].
It would be interesting to tease apart the scaffolding function of Bcr-Abl and to study this function in LSCs separately from the catalytic function of Bcr-Abl. Should the scaffolding function of Bcr-Abl be able to mediate activation of the Ras or PI3K pathway, one might envision a scenario in which kinases that are expressed exclusively in LSCs and not LPCs could enable scaffolding ability by effecting phosphorylation of Bcr-Abl Y177. In this scenario, the presence of TKIs that are currently used for the treatment of CML would not be effective because they would merely inhibit the catalytic ability of Bcr-Abl and hence would abrogate Bcr-Abl autophosphorylation. Nevertheless, other tyrosine kinases that are expressed in LSCs, which would not be targeted by present-day TKIs, would be able to phosphorylate Bcr-Abl, enabling its scaffolding activity. One such kinase is Hck, but there are likely others, perhaps even some that are LSC-specific. Bcr-Abl can bind to and activate Hck kinase, independent of the catalytic activity of the Abl domain [110]. Furthermore, activated Hck can phosphorylate Bcr-Abl at Y177 to induce binding of Grb-2 [110]. Although it has not been shown directly that kinase-dead Bcr-Abl can transform cells, one can appreciate why this could be cell-type specific: unless the target cell expresses the kinases that are expressed in the LSC, one would not expect to see activation of the scaffolding ability of Bcr-Abl when its autophosphorylation function is inhibited.
Hck and the related Src family kinase Lyn are highly activated in TKI-resistant patients but not in sensitive patients [120]. Perhaps Lyn and Hck are active in the stem cells of both patient populations, but in sensitive patients, Lyn and Hck activity is not detectable because the population of LSCs is so small. The acquisition of a secondary hit that blocks differentiation would effectively increase the quantity of LSCs (whether the LSCs are derived from a HSC-like LSC that acquires a block in differentiation or a LPC that acquires de novo stem-like abilities), hence allowing the detection of Lyn and Hck activity in resistant patients because of the increase of stem-like cells. In such a scenario, a kinase-independent but Bcr-Abl-dependent mechanism could be envisioned, although it is also possible that Lyn and Hck are activated independently of Bcr-Abl.
The importance of cell type-specific effects is highlighted by the work of Cortez et al. [121], who showed that creation of Bcr-Abl mutants that abrogate the scaffolding ability of Bcr-Abl (a point mutation in the Grb2 binding site, a point mutation in the SH2 domain, and a point mutation in the Y793F autophosphorylation site) had minimal impact on the effects of Bcr-Abl in the myeloid cell line 32D and in the murine bone marrow-derived pro-B-cell line Ba/F3 [121] even though these mutations have dramatic effects in fibroblasts [103]. Although it might be disconcerting that impairing the ability of Bcr-Abl to scaffold Grb2 had minimal effects on hematopoietic cells, it is important to note that the authors observed cell type-specific effects even between the two hematopoietic cell lines they used. It should also be noted that the cells used were not CML cells, nor were they LSCs. This study shows that in 32D and Ba/F3 cells, Ras can be activated independently of the ability of Grb2 to bind Bcr-Abl, but this does not preclude the possibility that Bcr-Abl can activate Ras via a scaffolding function in LSCs.
Protein Domains of the Bcr-Abl Protein May Mediate Nuclear Localization
Several domains in the Bcr-Abl protein are worth highlighting. Constitutive activation of the Abl kinase domain is mediated by dimerization via the coiled-coil domain of Bcr; this is required for transformation by Bcr-Abl [122, 123]. The last exon domain of Abl contains sites that mediate protein-protein interactions with such proteins as Crk, Grb, Nck, Atm, p53, and Rb. There are also three nuclear localization sequences as well as a DNA-binding domain. Whereas Abl is expressed in both the nucleus and cytoplasm, Bcr-Abl is thought to be expressed exclusively in the cytoplasm [124]. Hence, the precise function of these domains in Bcr-Abl is intriguing. Whereas deletions of regions within the last exon domain of Bcr-Abl results in increased tyrosine phosphorylation and transformation ability, deletion of the entire last exon prevents transformation. This leads to the question of whether Bcr-Abl, like Abl, is able to regulate gene transcription. For example, v-Abl response elements that are able to activate transcription have been identified in the E2F sites of the c-Myc gene promoter [125]. Furthermore, c-Abl is able to upregulate expression of the cyclin-dependent kinase inhibitor p21WAF-1 by recruitment to p53 binding sites in the p21 promoter [126]. The data suggesting that Bcr-Abl is cytoplasmic are not comprehensive: early studies were conducted in conditions that did not necessarily reflect localization in vivo, in patients: they were conducted using antibodies to either Abl or Bcr (not the Bcr-Abl fusion) in cell lines that overexpress the Bcr-Abl oncogene, in cell culture, under specific conditions [124, 127]. Whereas Bcr-Abl is cytoplasmic under basal conditions, it has been shown that Bcr-Abl is able to translocate to the nucleus under specific conditions. For example, following irradiation, Bcr-Abl translocates to the nucleus, where it increases genomic instability by causing a radioresistant DNA synthesis phenotype by interfering with ataxia-telangiectasia and rad 3-related protein (Atr)-dependent signaling [128]. Interestingly, another condition that induces the nuclear localization of Bcr-Abl is treatment with imatinib [129–131]. In fact, retention of Bcr-Abl outside the nucleus was shown to be dependent on a conformational change induced by autophosphorylation at three tyrosine moieties, as well as by the C-terminal region including its F-actin binding domain, which together act to inhibit nuclear import by regulating the function of the nuclear localization signal [129]. It is intriguing to speculate that imatinib might actually act to facilitate kinase-independent functions of Bcr-Abl by inducing the nuclear localization of this molecule. This idea does not contradict the observation that trapping Bcr-Abl in the nucleus induces apoptosis [130] as it is not clear whether the induction of apoptosis is due to Bcr-Abl acting as a tumor suppressor in a fashion similar to c-Abl or to depletion of cytoplasmic Bcr-Abl in cells that may be oncogene-addicted.
A Kinase-Independent Role for Bcr-Abl Signaling?
The cancer stem cell model proposes that cancer is composed of stem cells and nonstem cancer cells [132]. Oncogenesis, hence, is akin to a “Darwinian” process in which overexpression of oncogenes and silencing of tumor suppressor genes provide a malignant clone with a competitive advantage. Because natural selection is based on the ability to pass down traits to clonal progeny with proliferative ability, one may speculate that oncogenes enhance the fitness of stem cells given that the cancer stem cell is the only cell that is capable of unlimited proliferative ability.
Herein lies the conundrum: how is it possible that Bcr-Abl is necessary and sufficient for the initiation of CML, at least in animal models [133, 134], and yet inhibition of Bcr-Abl in stem cells is without consequence? From a Darwinian point of view, it would not make sense that this sine qua non oncogene would not enhance the clonal fitness of the CML stem cell. Hence, an alternative hypothesis is proposed. It is not that CML stem cells are independent of Bcr-Abl, but rather that CML stem cells are not dependent on the catalytic ability of Bcr-Abl, relying instead on its kinase-independent functions.
How Could a Kinase-Independent Role of Bcr-Abl Be Reconciled With Earlier Studies?
It is proposed that the role of the catalytic ability as well as the scaffolding ability of Bcr-Abl in defined highly purified hematopoietic cell populations be examined. Although early studies examining the phenotype of kinase-dead mutants of Bcr-Abl concluded that this mutant lacks the transforming ability of intact Bcr-Abl, these studies did not comprehensively examine the phenotype of kinase-dead mutants in primary stem versus progenitor cells. Rather, many studies looked at the ability of these mutants to confer growth ability to cell lines in SCID mice, and were therefore limited in the question that was asked and the phenotype that was examined. To elucidate this question, experiments must evaluate the effect of expression of kinase-inactive Bcr-Abl in primary HSCs, comprehensively, using hematological and biochemical assays that specifically examine HSC parameters such as self-renewal, dependence on niche, and propensity to differentiate. Furthermore, it is important to address the role of the scaffolding functions of Bcr-Abl in LSCs. Mutation of the Bcr-Abl kinase domain abrogates both its kinase activity and, via loss of autophosphorylation, some of its scaffolding activity [103]. It is easy to speculate that HSCs may express unique kinases that are able to use Bcr-Abl as substrate, thereby overcoming the reliance of Bcr-Abl on its kinase function for autophosphorylation. Therefore, it is important to determine whether the scaffolding function of Bcr-Abl is important for its LSC phenotype. Finally, earlier studies also did not accurately address a crucial feature of the disease: Bcr-Abl kinase activity is not necessary in the LSC compartment, but kinase activity is indeed necessary in the LPC compartment, based on the observation that LPCs undergo apoptosis upon treatment with TKIs, whereas LSCs do not. This point is important because any putative effect that a kinase-inactive mutant would have on the LSC would be obscured by the lack of effect in the LPC compartment; hence, kinase-inactive Bcr-Abl is not expected to result in full-blown disease.
There are, of course, other ways to interpret the clinical and molecular observations: it is possible that LPCs are addicted to Bcr-Abl, whereas LSCs are not. It is not entirely clear why inhibition of Bcr-Abl induces apoptosis in nonstem CML cells instead of merely reversing the phenotype and restoring normal, basal-level cellular functions. Indeed, a case that LSCs and LPCs exhibit differences in their addiction to the Bcr-Abl oncogene was made by Hamilton et al., who used a tetracycline-inducible mouse model of CML in which Bcr-Abl expression was shut off and then on again, demonstrating that survival of LSCs in this model is not dependent on expression of Bcr-Abl [135]. Although there are caveats to using mice to model human disease, and it is not likely that the short duration of oncogene expression and withdrawal would be sufficient to recapitulate the changes in nuclear organization and epigenetics that are likely present in patients and that contribute to oncogene addiction in vivo, nonetheless this study leads to a key question: why should it be that LPCs should acquire a dependence on expression of Bcr-Abl when LSCs do not? This study does not prove that Bcr-Abl is dispensable in LSCs for the maintenance of leukemia; it merely substantiates the point that Bcr-Abl signaling in LSCs and LPCs is distinct, to the point that LPCs are addicted to the oncogene, whereas LSCs are not. Further study is warranted of whether LSCs in patients respond to TKIs by modulating parameters other than survival. Therefore, in investigating the role of Bcr-Abl mutants deficient in catalytic activity, it would be interesting to assess the effects of not just a kinase-inactive mutation but of a conditional mutant that would lack kinase activity in LSCs but restore regular, kinase-dependent signaling in LPCs.
Earlier studies did not resolve the signaling of kinase-inactive Bcr-Abl mutants in specific hematopoietic compartments. Given the scarcity of the LSCs within the population of hematopoietic cells, previous studies lacked the potential to resolve the signaling in the LSCs as this cell population would have been contaminated with nonstem leukemia cells that are far more plentiful. Nevertheless, in the last decade, several advances have been made that invite a re-examination of earlier data. Specifically, the ability to prospectively isolate populations of LSCs, as well as the ability to apply biochemical techniques to samples containing as little as a single cell, will enable discovery of the mechanisms of the resistance of LSCs to TKIs.
At the time of submission of this article, no direct evidence existed suggesting that CML stem cells rely on the speculated scaffolding functions of Bcr-Abl. However, in a recent publication, Neviani et al. [136] showed for the first time that survival of quiescent CML stem cells requires Bcr-Abl expression but not Bcr-Abl kinase activity. In this seminal study, the authors showed that expression but not activity of the Bcr-Abl protein is necessary for the recruitment and subsequent activation of Jak2 and its downstream target β-catenin; that activation depends on reduced activity of the tumor-suppressor protein phosphatase 2A (PP2A). Reduction of PP2A is necessary for the maintenance of the LSCs of CML such that use of a PP2A-activating drug (FYT720) reduced survival of CML LSCs but not healthy control HSCs, both in vitro and in vivo. These findings are particularly interesting in light of the previous observation that PP2A acts to dephosphorylate Bcr-Abl [137, 138]. Given that activation of the Jak2/β-catenin pathway is independent of the kinase activity of Bcr-Abl and that Bcr-Abl acts as a molecular scaffold, it would be interesting to determine precisely how Bcr-Abl activates Jak2. Nevertheless, these data are consistent with a model in which phosphorylation of Bcr-Abl is necessary for activation of the scaffolding function of Bcr-Abl, which is then able to activate downstream pathways independent of its kinase activity (Fig. 1B).
A number of questions remain: it will be interesting to determine whether the tyrosine phosphatase activity of PP2A is the mechanism by which this protein is detrimental to the CML stem cell. If so, is Bcr-Abl indeed the critical substrate of PP2A or does PP2A act as a phosphatase to proteins that physically interact with Bcr-Abl, such as Jak2 (Fig. 1B)? Furthermore, it is important to identify the critical residues that are dephosphorylated by PP2A and to assess whether PP2A dephosphorylates residues directly involved in the tethering of proteins to the Bcr-Abl scaffold. Seven serine/threonine and three tyrosine phosphorylation sites, including the Y177 residue that is involved in binding to Grb2, can be identified on Bcr-Abl using PhosphoSitePlus (http://www.phosphosite.org) [139]. It will be interesting to see whether any of these sites are involved in the recruitment of signaling proteins to Bcr-Abl, and whether these sites are substrates of PP2A (Fig. 1C). In addition, it would be interesting to assess whether the kinases that phosphorylate the key target molecules are LSC-specific. Finally, it will also be interesting to determine whether there are other pathways in addition to the Jak2/β-catenin axis that may be activated by the scaffolding function of Bcr-Abl.
Proposed Research Questions
Investigation of the Transcriptional Modulatory Ability of Bcr-Abl
Many kinases including the epidermal growth factor receptor are able to translocate to the nucleus to modulate transcription in either a DNA-binding-dependent [140] or -independent manner [141–143]. Jak2 is another such kinase that can shuttle between the cytoplasm and the nucleus. Jak2 is a nonreceptor tyrosine kinase that canonically signals through the Stat proteins. Nevertheless, Jak2 is able to translocate to the nucleus to interact directly with nucleoproteins and induce epigenetic modifications independent of interaction with Stat family members [144–147]. Nuclear Jak2 is able to directly phosphorylate histone H3Y41 [144, 148], NF1C2 [145, 149], and Rush-1α [154], regulating important cellular functions such as cell cycle progression, differentiation, and apoptosis. Similarly, it is possible that Bcr-Abl may also have nuclear functions.
Bcr-Abl is able to translocate to the nucleus at least in conditions of genotoxic stress where it interacts with Atm and Atr and disrupts Atr signaling [128]. It has already been shown that Bcr-Abl is able to modulate transcription in a kinase-independent manner. Chen et al. identified 5-lipoxygenase (5-LO, Alox5) as a transcriptional target of Bcr-Abl in a screen of genes that were altered in LSCs by Bcr-Abl but not rescued by treatment with imatinib [150]. The study then showed both genetically and with small molecule inhibitors that 5-lipoxygenase was necessary for the transforming ability of Bcr-Abl in mice. Although this study is extremely exciting and is consistent with a kinase-independent role for Bcr-Abl, it does not address the question of whether the effect of Bcr-Abl on transcription is direct or indirect.
It would be interesting to investigate comprehensively whether Bcr-Abl has the ability to modulate transcription directly, as well as to establish the conditions needed to induce nuclear localization of Bcr-Abl in LSCs. The localization of Bcr-Abl is thought to be cytoplasmic [124], but whether this holds true in LSCs is unknown. It is worth examining the localization of Bcr-Abl in highly purified LSCs under basal conditions, as well as conditions of stress such as serum starvation, growth factor deprivation, and genotoxic stress.
Once conditions that localize Bcr-Abl to the nucleus are determined, it would be interesting to investigate the effects of nuclear Bcr-Abl on gene transcription to determine the downstream targets and to validate that Bcr-Abl actually regulates transcription.
Investigation of Bcr-Abl Signaling in Highly Purified Populations of LSCs and LPCs
The observation that LSCs are refractory to TKI therapy highlights that LSCs differ from nonstem leukemia cells in their fundamental signaling properties. Earlier studies examined the role of Bcr-Abl in hematopoietic cells, but they did not comprehensively contrast Bcr-Abl signaling in highly purified LSC and LPC populations. Corbin et al. commenced characterization of the differences in signaling in LSCs and nonstem leukemia cells; however, this work was far from comprehensive as they examined differences in the phosphorylation of only three proteins: Crk, Akt, and Stat5 [35]. It would be fascinating to contrast comprehensively Bcr-Abl signaling in LSC and LPC compartments to identify core differences in Bcr-Abl signaling. Jak/Stat, Ras/Raf/Mek/Erk, and PI3K/Akt would be of particular interest because there is reason to believe that these pathways might be activated in a kinase-independent manner by Bcr-Abl.
Investigation of the Transforming Ability and Cellular and Molecular Functions of Kinase-Dead Bcr-Abl
In addition to its cellular and molecular biological properties, the phenotype of kinase-dead and scaffolding-dead Bcr-Abl molecules needs to be thoroughly investigated with different mutations and different strains of animal models. Tissue-specific promoters would be useful to create animal models that express kinase-dead Bcr-Abl only in the stem cell compartment so that kinase-independent effects of Bcr-Abl on stem cell parameters might be investigated.
I hypothesize that a kinase-inactive mutant of Bcr-Abl would still be able to activate pathways important in cell growth by acting as a scaffold or by regulating transcription, at least in LSCs. Bcr-Abl acts as a docking site for the protein Grb-2 that is pivotal to the activation of the Ras pathway and PI3K pathway. The importance of the scaffolding function of Bcr-Abl in the activation of the Ras and PI3K pathways has not been investigated in LSCs, nor has the phenotype of a kinase-dead Bcr-Abl molecule ever been investigated in LSCs. Furthermore, although Bcr-Abl also activates the Src family member Hck through its ability to act as a molecular scaffold, the ability of Bcr-Abl to activate Hck and Stat5 through its scaffolding ability has never been investigated in LSCs, nor has the ability of a kinase-dead mutant of Bcr-Abl to activate Hck and Stat5 ever been shown in LSCs. Therefore, I propose that the signaling ability of kinase-dead mutants of Bcr-Abl be investigated in both LSCs and LPCs. As well, it is worth investigating the question of whether LSCs express kinases, not expressed in LPCs, which can phosphorylate Bcr-Abl and thus enable its scaffolding ability. LSC-specific kinases would be refractory to drugs such as imatinib that target Bcr-Abl, and since imatinib would not target the scaffolding function of Bcr-Abl, this could be one mechanism of resistance of LSCs to imatinib.
Other Kinase-Independent Functions
The spectrum of cellular and molecular properties of Bcr-Abl that are kinase-independent needs to be fully investigated. Many of the early studies that investigated the phenotype of kinase-inactivating mutations of Bcr-Abl used readouts that did not probe the function of the stem cell compartment per se, for example, acquisition of growth factor independence, apoptosis, and transformation (defined as the ability of hematopoietic primary cells or cell lines to induce leukemia in SCID mice [106, 151]). This early work was pivotal in illuminating the importance of the catalytic function of Bcr-Abl that enabled the development of drugs that have changed the clinical course of CML, but results from these investigations do not preclude the existence of more subtle, stem cell-specific, kinase-independent effects of Bcr-Abl. We know that imatinib and other TKIs show striking clinical efficacy; hence, it is not surprising that the majority of leukemia cells would be dependent on the catalytic ability of Bcr-Abl. However, using assays that did not specifically measure hematopoietic stem cell-specific parameters may have resulted in a missed chance to unravel the more subtle, stem cell-specific effects of Bcr-Abl.
For example, it is possible to envision scenarios in which altering the adhesion and migration properties of the cell can impinge on its propensity to be regulated by the niche. Expression of the Bcr-Abl oncogene alters the adhesive and migratory properties of hematopoietic progenitor cells, in some contexts reducing progenitor cell adhesion to stroma and fibronectin, as well as reducing chemotaxis to stroma-derived factor-1α [152–154], while in other contexts increasing adhesion to fibronectin [155, 156]. Kinase-inactive mutants of Bcr-Abl have been shown partially to retain this reduction in adhesion and chemotaxis [154], and Wertheim et al. showed that the increased adhesion to fibronectin observed in 32D cells expressing Bcr-Abl could not be rescued by imatinib or by the catalytically inactivating K1176R mutation [155].
Although the relevance of alterations in the adhesion properties of hematopoietic cells in the maintenance and clinical progression of leukemia may not be directly apparent, these features may be important to the clinical presentation of the disease contributing extramedullary hematopoiesis, the trafficking of immature blast cells from the bone marrow into the bloodstream, and stem cell-stroma interactions. In any case, this study is important because it highlights the identification of biologically relevant kinase-independent functions of Bcr-Abl.
In addition, genomic instability is a crucial element of leukemia progression that may be attributed to kinase-independent functions of Bcr-Abl. Despite the ability of imatinib to inhibit the kinase activity of Bcr-Abl, LSCs expressing Bcr-Abl that are treated with imatinib continue to accumulate oxidative damage and have elevated levels of reactive oxygen species compared with healthy controls [157]. This suggests that genomic instability might be a kinase-independent mechanism of Bcr-Abl. Although there is no way to predict the range of functions that could be kinase-independent in Bcr-Abl-expressing CML, it is important to investigate which functions are kinase-dependent, which functions depend on the scaffolding ability of Bcr-Abl, which depend on both, and which depend on neither. The conundrum remains: if Bcr-Abl is expressed in the LSCs of chronic phase CML and if the LSC is refractory to TKIs, what role if any does Bcr-Abl play in the LSCs?
Other Possibilities
In this Perspective, the idea has been brought forth that Bcr-Abl may signal differently in LSCs than in LPCs, and that LSC persistence may rely on Bcr-Abl expression instead of Bcr-Abl kinase activity. There are, of course, other plausible scenarios. TKIs do not inhibit 100% of kinase activity. One possibility is that LSCs simply require less Bcr-Abl kinase activity than LPCs. This scenario would not conflict with a scaffolding role for Bcr-Abl. For example, a lower threshold of kinase activity than required for activation of its other substrates could be able to activate the scaffolding function of Bcr-Abl that could then be sustained independent of Bcr-Abl's catalytic activity.
Another possibility is that inhibition of Bcr-Abl using imatinib mesylate may activate backup pathways. In LPCs, it could be that inhibition of Bcr-Abl induces activation of the stem cell factor/c-kit backup pathway. NaPP1, a specific monoinhibitor of Bcr-Abl, was unable to eliminate Bcr-Abl+, c-kit+ progenitor cells, in which imatinib mesylate, which is able to inhibit multiple signaling pathways in addition to Bcr-Abl, such as c-kit and PDGF, was able to eliminate said cells [158]. It is conceivable that in LSCs backup pathways specific to the LSC also exist that can substitute for Bcr-Abl signaling. One such pathway could be the β-catenin/Wnt pathway.
Conclusion
CML is a fatal bone marrow disease that invariably progresses from an indolent disorder to a drug-resistant phase that resembles acute leukemia. TKIs have dramatically improved the clinical outcome of CML but are not curative as they do not affect the LSCs. Hence, patients currently require lifetime TKI therapy. This is expensive and presents patients with side effects as well as medical complications.
In order to cure the disease, it is necessary to understand the fundamental biology underpinning the resistance of LSCs to TKIs. I have proposed a novel mechanism to explain why LSCs are refractory to TKIs: that Bcr-Abl signaling is fundamentally different in LSCs compared with nonstem leukemia cells, and that LSCs rely on kinase-independent Bcr-Abl signaling for survival. It is important to determine what role, if any, these kinase-independent functions have in the Bcr-Abl signaling of LSCs. Testing of this hypothesis will provide important insight in the Bcr-Abl signaling pathways in LSCs, and will elucidate how these pathways differ in LSCs versus LPCs. At the same time, the proposed work will evaluate the potential of Bcr-Abl to alter gene transcription directly and—of key importance if we are to cure this disease—further our understanding of the basic biology of LSCs in CML.
Acknowledgments
I thank Dr. Tony Hunter (Salk Institute for Biological Sciences), Dr. Walter Eckhart (Salk Institute for Biological Sciences), Dr. Richard A. Wells (Odette Cancer Research Program, Sunnybrook Research Institute, University of Toronto), and Dr. Jacinth Abraham (Canadian Intellectual Property Office) for critical review of the manuscript and the anonymous reviewers of drafts of the manuscript for their excellent comments and suggestions. This article is dedicated to the memory of Florica Batu Ichim, who was diagnosed with CML in 1987, in an era when survival was less than 5 years and busulfan was the standard of care, and to the doctors and basic scientists who worked long nights to develop the science behind TKIs, drugs that extended her overall survival to 23 years.
Author Contributions
C.V.I.: conception and design, analysis and interpretation of the literature, manuscript writing, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
The author indicates no potential conflicts of interest.

{kind=link}