





Abstract
Polyarteritis Nodosa (PAN), is the firstly described vasculitis and can be seen in paediatric and adult age. PAN has a heterozygous clinical picture including cutaneous, constitutional, musculoskeletal, gastrointestinal, and renal involvement. Description and splitting of other vasculitis, makes this medium vessel vasculitis, a very rare disease. Additionally, many subgroups of PAN have been defined and this effort let to move Hepatitis B virus-PAN to Vasculitis with probable aetiology. Anyhow, idiopathic PAN still exists and cohorts from various countries such as France, India, and Japan have been published. Rarity of PAN necessities global collaboration to highlight clinical features and genetics studies. GLOBAL-PAN is an ongoing collaborative project of EUVAS, VCRC and many national cohorts. This review covers the recent epidemiological data of PAN along with demographic and clinical characteristics of cohorts from all-over the world and GLOBAL-PAN.
Idiopathic PAN still exists and recent cohorts from various countries such as France, India and Japan have been published.
Rarity of PAN necessities global collaboration to highlight clinical features and genetics studies.
GLOBAL-PAN is an ongoing collaborative project of EUVAS, VCRC and many national cohorts.
Since the first description of systemic necrotizing vasculitis in 1866, numerous efforts have been made to establish the nosology of classification criteria for vasculitis [1]. The term periarteritis nodosa was revised into polyarteritis nodosa (PAN). Over the years, with further description and differentiation of other vasculitis, this medium-vessel vasculitis has become a very rare disease with an estimated prevalence of 30/million in Europe [2].
In 1990, ACR suggested a Classification Criteria for PAN [3] including 10 items: weight loss ≥4 kg, livedo reticularis, testicular pain or tenderness, myalgias, mononeuropathy or polyneuropathy, diastolic blood pressure >90 mm, elevated blood urea nitrogen or serum creatinine levels, presence of hepatitis B reactants in the serum, arteriographic abnormality and presence of granulocyte or mixed leucocyte infiltrate in an arterial wall on biopsy. The presence of three or more criteria has a sensitivity of 82.2% and a specificity of 86.6%, representing the lowest sensitivity and specificity among all 1990 ACR Classification Criteria for systemic vasculitides.
The Chapel Hill Consensus Conference (CHCC) 2012 has revised the definition of PAN, emphasizing that this definition was not intended to serve as either classification or diagnostic criteria. The jury explained the terminology of these criteria. PAN was defined as medium artery necrotizing arteritis seen on biopsy, negative ANCA, absence of mucocutaneous lymph node syndrome and no evidence of glomerulonephritis [4]. In addition to the description of classification criteria, a medium artery aneurysm seen on imaging was added for diagnostic criteria. Another important update in this consensus paper was the reclassification of HBV-PAN, categorized under ‘Vasculitis With Probable aetiology’ as Hepatitis B Virus-Associated Vasculitis [5]. In 2014, a monogenic disease was introduced as Deficiency of Adenosine Deaminase-2 (DADA2) [6]. Patients with DADA2 can also have polyarteritis proven by objective findings such as microaneurysms observed in radiologic evaluations and medium-vessel vasculitis in biopsy. Furthermore, another monogenic disease, VEXAS syndrome, was introduced in 2020, and three (12%) of the patients in this paper had PAN [5]. Some authors suggested the term as ‘secondary PAN’ for the cases presenting with objective (histopathologic/radiologic) findings of PAN, which should be discussed in detail.
Diagnostic and Classification Criteria for Vasculitis (DCVAS) is the largest vasculitis study to date, aiming to develop criteria for granulomatosis with polyangiitis, microscopic with polyangiitis, eosinophilic granulomatosis with polyangiitis, PAN, Takayasu’s arteritis and GCA. Between 2011 and 2017, ∼7000 patients enrolled in this study, with the total number of PAN cases being fewer than 200. The study group published new classification criteria for eosinophilic granulomatosis with polyangiitis, granulomatosis with polyangiitis, microscopic polyangiitis, GCA, and Takayasu’s arteritis. However, classification criteria for PAN based on DCVAS data is still pending. We hope it will be established in the near future.
PAN cohorts from different parts of the world have been published in recent years. Different diagnostic criteria and algorithms for PAN have been used in these papers. Kanecki et al. showed a decline in the frequency of patients diagnosed as PAN during the years between 2008 and 2013 based on Polish hospital morbidity records and ICD-10 codes. In total, 557 unique individuals were found and they reported an overall incidence of 2.4 million/year [7].
Naidu et al. [8] presented data on Indian patients with systemic PAN (n = 37) and DADA2 (n = 14) seen in a tertiary care centre between 2005 and 2020. The classification of patients as PAN was based on the European Medicines Agency algorithm. The median age was 33, and male predominance was observed in both groups. Seven of 37 systemic PAN patients had Hepatitis B positivity. Approximately 60% of patients had positive angiography or histopathology in both groups.
In a study based on survey analysis of country-wide hospitals from Japan, Ministry of Health, Labor and Welfare of Japan (MHLW) diagnostic criteria was used (fever for 1 week or weight loss ≥4 kg, mononeuropathy multiplex, gastrointestinal involvement, absence of MPO-ANCA, urine protein <2+, angiographic abnormality, histologic evidence on biopsy) [9]. A total of 868 patients with PAN have been reported, which corresponds to an estimated incidence of 17.6/million in Japan.
Last year, the French Vasculitis Study Group presented a paper on PAN, including patients seen since 2005 [10]. They used a diagnostic criteria paradigm that requires the presence of one of three positive predictive parameters (hepatitis B virus antigen and/or DNA in serum, arteriographic anomalies, mononeuropathy or polyneuropathy) and the exclusion of five negative parameters (asthma, ear–nose–throat sign, immunofluorescence assay (IFA) ANCA, glomerulopathy, cryoglobulinemia). The number of newly diagnosed patients was 14/year. Of the presented 196 patients, 55 were reported as secondary PAN. The distribution of conditions associated with secondary PAN included myelodysplastic syndrome (n = 17 including 7 VEXAS), solid cancer (n = 13), lymphoproliferative disorders (n = 9), autoinflammatory disorders (n = 8), DADA2 (n = 3), hypereosinophilic syndrome (n = 5) and viral infections (no HBV, 1 HCV and 2 HIV).
Overall, the idiopathic PAN has shown a decreasing trend attributed to several factors. First, the evolution of classification criteria for vasculitides has led to the reclassification of diseases that were once considered PAN. For example, HBV-associated PAN was moved into the category of vasculitis with a probable aetiology, while monogenic diseases like DADA2 and VEXAS syndrome have been identified as distinct entities, though they present with similar findings. The differences in diagnostic approaches, combined with the identification of mimicking conditions, likely explain the observed decline in idiopathic PAN diagnoses over time.
GLOBAL-PAN is a worldwide initiative to clarify the characteristics and outcome of PAN and has been working since 2017. As a collaboration of EUVAS, VCRC and national networks in many countries, we are merging cohorts together to generate large cohorts of patients and facilitate global collaborations. GLOBAL-PAN projects include three sub studies, and the first study aims to identify clinical characteristics and outcomes of idiopathic PAN and was recently published [11]. In total, 460 case report forms from observational cohorts of nine countries across Europe, Japan and North America, including patients seen from 1990 to 2020, have been collected for GLOBAL-PAN I. EMA algorithm was applied, and 63 patients were excluded (4 patients due to missing data and 59 patients for not fulfilling EMA). To identify idiopathic PAN, we also excluded 12 patients with HBV, 11 patients with FMF and 16 patients with DADA2. In the final analysis, 76 patients with cutaneous PAN and 282 patients with systemic PAN were reported.
PAN typically occurs in individuals aged 40–60 years, but it can also be seen in paediatric age. In our GLOBAL-PAN combined cohort, even after excluding patients with monogenic diseases, paediatric PAN patients still frequently exhibit traits similar to PAN-mimickers, including a higher incidence of fever, abdominal pain, and central nervous system involvement, compared with adult-onset systemic PAN patients. This suggests that there may be additional genetic factors yet to be identified in children with PAN [11].
Relapse is a major concern and differs among subgroups of PAN. HBV-related PAN rarely relapses, whereas non-HBV PAN relapsed more. In GLOBAL-PAN, during a median (interquartile range) disease duration of 59.6 (99.5) months, relapse occurred in 48.5% of patients, with 78.0% of these relapses occurring within the first 2 years. The survival rates of the patients with PAN were 97.1% at 1 year, 94.0% at 5 years, and comparable to other vasculitides.
Glucocorticoids and immunosuppressive drugs are the mainstay of PAN treatment. However, there are extremely significant differences between the treatments of DADA2 (e.g. TNF inhibitors), HBV-related PAN (e.g. antiviral agents) and VEXAS (e.g. azacytidine). On the other hand, physicians must carefully balance the risks of undertreatment and overtreatment, especially in these types of chronic diseases.
GLOBAL-PAN II is still ongoing. The plan is to enrol additional patients, including viral-related PAN, DADA 2, etc. from all over the world. This study will focus on latent class analysis and target organ involvement. Better identification of these subgroups will be useful not only to reveal idiopathic and secondary PAN but also to guide rational treatment of these patients. As observed in the experiences with DADA2 and VEXAS, there may be additional genetic factors contributing to the aetiopathogenesis of PAN (Fig. 1). For GLOBAL-PAN III, DNA samples are being collected in each country to make a worldwide genetics study. One of the main aims of GLOBAL-PAN III is to manage this project in a similar manner.
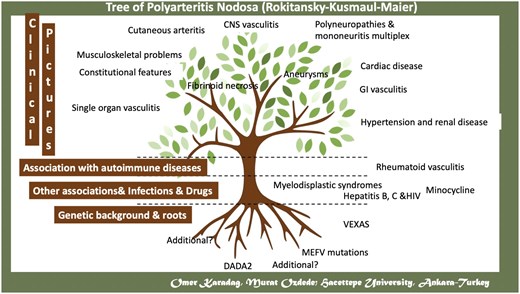
A schematic representation of aetiologic factors and clinical features of polyarteritis nodosa
As a conclusion, PAN is very rare but still can be seen in both adults and paediatric age worldwide. We are now witnessing the disappearance of HBV-PAN and the emergence of secondary PAN (see Fig. 1). Additionally, systemic PAN can be organ/life threatening; thus, we recommend conducting high-quality imaging and biopsy for patients suspected of having PAN. Worldwide collaboration, as exemplified by GLOBAL-PAN, helps to better discern this rare disease. Future omics studies may enlighten the pathogenesis, enabling a clearer specification of the disease spectrum and enhancing diagnostic accuracy.
Data availability
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Funding
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this article.
Disclosure statement: The author has declared no conflicts of interest.
Acknowledgements
My mentors David Jayne, Peter Merkel and Seza Ozen deserve special thanks. I also want to thank my fellows Ertugrul Cagri Bolek and Gizem Ayan, and all GLOBAL-PAN collaborators, patients and the study support team.

{kind=link}
Comments