





Abstract
Evidence shows that dysfunctional SSc keratinocytes contribute to fibrosis by altering dermal homeostasis. Whether IL-25, an IL-17 family member regulating many epidermal functions, takes part in skin fibrosis is unknown. Here we address the role of IL-25 in skin fibrosis.
The expression of IL-25 was evaluated by immunofluorescence and in situ hybridization in 10 SSc and seven healthy donor (HD) skin biopsies. Epidermal equivalents (EE) reconstituted by primary HD keratinocytes were used as a model to study transcriptomic changes induced by IL-25 in the epidermis. RNA expression profile in EEs was characterized by RNAseq. The conditioned medium (CM) from primary SSc and HD keratinocytes primed with IL-25 was used to stimulate fibroblasts. IL-6, IL-8, MMP-1, type-I collagen (Col-I), and fibronectin production by fibroblasts was assessed by ELISA.
SSc epidermis expressed lower levels of IL-25 compared with HDs. In EEs, IL-25 regulated several molecular pathways related to wound healing and extracellular matrix remodelling. Compared with control CM, the CM from IL-25-primed keratinocytes enhanced the fibroblast production of MMP-1, IL-6 and IL-8, but not of Col-I nor fibronectin. However, IL-25 significantly reduced the production of Col-I when applied directly to fibroblasts. The activation of keratinocytes by IL-25 was receptor-dependent and evident after a very short incubation time (10 min), largely mediated by IL-1, suggesting enhanced and specific release of preformed mediators.
These results show that IL-25 participates in skin homeostasis, and its decreased expression in SSc may contribute to skin fibrosis by favouring extracellular matrix deposition over degradation.

Rheumatology key messages
SSc epidermis is characterized by a reduced expression of IL-25.
The relative lack of IL-25 may alter dermal homeostasis favouring a reduced extracellular matrix reabsorption.
This perspective may contribute to better understanding of SSc and other fibrotic skin disorders.
Introduction
Skin fibrosis, a hallmark of SSc, is characterized by excessive deposition of extracellular matrix (ECM) in the dermis [1]. The mechanisms involved in skin fibrosis remain poorly understood, but it is commonly accepted that inflammation due to vasculopathy and immune activation concurs to abnormal fibroblast activation leading to fibrogenesis [2]. Recent evidence suggests that keratinocytes may also take part in skin fibrosis [3]. Interestingly, SSc keratinocytes show a dysfunctional phenotype with aberrant differentiation and enhanced production/accumulation of inflammatory mediators [4–6]. In SSc and keloids, dysfunctional keratinocytes may establish pathological interactions with fibroblasts skewing ECM turnover in favour of enhanced deposition, in addition to increased production of inflammatory mediators and growth factors by fibroblasts [6–8].
IL-25, also known as IL-17E, is a low-homology member of the IL-17 family, which uses the heterodimer IL-17RA–IL-17RB to signal [9, 10]. Originally described as a Th2-type cytokine produced by hematopoietic cells [11], IL-25 is currently considered important in maintaining barrier integrity and homeostasis at mucosal and cutaneous surfaces when produced by epithelial cells. Further, IL-25 produced by keratinocytes appears to be a key inflammatory mediator in diseases as diverse as psoriasis [12–14], atopic dermatitis [15, 16] and contact dermatitis [17].
Little is known about the role of IL-25 in fibrosis and particularly in dermal fibrosis. In the lung, however, epithelium-derived IL-25 is thought to participate in fibrosis and airway remodelling both by its action on innate lymphoid cells 2 (ILC2) and macrophages and by modulating epithelial–mesenchymal transition in the context of allergic lung inflammation and in idiopathic lung fibrosis [18–20]. Furthermore, IL-25 in conjunction with IL-33 and thymic stroma lymphopoietin (TSLP) was identified as a key mediator in various murine models of organ fibrosis [21]. Serum levels of IL-25 have been reported to be increased in SSc [22] and the number of IL-25 positive cells increased in SSc dermis [23], but no study has addressed the potential role of IL-25 within SSc skin fibrosis. With this aim, we assessed the presence of IL-25 in SSc epidermis and the autocrine effect of IL-25 on keratinocytes as well as the effect of IL-25 on keratinocyte-driven fibroblast activation. Our results indicate that IL-25 is decreased in SSc epidermis. Further, under the influence of IL-25, healthy keratinocytes enhance inflammatory cytokine and matrix metalloproteinase-1 but not type I collagen production by fibroblasts, consequently acting as negative regulators of ECM deposition. Thus, IL-25 deficiency in SSc may contribute to dermal fibrosis.
Methods
Detailed materials and methods are provided as in Supplementary Data S1, available at Rheumatology online.
Human samples
All SSc patients fulfilled the 2013 ACR/EULAR classification criteria. Their clinical characteristics are reported in Supplementary Table S6, available at Rheumatology online. Punch biopsies were obtained from SSc affected skin. Surgical biopsies were obtained from age- and sex-matched healthy donors (HD) undergoing aesthetic surgery (abdominoplasty) at the Department of Plastic, Reconstructive, and Aesthetic Surgery of Geneva University Hospital (Geneva, Switzerland). None of the healthy donors had dermatological disorders. This study was approved by the local ethical committee (06-063, Commission cantonale d’éthique de la recherche, Geneva, Switzerland) and was conducted according to the Declaration of Helsinki. Written informed consent was obtained from each individual.
Cell cultures and epidermal equivalent engineering
Primary human dermal fibroblasts and keratinocytes were isolated from HD and SSc skin, as described [24]. Epidermal equivalents (EE) were generated according to [14]. Briefly, 5 × 105 HD keratinocytes were plated onto ThinCert cell culture inserts (Greiner Bio-One GmbH, Kremsmünster, Austria) and grown to confluence in CnT-Prime medium (CELLnTEC AG, Bern, Switzerland). After 3 days, the medium was switched to the CnT-Prime 3D Barrier medium (CELLnTEC AG, Bern, Switzerland), and the cells cultured at the air–liquid interface for 11 days. Fresh medium containing or not IL-25 (100 ng/ml) was replaced every other day. On the last day of culture, conditioned medium was collected and immediately frozen. EEs were harvested and used for mRNA isolation or histology.
RNA sequencing analysis
Library preparation, sequencing and read mapping to the reference genome were performed by the Genomics Platform at the University of Geneva (Switzerland) as described in Supplementary Material and Methods, available at Rheumatology online.
The differential expression analysis was performed using R/Bioconductor package edgeR v.3.18.1. A paired Student’s t-test was used to assess the differentially expressed genes (DEG), and P-values were corrected for multiple testing errors with 5% false discovery rate (FDR) according to Benjamini–Hochberg procedure.
The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was used for functional analysis of DEGs and the Gene Ontology (GO) database queried for DEGs with FDR <0.05 and log fold change (logFC) ≤2 or ≥2. The functional annotation clustering method was used to perform cluster analysis, selecting high classification stringency. Pathway and network analysis was performed using MetaCore (Cortellis Solution software; Clarivate Analytics, London, UK) or g: GOSt (g: profiler, by ELIXIR) with an FDR <0.05. Raw data are stored under GSE168312.
Statistical analysis
Statistical analysis was performed with GraphPad Prism, version 8 (Graphpad Software, La Jolla, CA, USA). The Shapiro–Wilk normality test was used to evaluate normal distribution. Accordingly, statistical significance was assessed by paired Student’s t-test or Wilcoxon test. The non-parametric, two samples, Kolmogorov–Smirnov test was used to assess differences in IL-25 epidermal distribution between groups. P-values ≤0.05 were considered statistically significant.
Results
IL-25 expression is decreased in the epidermis of SSc and scleroderma-like disorders
In healthy skin, IL-25 mostly accumulates in the granular layer of the epidermis [12]. We asked the question whether this was the case also in SSc-involved skin. By immunofluorescence we found that IL-25 was enriched in the granular layer of SSc epidermis, but the intensity of the signal was much fainter in SSc than in HDs (Fig. 1A). The fluorescence intensity of IL-25, as assessed by automated quantification, was significantly lower in 10 SSc than in seven HDs, specifically in the granular layer of the epidermis (P < 0.0001) (Fig. 1B). Individuals with diffuse cutaneous SSc (n = 5) and limited cutaneous SSc (n = 5) exhibited similar IL-25 decreased expression (not shown), indicating that IL-25 is reduced in involved SSc epidermis. To substantiate our findings, we assessed the presence of IL-25 mRNA within the skin of three SSc patients and three HDs by chromogenic in situ hybridization. Keratinocytes were found positive for IL-25 mRNA, and consistent with the immunofluorescence data, SSc keratinocytes expressed lower levels of IL-25 mRNA compared with HDs (Fig. 1C). We then tested whether the common receptor IL-17RA and the IL-25-specific receptor IL-17RB were expressed in skin keratinocytes. This was the case both in SSc and HD skin. Moreover, no differences were readily apparent in staining intensity of IL-17RB between SSc and HD skin (Supplementary Fig. S1, available at Rheumatology online).
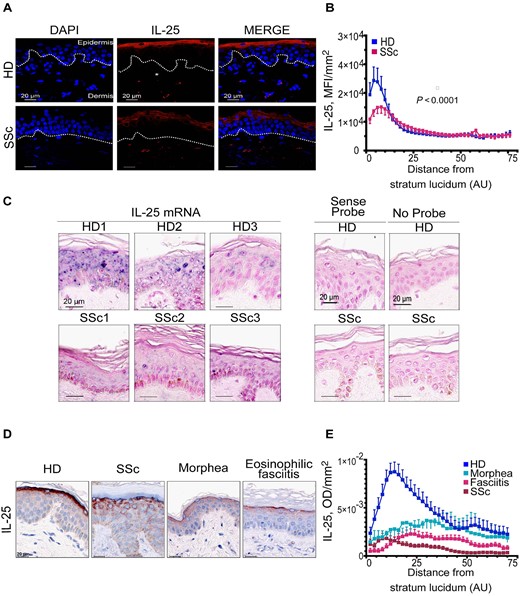
IL-25 expression is decreased in the epidermis of SSc and scleroderma-like disorders
(A, D) Representative detection of IL-25 by immunofluorescence (A) and immunohistochemistry (D). Dotted line depicts basement membrane (BM). (B) IL-25 mean fluorescence intensity (MFI) as function of distance from BM. Symbols represent the mean MFI (s.e.m.) of 10 SSc and seven HDs. (C) IL-25 mRNA (blue) in situ hybridization. (E) IL-25 optical density (OD) as function of distance from BM. Symbols represent the mean OD (s.e.m.) of 10 HDs (blue), six SSc (fuchsia), four eosinophilic fasciitis (brown), and six morphea (light-blue). Nuclear counterstaining by DAPI (A), Fast Red (C) and haematoxylin (D). (B, E) P < 0.0001 between each pathology and HD; significant difference assessed by Kolmogorov–Smirnov test. AU: arbitrary units; DAPI: 4′,6-diamidino-2-phenylindole; HD: healthy donor.
We then asked whether IL-25 was also dysregulated in the epidermis of other skin disorders characterized by dermal fibrosis including morphea (n = 6) and eosinophilic fasciitis (n = 4) in addition to SSc (n = 6) and compared with HDs (n = 10). By immunohistochemistry we observed that IL-25 was mainly localized in the granular layer of the epidermis in all four conditions. However, the levels of IL-25 were lower in all analysed diseases when compared with HD (P < 0.0001 for comparison with HDs for each condition (Fig. 1D and E). These results suggest that decreased IL-25 in the epidermis is a common feature of fibrotic skin conditions.
IL-25 regulates tissue remodelling related pathways in keratinocytes
To investigate the effects IL-25 on keratinocyte functions, we generated EEs from three distinct HD primary keratinocytes (Supplementary Fig. S2, available at Rheumatology online) and cultured them in the presence or not of IL-25 for 11 days.
Unsupervised analysis of expressed genes identified by RNAseq shows that IL-25 critically shapes keratinocytes expression profiles (Supplementary Fig. S3, available at Rheumatology online).
We, then identified 1491 DEGs in EEs exposed to IL-25 compared with not exposed EEs, 949 of which were downregulated and 542 were upregulated with a FDR ≤0.05 (Fig. 2A;Supplementary Tables S1 and S2, available at Rheumatology online).
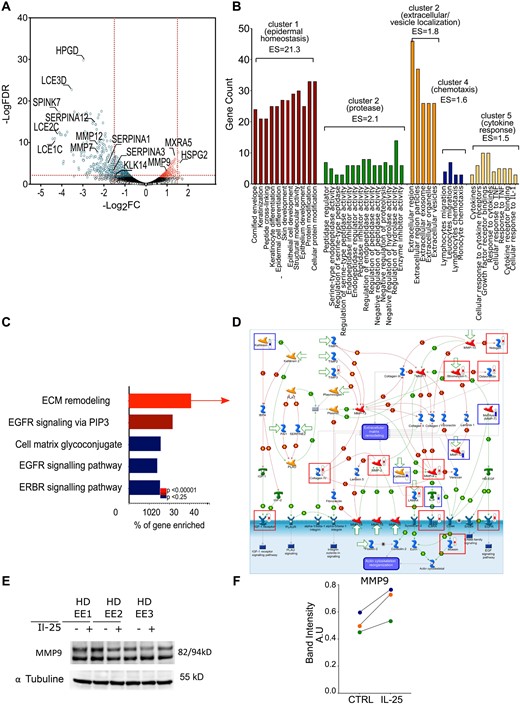
IL-25 regulates genes involved in epidermal homeostasis and ECM remodelling
Gene expression in EEs treated or not with IL-25 analysed by RNAseq. (A) DEGs in IL-25 compared with control EEs. (B) Functional annotation clustering of GO terms for genes with a logFC ≥2 or ≤2 and an FDR ≤0.01 (gene counts >3 and EASE Score <0.01), computed by DAVID. (C) Top five pathways from the significantly enriched (P < 0.00001) folder: ‘tissue remodelling and wound healing’ from MetaCore database. (D) Representation of ‘ECM remodelling’ map in MetaCore. Thermometers: gene upregulated (red) and downregulated (blue) in EEs treated with IL-25. Squares represent relevant upregulated (red) or downregulated (blue) genes. Detailed legend is in Supplementary Data S2, available at Rheumatology online. (E) Protein levels of MMP-9 were quantified by western blotting in EEs reconstituted from three independent healthy donor keratinocytes cultured 11 days in presence or not of IL-25. (F) In western blotting quantification, the numbers indicate the densitometric quantifications of MMP-9 bands normalized to the corresponding α-tubulin band. Each circle represents an independent EE. AU: arbitrary units; DAVID: Database for Annotation, Visualization and Integrated Discovery; EASE: Expression Analysis Systematic Explorer; ECM: extracellular matrix; EE: epidermal equivalent; EGFR: epidermal growth factor receptor; ES: EASE score; ERBR (HER2): human epidermal growth factor receptor 2; FC, fold change; FDR: false discovery rate; GO: Gene Ontology; IBP4: Insulin Like Growth Factor Binding Protein 4; IGF: Insulin growth factor; PAI1: plasminogen activator inhibitor-1; PIP3: Phosphatidylinositol-3,4,5-trisphosphate; PLAT: Plasminogen Activator, Tissue Type; PLAU: Plasminogen Activator, Urokinase.
Functional annotation clustering of the GO term enriched for the genes with a logFC ≥±2 revealed that IL-25 mainly regulated genes participating in epidermal homeostasis (cluster 1), proteolysis (cluster 2), products located in vesicles or extracellular compartments (cluster 3), chemotaxis (cluster 4) and cytokine responses (cluster 5) (Fig. 2B). To further understand physiological or pathological processes enriched in IL-25 exposed EEs we used the Map Folders ontology from MetaCore database. Of note, tissue remodelling and wound healing were among the top 10 most significantly enriched processes (P = 8.533 × 10–10; Supplementary Table S3, available at Rheumatology online) and the top five enriched pathways in this folder include ECM remodelling, cell matrix glycoconjugate, epidermal growth factor receptor (EGFR) signalling, and ERBB (also known as HER2 (human epidermal growth factor receptor 2)) signalling (Fig. 2C). Importantly, the detailed representation of the pathways most significantly enriched suggests that keratinocytes cultured in the presence of IL-25 may participate in enhanced ECM remodelling by regulating protease activations (Fig. 2D). To substantiate some of these findings, we tested whether MMP-9, upregulated at the mRNA level by IL-25, was also regulated at the protein level. Indeed, we found that MMP-9 was upregulated at protein level (Fig. 2E), thus mimicking the results observed by RNAseq (Fig. 2A), and highlighted when IL-25-regulated pathways were analysed by MetaCore (Fig. 2D).
Enrichment analysis of DEGs in EEs treated with IL-25 using more pathway databases (Kyoto Encyclopedia of Genes and Genomes (KEGG), REACTOME, GO) also revealed that several ECM-related pathways are regulated by IL-25 in keratinocytes (Supplementary Fig. S3 and Supplementary Table S4, available at Rheumatology online). Altogether these data suggest that IL-25 is involved in modulation of keratinocyte–ECM crosstalk.
IL-25 priming of keratinocytes enhances paracrine fibroblast inflammatory responses
We and others have previously demonstrated that keratinocytes, by releasing soluble mediators, may affect the functional capabilities of dermal fibroblasts [6–8, 24]. Thus, we investigated to what extent IL-25 modulates the interaction between keratinocytes and fibroblasts. With this aim, we established several culture conditions (Fig. 3A, Supplementary Fig. S4, available at Rheumatology online) using HD or SSc keratinocytes primed or not with IL-25 for 24 h to generate keratinocyte conditioned medium (KCM). KCMs were then added to HD dermal fibroblasts (HDF) and their production of inflammatory mediators (IL-6, IL-8), MMP-1 and the matrix proteins type-I collagen (Col-I) and fibronectin was then assessed as read-out. KCMs were added at 12.5% in fibroblast cultures. We observed that at the dose contained in KCMs, when applied directly to fibroblasts, IL-25 did not induce substantial responses (grey symbols in Fig. 3B–E). As expected, KCM strongly (by 10–1000 fold) enhanced the production of IL-6, IL-8 and MMP-1 (Fig. 3B–D; Supplementary Table S5, available at Rheumatology online). However, no major effects were observed for Col-I (Fig. 3E) and fibronectin (Supplementary Fig. S5, available at Rheumatology online) production. The magnitude of these responses was in general similar when KCMs were generated from HD (blue) or SSc (pink) keratinocytes, but the magnitude of IL-6 and MMP-1 production was greater in the presence of KCM generated from SSc keratinocytes. Interestingly, KCM generated from IL-25-primed keratinocytes (IL-25+KCM) induced—or tended to induce—stronger inflammatory responses by fibroblasts compared with IL-25 non-primed keratinocytes. The production of fibronectin and Col-I was similar in HDFs cultured with IL-25+ or IL-25− KCM either from HD or SSc keratinocytes. However, KCM skewed the ratio of collagen-I to MMP-1, which was in favour of collagen degradation, more so in the presence of IL-25+KCM (Fig. 3F). Of note, none of the biomarkers we assessed reached the assay detection level in KCM, which indicates that fibroblasts were the only or predominant producing cells in our assays. Overall, these results indicate that IL-25-primed keratinocytes enhance the inflammatory IL-6, IL-8 and MMP-1 fibroblast responses. In addition, by favouring MMP-1 over Col-I production they may tip the balance in favour of ECM degradation rather than deposition.
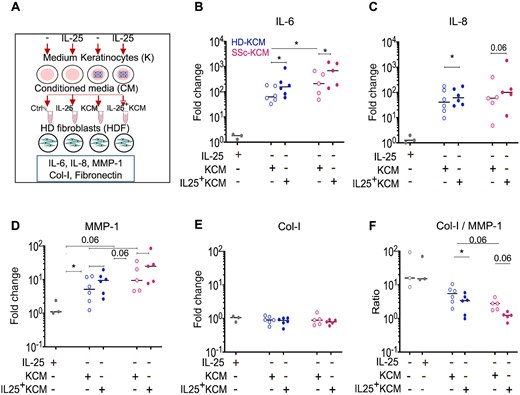
IL-25 keratinocyte priming enhances paracrine fibroblast inflammatory responses
(A) Experimental design. HD fibroblasts (HDF) were cultured in the presence of 12.5% conditioned medium (CM) from HD (blue) or SSc (pink) keratinocytes primed or not with IL-25 (IL-25+KCM). Keratinocyte culture medium ±IL-25, with no cells, was used as negative control, and HDF cultured with IL-25 (12.5 ng/ml) as control for possible carryover. IL-6, IL-8, MMP-1 and collagen-I levels assessed by ELISA in HDF supernatants. Circles represent KCM from independent donors. Horizontal lines represent medians. Wilcoxon signed-rank-test, *P ≤ 0.05. Full data set reported in Supplementary Tables S3A and B, available at Rheumatology online. Col I: type-I collagen; HD: healthy donor; IL-25+KCM: CM from keratinocytes primed with IL-25; KCM: CM from keratinocytes.
Given these findings, we assessed the effect of IL-25 to directly prime dermal fibroblasts. In these assays, where TGF-β as expected enhanced Col-I production, IL-25 at 100 ng/ml significantly reduced the Col-I production by 1.8-fold (P = 0.02) in HD fibroblasts, a reduction similar to that reached by TNF (Fig. 4). Findings at the mRNA level are shown in Supplementary Fig. S6, available at Rheumatology online. Intriguingly, SSc fibroblasts did not show a significantly reduced production of collagen in response to both IL-25 and TNF. Whereas TNF powerfully enhanced MMP-1 production, IL-25 did not modify the levels of MMP-1 produced by fibroblasts. However, the overall result of the direct priming of dermal fibroblasts with IL-25 was a decreased Col-I to MMP-1 ratio (Fig. 4). This was distinctly different from the pro-fibrotic effect observed in the presence of TGF-β with enhanced Col-I to MMP-1 ratio and supports the contention that IL-25 by its direct effects on fibroblasts as well as by its indirect effects through keratinocytes tips the Col-I to MMP-1 balance favouring enhanced ECM degradation, thus exerting, at least in vitro, an anti-fibrotic effect.
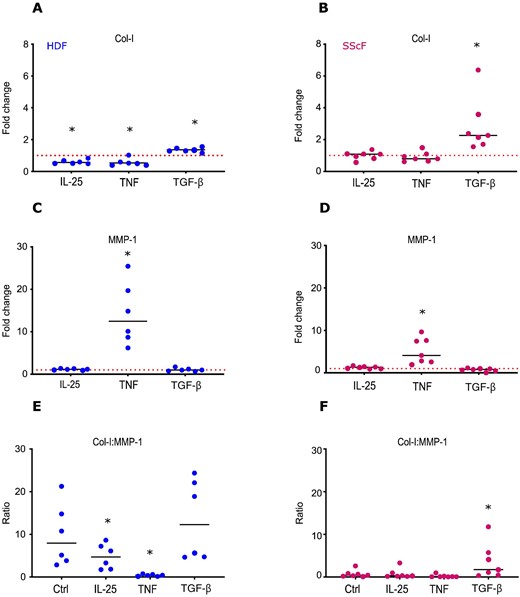
IL-25 reduces type-I collagen production by HD fibroblasts
HD (blue circles) or SSc (pink circles) fibroblasts were cultured in the presence of IL-25 (100 ng/ml), TGF-β (10 ng/ml), TNF (1 ng/ml) or control medium (Ctrl). MMP-1 and Col-I levels assessed by ELISA in fibroblast supernatants. Results are expressed as fold change from spontaneous production (Ctrl). Each circle represents a distinct donor. Horizontal continuous lines depict the mean. Red dotted lines depict control. Statistical differences were assessed by paired t-test. *P ≤ 0.05. Col I: type-I collagen; HD: healthy donor; HDF: HD fibroblast; SScF: SSc fibroblast.
In addition, we observed that IL-25 partially inhibited α-smooth muscle actin (α-SMA) expression promoted by TGF-β in dermal fibroblasts (Supplementary Fig. S7, available at Rheumatology online), further suggesting that IL-25 may preferentially restrain fibrosis.
Early keratinocyte responses to IL-25 suggest the release of preformed mediators
To communicate with neighbouring cells, including dermal fibroblasts, keratinocytes may release preformed and stored material [25]. We conducted time-dependent experiments to test the hypothesis that IL-25 could enhance this phenomenon. We observed that the production of IL-6 by dermal fibroblasts was significantly enhanced when fibroblasts were exposed to conditioned medium generated from keratinocytes primed by IL-25 (IL-25+KCM) for 10 min only. Similar results were obtained when keratinocytes were primed with IL-25 for 30 min (Fig. 5A). The enhancing effect over control KCM was present also when keratinocytes were primed with IL-25 for 4 and 24 h. However, the effect over background of non-primed KCM became evident at these later time points (Fig. 5A). The enhancing effect of 10 min IL-25+KCM was not due to IL-25 carryover since the neutralization of IL-25 in IL-25+KCM added to fibroblasts did not modify the results (Fig. 5B). Furthermore, the effect of 10 min priming of keratinocytes with IL-25 was specific, since we did not observe such an effect when keratinocytes were primed with TNF (Fig. 5C). This is relevant, since the conditioned medium of keratinocytes primed with TNF for 24 h induced a robust IL-6 production by fibroblasts (Fig. 5C). Thus, the differential effects of short (10 min) vs long (24 h) priming distinguish the activities of IL-25 and TNF on keratinocytes. Of interest, IL-17A shared with IL-25 the capacity to prime keratinocytes in 10 min time, resulting in higher IL-6 production by fibroblasts (Fig. 5D).
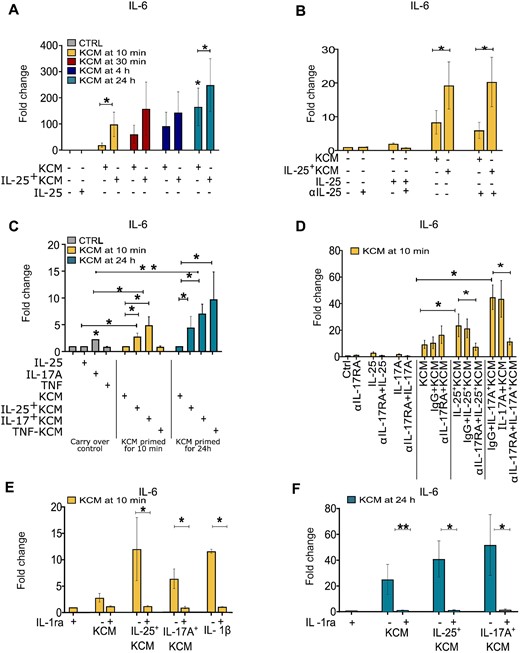
IL-25 promotes the release of fibroblast-activating factors by keratinocytes at early time points
(A) IL-6 production by HDFs cultured with 12.5% control keratinocyte conditioned medium (KCM) or IL-25+KCM harvested at the indicated time points. (B, D) anti-IL-25 polyclonal-Ab, anti-IL-17RA mAb, and irrelevant Ab (IgG) added to KCM (B) or keratinocytes (D) prior to HDF stimulation. (C) HDFs cultured with KCM, IL-25+KCM, IL-17A (IL-17A+KCM), or TNF (TNF+KCM) harvested after 10 min. (E, F) IL-1ra (100 ng/ml) added to HDF before priming with KCM or IL-25+KCM collected at 10 min (E) or 24 h (F). Results are expressed as fold change from spontaneous production (Ctrl). For all panels n ≥ 3; bars represent the mean (s.e.m.). Wilcoxon signed-rank test was used to assess statistical significance. *P ≤ 0.05; **P ≤ 0.01. HDF: healthy donor fibroblast.
We then wondered whether the effects of IL-25 and IL-17A on keratinocytes were mediated by receptor binding. We observed that blockade of IL-17RA on keratinocytes resulted in the abrogation of the enhancing effect due to priming with IL-25 and IL-17A (Fig. 5D). Finally, guided by the results obtained in similar settings [5, 6, 24], we tested whether IL-1 could be mediating the early as well as late effects of KCM. This was indeed the case since IL-1ra abrogated the IL-6 production by fibroblasts in response to IL-25+KCM obtained after 10 min and 24 h (Fig. 5E and F). Overall, these results suggest that IL-25 induces early keratinocyte responses (within 10 min) capable of activating fibroblasts, a characteristic shared with IL-17A.
Discussion
When initiating this work, we hypothesized that epidermal IL-25 would be increased in SSc. Against our hypothesis we found that SSc keratinocytes ex vivo express significantly lower amounts of IL-25 than healthy keratinocytes, a characteristic shared by other fibrotic skin disorders including morphea and eosinophilic fasciitis. There were several supportive elements for our original hypothesis. First, serum IL-25 was reported to be increased in SSc [22], and IL-25 positive cells increased in SSc dermis [23]. Second, IL-25, in conjunction with IL-33 and TSLP, was associated with fibrosis in several mouse models of fibrosis, particularly mediated by Th2 responses [21]. Indeed, a predominant Th2-like environment characterizes SSc in which T cells producing IL-4 and/or IL-13, ILC2 and IL-33 as well as TSLP positive cells have been documented in SSc skin [26–31]. Third, enhanced IL-25 expression by epithelial airway and alveolar cells was linked to lung fibrosis in asthma and idiopathic pulmonary fibrosis [18–20]. Vice versa, for the first time, our work documents that a relative lack of IL-25 rather than its excess characterizes SSc epidermis, which is distinctly different from psoriasis, atopic dermatitis and contact dermatitis in which IL-25 epidermal expression is increased [12, 13, 15–17]. Such an observation supports the hypothesis that an IL-25 epidermal deficiency could be implicated in skin fibrosis. However, we did not investigate the mechanism underlying the IL-25 dysregulation in SSc epidermis and further research is needed to address this finding.
The constitutive expression of IL-25 in healthy keratinocytes substantiates its role in skin homeostasis and is consistent with previous findings [32, 33]. The information we obtained by assessing the effect of IL-25 on gene transcription in EEs generated form healthy keratinocytes shows that IL-25 may regulate a wide variety of gene products located in vesicles or the extracellular compartment and participating in epidermal homeostasis, proteolysis, chemotaxis, and IL-1 or TNF family member signalling. An important finding was that IL-25 regulates molecular pathways involved in ECM turnover and remodelling by coordinating the expression of several proteases and antiproteases (such as MMP-9, MMP-7, MMP-12, TIMP3, SERPIN12 and SERPIN1) (Fig. 2A and D). This is intriguing because it is increasingly clear that the ECM architecture affects the availability of growth factors and cytokines and determines tissue stiffness, and that an altered ECM remodelling participate in fibrosis [34, 35]. Furthermore, proteases may participate in vascular damage, crucial in SSc pathogenesis. For example, MMP-12 and MMP-7, both downregulated by IL-25, are increased in SSc skin and lung, and their levels correlate with fibrosis severity and vascular damage [36]. Consistently, there is evidence that MMP-12 deficiency attenuate skin fibrosis and angiotensin-II-mediated vascular injury [37]. Transcriptomic analysis also revealed that IL-25 negatively regulates a number of serin proteases, involved in fibrinolysis (such as SERPIN12 and SERPIN1). This is captivating because fibrinolysis is increased in SSc and is associated to vascular dysfunction and fibrosis in SSc [38]. To complement RNAseq data, we also found that MMP-9 protein levels were increased in EE by IL-25. Thus, a relative lack of IL-25 in SSc skin may result in less MMP-9 available, which is consistent with the reported reduced MMP-9 activity detected in SSc sera [39]. Furthermore, we showed that, through its action on keratinocytes, IL-25 indirectly increases MMP-1 secretion by fibroblasts while it directly decreases their Col-I production, supporting the hypothesis that IL-25 may favour ECM resorption over deposition in the skin. In this context, IL-25 deficiency in SSc epidermis may alter the protease–antiprotease balance and ECM composition, favouring the activation of some pathways crucial in SSc pathogenesis. Similarly to our results, previous studies on lung fibrosis showed that IL-25 promoted protease activity, ECM remodelling, inflammatory responses and regulated epithelial–mesenchymal cross-talk [18–20].
Of interest, our data suggest that IL-25-priming of keratinocytes could be, at least in part, associated with a receptor-mediated, enhanced secretion of pre-stored mediators, as supported by the observation that keratinocyte conditioned medium collected after only 10 min was sufficient to promote a significant production of IL-6 by fibroblasts. We have not investigated the mechanisms underlying such an effect, but it appeared to be specific to IL-17 family members since TNF did not induce early keratinocyte responses while both IL-17A and IL-25 did, and an anti-IL-17AR mAb blocked their effect. The capacity of keratinocytes to release prestored mediators, eventually in extracellular vesicles of various size, has been reported in the literature [25]. Cellular stress induced by hypoxia, serum starvation, UV exposure, as well as keratinocyte stimulation by TGF-α, and poly I: C were described as factors favouring extracellular vesicle release (reviewed in [25]).
This paper has some limitations. To assess the differential expression of IL-25 between healthy and fibrotic skin we used immunohistology, which may be limited by being semiquantitative. However, the use of software-based signal quantification and the use of different techniques to assess the presence of IL-25 in keratinocytes permitted minimization of this point. We did not investigate why IL-25 is reduced in SSc patients. RNAseq analysis results were not validated at the protein level, and we did not successfully knock out the IL-25 gene in primary human keratinocyte to assess their function in the absence of the cytokine. However, we engineered epidermal equivalents allowing both keratinocyte proliferation and differentiation to study the effects of IL-25 on keratinocytes and focused on the role of IL-25 to modulate keratinocyte-generated mediators influencing fibroblast responses. By using larger numbers of keratinocytes—and biopsies—obtained from both limited and diffuse as well as early vs late disease would increase the breath of our findings, possibly identifying subset-related functions of IL-25. Further, the effect of IL-25-primed KCM on fibroblasts was assessed after 48 h of culture. This may hinder the possible secondary activities of cytokines induced by KCM, such as IL-6, which may secondarily result in enhanced collagen production. However, in other experimental systems in which IL-17 was added to organotypic skin cultures for 9 days, no such secondary effects were seen [24].
In conclusion, reduced expression of IL-25 characterizes SSc epidermis, and the relative lack of IL-25 may alter dermal homeostasis favouring deposition rather than ECM resorption. This perspective may contribute to better understanding SSc and other fibrotic skin disorders.
Acknowledgements
We thank Natacha Civic and Mylène Docquier (Genomics Platform) for performing the RNA sequencing; Nicolas Laudet (Bioimaging Core Facility) for quantification of immune-histology, François Prodon and Olivier Brun for microscope imaging (Bioimaging Core Facility), Marie Ebrahim Malek and Laura de Luca (Histology Core Facility) for contribution to tissue preparation (all at University Medical Center, Geneva, Switzerland). Drowings were created with BioRender.com and https://inkscape.org/.
Funding: This work was supported in part by grant 310030_159999 from the Swiss National Science Foundation (SNSF) to C.C., by a grant from the Swiss Scleroderma Patient organization (sclerodermie.ch) to C.C.; by grant 310030_152680 and 310030_175470/1 from SNSF to W.H.B., by grant 310030_184814 from SNSF to J.B.; and by a grant from the Ernst & Lucie Schmidheiny foundation to B.R.
Disclosure statement: M.E.T. has received honoraria as an advisor or invited speaker from Abbvie, Lilly, SOBI and Boehringer Ingelheim. W.H.B. has received honoraria as an advisor or invited speaker from Abbvie, Almirall, BMS, Celgene, Leo, Lilly, Novartis and UCB. C.C. has received honoraria as an advisor or invited speaker from GSK, Roche and Boehringer Ingelheim. The other authors have disclosed no conflicts of interest.
Data availability statement
The data underlying this article will be shared on reasonable request to the corresponding author.
Supplementary data
Supplementary data are available at Rheumatology online.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments