





Systemic sclerosis (SSc) is an autoimmune connective tissue disease that involves multi-organ dysfunction [1]. To date, gastrointestinal (GI) complications of SSc remain highly challenging to treat and significantly contribute to the lived patient morbidity and mortality [2, 3]. Treatment of major end-organ complications often involves the application of various immunosuppressive/immunomodulatory regimens that target affected organs. However, current data suggest that not all organ systems respond equally well to each immunosuppressive/immunomodulatory and other (e.g. antifibrotic) therapies. For example, nintedanib was recently found to positively impact scleroderma-related interstitial lung disease, as measured by pulmonary function testing, but was not demonstrated to significantly impact cutaneous fibrosis [4]. The differential response to therapy across organ systems suggests that there the tissue’s response to the SSc disease process (including autoimmunity), and the overall response to immunosuppression/modulation may vary across organ systems. Furthermore, the timing of such therapies is also likely important, highlighting the need for biomarkers that distinguish between GI disease activity and GI dysfunction from prior organ damage.
The heterogeneity of the gastrointestinal tract manifestations in SSc challenges the concept of a single unified mechanism and suggests that immune-mediated mechanisms are likely more relevant in some aspects or stages of the disease than in others. However, our current inability to measure disease activity and objectively assess GI response to immunosuppression/modulation is a major challenge. Such measurements are limited by our understanding of underlying GI disease mechanism(s), a notable absence of biomarkers of disease activity, and the costly and often invasive nature of objective GI testing. Without markers of disease activity and response to treatment, it is difficult to design studies that objectively measure the effects of therapy and hard for physicians to justify treatment of the GI tract with immunosuppression. In such cases (i) there is no optimal time of drug initiation or clear end point for therapy; (ii) immunosuppressive agents are often costly; and (iii) the potential benefits do not clearly outweigh known risks.
The complexity of contributors to SSc-GI symptoms further complicates clinical decision-making, as the symptoms of patients with SSc GI disease likely stem from a variety of interrelated factors (Fig. 1). At the tissue level, SSc GI disease mechanisms are poorly defined but associated with smooth muscle atrophy, loss of interstitial cells of Cajal, abnormal neural reflexes, and varying degrees of fibrosis (none/minimal to more significant), all of which are thought to contribute to GI dysfunction [2, 5]. Slow transit throughout the GI tract and sphincter dysfunction are thought to reflect SSc-related tissue damage. However, the aetiology of these pathological findings is likely diverse and driven by a combination of autoimmunity [e.g. anti-muscarinic 3-Receptor (M3R) antibodies], dysbiosis, dysautonomia, and other factors among patient subsets. Furthermore, environmental factors such as diet and the side-effects of medications are also known contributors to patient symptoms. This significantly complicates treatment strategies and does not result in a clear case for immunosuppression across all patients. For example, it is unclear whether the slow transit leads to dysbiosis or vice versa, and whether immunosuppression impacts (including worsening of) both scenarios, such as increasing risk of small intestinal bacterial overgrowth (SIBO). In fact, prior data suggest that gut immunity may induce tolerance and benefit patients with systemic sclerosis, leaving many unanswered questions about the overall impact of immunosuppression/modulation on disease activity and long-term outcomes.
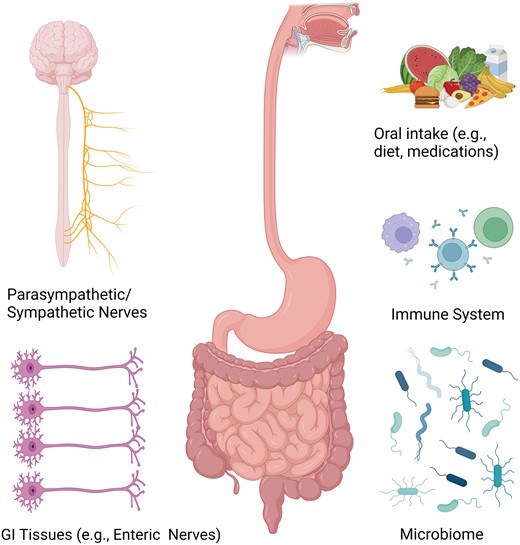
Contributors to GI symptoms and dysfunction in patients with systemic sclerosis. Gastrointestinal symptoms and dysfunction in systemic sclerosis are likely mediated by several factors in the body including the autonomic nervous system, gastrointestinal tissues (e.g. enteric nerves, smooth muscles), the immune system and the GI microbiome. Furthermore, external factors such as intake of food, drink and medications also likely play a role. Identifying the patients in which the autoimmune response is the primary driver of symptoms and damage is key to identifying the patients who are optimal candidates for immunosuppression
However, preclinical data and smaller studies in the literature do suggest that immunomodulation with IVIG may benefit a subset of SSc patients with anti-M3R antibodies and/or pseudo-obstruction. Anti-M3R antibodies target the muscarinic 3 receptors which are predominantly expressed on smooth muscle and play an important role in GI motility. These antibodies are demonstrated to negatively impact receptor function and impede smooth muscle contraction, which is reversible with the application of human immunoglobulin in vitro. SSc patients with anti-M3R antibodies are reported to have rapidly progressive severe lower GI disease and pseudo-obstruction and reports in the literature note a clinical response to IVIG, suggesting that immunomodulation may benefit some patients. Identifying other autoantibodies that may impact SSc GI function and determining which SSc subgroups benefit from immunosuppression/modulation for treatment of their GI disease are also important priorities for future research.
The autonomic nervous system is also a key regulator of normal GI motility, and it is now well recognized that patients with SSc who have more prominent upper GI dysmotility are more likely to have a higher burden of autonomic symptoms across multiple organ systems [6, 7]. However, it remains unclear as to why autonomic dysfunction occurs in SSc and whether the immune system plays a significant role in its dysfunction. Therefore, the timing and application of immune modulating/suppressive therapies (IVIG, anti-CD19, MMF, etc) vs non-immunomodulating therapies (e.g. direct autonomic stimulation, transcutaneous electrical stimulation/TEA, dietary modification, etc) among different patient subgroups is another important focus of future work.
Limited (and conflicting) data in the literature exist pertaining to the use of immunosuppression/modulation for GI involvement in SSc patients, and the optimal timing of intervention is yet to be defined. Stamm and colleagues [8] describe their single centre experience (currently published in abstract form) which included 219 patients (19.1% dcSSc) with established disease [median SSc disease duration of 6.0 (IQR 2.7, 12.5) years]. Patients completed the UCLA GIT 2.0 questionnaire on two occasions with an interval of 12 ± 3 months between study visits. Around one-third (n = 71) were exposed to immunosuppressive treatment during the observation period: MTX (n = 27), MMF (n = 17), TCZ (n = 18), RTX (n = 16), glucocorticoids >10 mg/day (n = 6), azathioprine (n = 3), leflunomide (n = 3) and CYC (n = 1). Multivariable linear regression revealed that immunosuppressive therapy during (but not before) the observation period was predictive (along with lower BMI, long disease duration and lower baseline UCLA GIT 2.0 score) were associated with better (lower) UCLA GIT 2.0 scores at follow-up.
In contrast, Richard et al. [9], report findings from a combined early dcSSc (<3 years disease duration) longitudinal cohort (n = 762, 42.8%) obtained from the Canadian Scleroderma Research Group (CSRG) and Australian Scleroderma Interest Group (ASIG). During a mean follow-up of 4.0 ± 2.6 years, severe GI involvement was defined as malabsorption, hyperalimentation, pseudo-obstruction, and/or 4–≥10% weight loss in association with the use of antibiotics for bacterial overgrowth or oesophageal stricture [9]. Then previous (ever) immunosuppressive treatment exposures studied were limited to AZA, MMF, MTX and/or CYC. Severe GI involvement occurred in similar proportions of those patients exposed (n = 319) compared with those not (n = 443) exposed to immunosuppression (11.6% vs 6.8%, respectively). In their IPTW-adjusted analysis, exposure to immunosuppression was not associated with severe GI disease (weighted hazard ratio of 0.91, 95%CI 0.52–1.58).
In both of the aforementioned studies, the safety of immunosuppressive therapies concerning GI involvement in patients with SSc is not apparent. However, it could be assumed that no major unexpected findings were observed.
There could also be important differences between specific approaches to treatment (e.g. csDMARDs vs bDMARDs). For example, positive effects on overall GI symptoms (UCLA GIT 2.0) and GERD (frequency and severity) were reported in 15 SSc patients with IVIG treatment (over a mean treatment duration of 2.3 years, frequency 6 weekly to 4 months). Furthermore, there are reports in the existing literature of benefit from stem cell transplantation in some cases, especially in childhood-onset SSc, which may suggest that the stage and/or type of SSc is highly relevant. Additionally, Vigone and Beretta [10] report their successful experience (after initial response to intramuscular methyl prednisolone) using abatacept to treat chronic intestinal pseudo-obstruction in five SSc patients, postulating an ‘inflammatory neuropathy resembling myenteric ganglionitis’ [10].
In conclusion, despite a potentially strong therapeutic rationale, there is currently limited evidence to support the use of immunosuppressive treatment for GI involvement in SSc patients. Further research is needed to understand the immunoinflammatory effector mechanisms to rationalize therapeutic targets, including targeting the use of exisiting drug therapies. Specifically, the ability of the gut immunity to induce tolerance and benefit SSc is another aspect that could be explored; for example, drawing on the rationale and findings of the oral type I collagen trials in SSc, which were completed some years ago. Furthermore, the impact of immunosuppression on the frequency and consequences of small intestinal bacterial overgrowth needs to be defined. Individual patient selection will be essential (balancing the risks and benefits) for immunosuppressive treatment, and well-designed RCTs are needed to assess the safety and efficacy of this novel therapeutic approach.
Data availability
No new data were generated or analysed in support of this research.
Funding
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this article.
Disclosure statement: Z.H.M. is supported by NIH/NIAMS R01 AR081382-01A1. M.H. reports speaking fees from Actelion pharmaceuticals, Eli Lilly, Pfizer, and Janssen; Research funding from Janssen; Member of a Data and Safety Monitoring Board for Certa Therapeutics, all outside of the submitted work.

{kind=link}
Comments