





Abstract
To investigate the efficacy and safety of rituximab + LEF in patients with RA.
In this investigator-initiated, randomized, double-blind, placebo-controlled phase 3 trial, patients with an inadequate response to LEF who had failed one or more DMARD were randomly assigned 2:1 to i.v. rituximab 1000 mg or placebo on day 1 and 15 plus ongoing oral LEF. The primary efficacy outcome was the difference between ≥50% improvement in ACR criteria (ACR50 response) rates at week 24 (P ≤ 0.025). Secondary endpoints included ACR20/70 responses, ACR50 responses at earlier timepoints and adverse event (AE) rates. The planned sample size was not achieved due to events beyond the investigators’ control.
Between 13 August 2010 and 28 January 2015, 140 patients received rituximab (n = 93) or placebo (n = 47) plus ongoing LEF. Rituximab + LEF resulted in an increase in the ACR50 response rate that was significant at week 16 (32 vs 15%; P = 0.020), but not week 24 (27 vs 15%; P = 0.081), the primary endpoint. Significant differences favouring the rituximab + LEF arm were observed in some secondary endpoints, including ACR20 rates from weeks 12 to 24. The rituximab and placebo arms had similar AE rates (71 vs 70%), but the rituximab arm had a higher rate of serious AEs (SAEs 20 vs 2%), primarily infections and musculoskeletal disorders.
The primary endpoint was not reached, but rituximab + LEF demonstrated clinical benefits vs LEF in secondary endpoints. Although generally well tolerated, the combination was associated with additional SAEs and requires monitoring.
EudraCT: 2009-015950-39; ClinicalTrials.gov: NCT01244958.
The addition of MTX improves rituximab effectiveness in RA, but many patients are intolerant.
Addition of rituximab to LEF improved outcomes, but the primary endpoint was not met.
The combination was generally well tolerated, but had an increased rate of serious adverse events.
Introduction
In patients with RA, combination therapy with biologic agents plus MTX is superior to monotherapy with either agent alone [1]. Although the majority of studies comparing monotherapy with MTX combination therapy have investigated TNF inhibitors, B-cell-depleting therapy with rituximab also achieves higher response rates in combination with MTX compared with rituximab monotherapy [2].
In daily clinical practice, MTX intolerance and contraindications can be critical barriers to the use of combination treatment regimens. Between 11 and 29% of patients are intolerant to MTX [3, 4], and >40% experience at least occasional gastrointestinal adverse events (AEs) [3]. Physicians routinely overestimate the proportion of patients who are taking MTX. In one study, ∼20% of patients whom physicians believed were adherent to MTX therapy were either not taking the drug at all (8%) or had missed one or more doses in the previous 4 weeks (11%) [5].
Little is known about the effectiveness of biologic treatment in combination with DMARDs other than MTX, such as LEF, a frequently used alternative to MTX in RA treatment regimens. Expert consensus and population-based data support the use of LEF as an alternative to MTX in combination regimens with biologic therapies [6–9]. Moreover, the combination of LEF and rituximab has produced promising results in RA registries [10, 11] and small observational studies [12, 13]. However, the efficacy and safety of this combination therapy has not been formally studied.
In recognition of the need for reliable evidence on this potentially useful combination regimen, we conducted a randomized clinical trial to evaluate the efficacy and safety of rituximab + LEF compared with placebo + LEF in patients with active RA.
Methods
Study design
The Addition of MabThera to Arava in RA (AMARA) study was a prospective, investigator-initiated, randomized, double-blind, placebo-controlled, phase 3 clinical trial conducted at 33 clinical centres in Germany (Supplementary Material Section 1, available at Rheumatology online) between 8 August 2010 and 28 January 2015. The study protocol was approved by the ethics committee of Goethe University (Ethikkommission des Fachbereichs Medizin der Goethe Universität) and by local ethics committees at participating sites. The protocol is available from the corresponding author upon request. The AMARA study was registered with the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT 2009-015950-39) on 28 December 2009, prior to submission to ethical committees and prior to inclusion of the first subject as required by International Committee of Medical Journal Editors guidelines; subsequently it was additionally registered with ClinicalTrials.gov (NCT01244958) to provide broader access to the protocol. The study was conducted in accordance with the declaration of Helsinki. All patients gave written informed consent for study participation.
Randomization at study entry was performed by use of a computer-prepared randomization list. All investigators and patients were blinded to the treatment arm. Masking/blinding was accomplished through the use of placebo vials of identical appearance to the active drug.
Patients were randomized 2:1 to 1000 mg rituximab or placebo administered as an i.v. infusion on day 1 and day 15. Both groups also received rituximab-associated pre-medication (steroids, antipyretics and antihistamines) 30 min prior to rituximab/placebo infusion. Throughout the study, patients continued oral LEF treatment at the pre-enrolment dose (10–20 mg/day). Rituximab or placebo was administered under the supervision of study staff at the clinical centre. Adherence to LEF therapy was not formally assessed. Rescue treatment with any standard of care treatment (at the discretion of the investigator) was allowed between weeks 16 and 24 for patients with a DAS–28 joints (DAS28) change <0.6 or <20% improvement in both tender and swollen joint counts. Patients receiving rescue treatment were considered to have discontinued the study.
The primary efficacy analysis was based on ACR criteria for 50% improvement (ACR50) response rates at week 24. Patient visits were also conducted at weeks 2 and 4 and every 4 weeks thereafter through week 24, and secondary efficacy data were collected at weeks 8, 12, 16 and 24. A second part of this study (not reported here) involved follow-up to week 52 following re-randomization to treatment with two different doses of rituximab.
Patients
The trial enrolled adult patients (18–75 years) with a diagnosis of RA according to the revised 1987 ACR criteria [14]. Patients with active RA, defined as DAS28 >3.2 with at least three tender and three swollen joints based on a 28-joint count, and an inadequate response to LEF therapy (at least 3 months of stable therapy prior to randomization) were eligible. Exclusion criteria included prior treatment with more than three conventional DMARDs (including LEF), use of DMARDs other than LEF in the previous 4 weeks and treatment with biologic DMARDs other than anti-TNF agents. Previous anti-TNF therapy (maximum of two agents) was allowed, but only one could have been terminated due to inadequate response. Ongoing CS therapy was allowed at stable doses of ≤10 mg/day prednisolone equivalent. Other major exclusion criteria included RA functional class IV, chronic inflammatory articular disease or systemic autoimmune disease, active or recurrent infections, and previous exposure to rituximab or biologic agents other than TNF inhibitors.
Outcomes
The primary efficacy outcome was the difference in the proportion of patients who achieved ACR50 responses [15] at week 24. Secondary efficacy outcomes included ACR50 responses at visits other than week 24 and ACR20 or ACR70 responses. Additional secondary objectives included changes in disease activity and patient-reported outcomes. DAS28 and the Clinical Disease Activity Index (CDAI) were used to document disease activity. Health-related quality of life was assessed by the patient-reported Short Form-36 (SF-36) [16]. Other outcomes included the HAQ-Disability Index (HAQ-DI) and the Functional Assessment of Chronic Illness-Therapy (FACIT) fatigue score [17]. Exploratory analyses of ACR response rates based on CCP serostatus and regression analysis of factors potentially influencing the primary objective were also conducted.
Safety analyses included AEs reported by Medical Dictionary for Regulatory Activities system organ class and preferred term, and serious adverse events (SAEs), including deaths. AEs and SAEs considered to be treatment-related were based on the judgement of the investigator. Investigators were requested to report all potentially treatment-related AEs that occurred within 1 year of the last administration of study drug. Peripheral CD19+/CD20+ cells were measured by FACS to evaluate B-cell depletion. IgG levels were monitored at baseline, before retreatment and at study discontinuation.
Statistical analysis
Based on previous studies of rituximab + MTX [2, 18], and estimating a 10% discontinuation rate, initial sample size calculations determined that 270 patients would be required to detect superiority in ACR50 response of an estimated 40% for rituximab vs 15% for placebo at week 24 with 90% power and a Fisher’s exact test with a 0.025 one-sided significance level. Due to subsequent events, including a publication on the potential risk of pancreatic cancer in patients treated with LEF [19] and the manufacturer’s decision to not apply for rituximab licensing as a first-line biologic agent for RA in Germany, trial enrolment was much lower than anticipated, even though the association between pancreatic cancer and LEF was not confirmed [20]. Once enrolment issues became apparent, we reassessed the sample size calculations and found that for the expected effect size and discontinuation rate, a study cohort of 140 was sufficient for statistical power of 80% for the primary outcome.
Efficacy analyses were conducted on the intention-to-treat population, defined as all patients who received at least one dose of study medication and had at least one assessment under study medication, with imputation using last observation carried forward for the primary and key secondary outcomes (ACR response rates). Analyses of ACR response rates were performed using a one-sided Fisher test with a significance level alpha of 2.5% (corresponding to a two-sided Fisher test with a significance level alpha of 5%). Further efficacy parameters (DAS28, SF-36, HAQ-DI, and FACIT) were analysed using two-sided t-tests (significance level alpha of 5%). DAS28 and CDAI remission rates were compared using a two-sided Fisher test. A multivariable regression analysis was conducted to evaluate the impact of patient and disease characteristics on the primary outcome. Safety analyses were performed on the full analysis set, defined as all patients who received at least one complete dose of study medication.
Statistical analysis was performed with R version 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients
Between 13 August 2010 and 28 January 2015, 148 patients were randomized to treatment (Fig. 1). Seven patients did not receive treatment. In one additional patient, the first rituximab infusion was stopped and never completed; this patient did not receive further rituximab and was not included in efficacy or safety analyses. Of 140 patients who received treatment, 93 received rituximab and 47 received placebo in addition to ongoing stable therapy with LEF. All 140 patients fulfilled criteria for both the full analysis set for safety analyses and the intention-to-treat population for efficacy analyses. As a result, the same patient cohort was included in both analyses. During the 24-week study, 21 patients (23%) discontinued treatment in the rituximab + LEF arm and 14 (30%) in the placebo + LEF arm. Rescue treatment was received by 21 patients (11 and 10 in the rituximab and placebo arms, respectively). Of patients assigned to rituximab + LEF, 61/94 (65%) completed the 24-week study compared with 23/47 (49%) patients in the placebo + LEF arm.

Patient disposition through week 24
AE: adverse event; RTX: rituximab.
Patients enrolled in this study had moderate to severe RA, as indicated by a mean DAS28 of ∼5.6, and extensive joint involvement. Baseline characteristics of the two arms were generally comparable (Table 1).
Patient characteristics at baseline
| Characteristic | Rituximab + LEF (n = 93) | Placebo + LEF (n = 47) |
|---|---|---|
| Age, years | 56.7 (11.7) | 56.1 (9.9) |
| Females, n (%) | 66 (71.0%) | 38 (80.9%) |
| BMI, kg/m2 | 27.3 (5.8) | 27.1 (5.5) |
| Disease duration, years | 7.44 (8.15) | 5.82 (7.67) |
| DAS28 | 5.55 (0.99) | 5.53 (1.09) |
| CDAI | 25.8 (8.4) | 27.0 (7.2) |
| Tender joint count (68 joints) | 17.1 (11.4) | 18.0 (11.7) |
| Swollen joint count (66 joints) | 10.4 (5.0) | 10.2 (4.9) |
| CRP, mg/l | 8.6 (14.0) | 9.9 (17.3) |
| RF seropositive, n (%) | 55 (59.1) | 25 (53.2) |
| Anti-CCP seropositive,an (%) | 53 (57.0) | 28 (59.6) |
| Patient global assessment (10-cm VAS) | 55.5 (22.7) | 58.3 (24.6) |
| Physician global assessment (10-cm VAS) | 54.0 (16.9) | 58.8 (16.3) |
| Number of previous conventional DMARDs | 2.2 (0.7) | 2.3 (0.6) |
| Previous anti-TNF therapy, n (%) | 10 (10.8) | 6 (12.8) |
| CS dose, mg/day | 7.2 (8.2) | 6.1 (2.8) |
| Characteristic | Rituximab + LEF (n = 93) | Placebo + LEF (n = 47) |
|---|---|---|
| Age, years | 56.7 (11.7) | 56.1 (9.9) |
| Females, n (%) | 66 (71.0%) | 38 (80.9%) |
| BMI, kg/m2 | 27.3 (5.8) | 27.1 (5.5) |
| Disease duration, years | 7.44 (8.15) | 5.82 (7.67) |
| DAS28 | 5.55 (0.99) | 5.53 (1.09) |
| CDAI | 25.8 (8.4) | 27.0 (7.2) |
| Tender joint count (68 joints) | 17.1 (11.4) | 18.0 (11.7) |
| Swollen joint count (66 joints) | 10.4 (5.0) | 10.2 (4.9) |
| CRP, mg/l | 8.6 (14.0) | 9.9 (17.3) |
| RF seropositive, n (%) | 55 (59.1) | 25 (53.2) |
| Anti-CCP seropositive,an (%) | 53 (57.0) | 28 (59.6) |
| Patient global assessment (10-cm VAS) | 55.5 (22.7) | 58.3 (24.6) |
| Physician global assessment (10-cm VAS) | 54.0 (16.9) | 58.8 (16.3) |
| Number of previous conventional DMARDs | 2.2 (0.7) | 2.3 (0.6) |
| Previous anti-TNF therapy, n (%) | 10 (10.8) | 6 (12.8) |
| CS dose, mg/day | 7.2 (8.2) | 6.1 (2.8) |
Data are mean (s.d.) unless otherwise stated. aAnti-CCP levels ≥7 relative units/ml. CDAI: Clinical Disease Activity Index; VAS: visual analogue scale.
Patient characteristics at baseline
| Characteristic | Rituximab + LEF (n = 93) | Placebo + LEF (n = 47) |
|---|---|---|
| Age, years | 56.7 (11.7) | 56.1 (9.9) |
| Females, n (%) | 66 (71.0%) | 38 (80.9%) |
| BMI, kg/m2 | 27.3 (5.8) | 27.1 (5.5) |
| Disease duration, years | 7.44 (8.15) | 5.82 (7.67) |
| DAS28 | 5.55 (0.99) | 5.53 (1.09) |
| CDAI | 25.8 (8.4) | 27.0 (7.2) |
| Tender joint count (68 joints) | 17.1 (11.4) | 18.0 (11.7) |
| Swollen joint count (66 joints) | 10.4 (5.0) | 10.2 (4.9) |
| CRP, mg/l | 8.6 (14.0) | 9.9 (17.3) |
| RF seropositive, n (%) | 55 (59.1) | 25 (53.2) |
| Anti-CCP seropositive,an (%) | 53 (57.0) | 28 (59.6) |
| Patient global assessment (10-cm VAS) | 55.5 (22.7) | 58.3 (24.6) |
| Physician global assessment (10-cm VAS) | 54.0 (16.9) | 58.8 (16.3) |
| Number of previous conventional DMARDs | 2.2 (0.7) | 2.3 (0.6) |
| Previous anti-TNF therapy, n (%) | 10 (10.8) | 6 (12.8) |
| CS dose, mg/day | 7.2 (8.2) | 6.1 (2.8) |
| Characteristic | Rituximab + LEF (n = 93) | Placebo + LEF (n = 47) |
|---|---|---|
| Age, years | 56.7 (11.7) | 56.1 (9.9) |
| Females, n (%) | 66 (71.0%) | 38 (80.9%) |
| BMI, kg/m2 | 27.3 (5.8) | 27.1 (5.5) |
| Disease duration, years | 7.44 (8.15) | 5.82 (7.67) |
| DAS28 | 5.55 (0.99) | 5.53 (1.09) |
| CDAI | 25.8 (8.4) | 27.0 (7.2) |
| Tender joint count (68 joints) | 17.1 (11.4) | 18.0 (11.7) |
| Swollen joint count (66 joints) | 10.4 (5.0) | 10.2 (4.9) |
| CRP, mg/l | 8.6 (14.0) | 9.9 (17.3) |
| RF seropositive, n (%) | 55 (59.1) | 25 (53.2) |
| Anti-CCP seropositive,an (%) | 53 (57.0) | 28 (59.6) |
| Patient global assessment (10-cm VAS) | 55.5 (22.7) | 58.3 (24.6) |
| Physician global assessment (10-cm VAS) | 54.0 (16.9) | 58.8 (16.3) |
| Number of previous conventional DMARDs | 2.2 (0.7) | 2.3 (0.6) |
| Previous anti-TNF therapy, n (%) | 10 (10.8) | 6 (12.8) |
| CS dose, mg/day | 7.2 (8.2) | 6.1 (2.8) |
Data are mean (s.d.) unless otherwise stated. aAnti-CCP levels ≥7 relative units/ml. CDAI: Clinical Disease Activity Index; VAS: visual analogue scale.
ACR response rates
The primary outcome, ACR50 response rates at week 24, was not significantly different between the rituximab + LEF arm (25/93; 27%) and the placebo + LEF arm (7/47; 15%); the treatment difference was 12.0 (95% CI −3.1, 24.8; P = 0.081). However, analyses of secondary endpoints supported a benefit in adding rituximab to LEF. Significant differences for rituximab compared with placebo were observed in ACR20 rates at weeks 12, 16 and 24, ACR50 rates at week 16, and ACR70 rates at weeks 8 and 16 (Fig. 2).
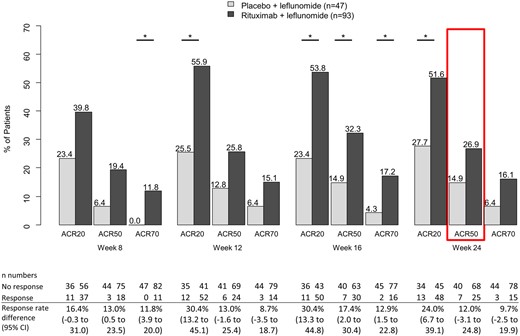
ACR response rates
The red box indicates the primary end point of ACR50 response at week 24. *P < 0.025 for rituximab + LEF vs placebo + LEF as assessed by one-sided Fisher’s exact test. ACR20/50/70: ACR criteria for 20/50/70% improvement.
More than half of the patients in each treatment arm were seropositive for antibodies to CCP (anti-CCP) (Table 1). In the rituximab + LEF arm, anti-CCP seropositive patients had higher ACR response rates than seronegative patients; CIs for most response ratios did not cross the null effect line, indicating a statistically significant difference (supplementary Fig. S1, available at Rheumatology online). However, the ACR50 response rates at 24 weeks in the rituximab + LEF group did not differ significantly between seropositive and seronegative patients: rates were 32% (17/53) in the seropositive group and 20% (8/40) in the seronegative group (response ratio 1.6; 95% CI 0.8, 3.3). In the placebo + LEF arm, differences in response rates between anti-CCP seropositive and seronegative patients were variable, and all CIs crossed the null effect line (supplementary Fig. S1, available at Rheumatology online).
An exploratory multivariable logistic regression analysis of factors potentially influencing the primary objective, including prior anti-TNF treatment, number of prior conventional DMARDs, age and anti-CCP seropositivity, did not identify any significant variables.
Additional efficacy assessments
DAS28 and CDAI assessments supported the beneficial effects of rituximab when added to ongoing LEF (Fig. 3). Mean (s.d.) DAS28 values improved from 5.6 (1.0) at baseline to 3.7 (1.4) at week 24 in the rituximab + LEF arm compared with a change from 5.5 (1.1) at baseline to 4.4 (1.3) in the placebo + LEF arm. Differences between mean DAS28 values in the two treatment arms were statistically significant from week 12 through week 24 (P ≤ 0.05). Significant differences were also observed in the proportion of patients in DAS28 remission (DAS28 <2.6) at week 24 (28.0 vs 6.4%; P = 0.004) and the proportion of patients in CDAI remission (CDAI ≤2.8) at week 24 (17.4 vs 0%; P = 0.002).
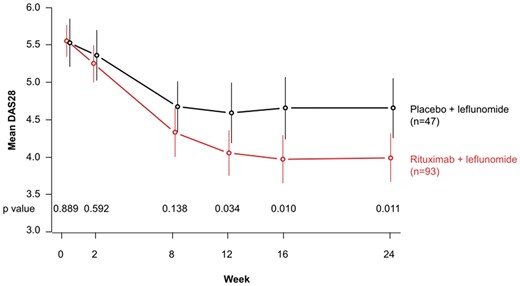
Mean DAS28 values; horizontal bars indicate 95% CIs
Stated P-values for rituximab + LEF vs placebo + LEF are two-sided values as determined by t-test (significance level of 0.05).
From baseline to week 24, significant improvements in SF-36 scores for the rituximab + LEF arm relative to the placebo + LEF arm were observed for the dimensions of bodily pain (mean improvement of 18.3 vs 7.4; P = 0.04) and emotional role functioning (mean improvement of 17.4 vs 8.9; P = 0.02) (Fig. 4 and supplementary Table S1, available at Rheumatology online). Changes in other dimensions were not statistically different between treatment arms. Improvements for rituximab + LEF were observed for the summary FACIT fatigue score, but the difference was not statistically significant (P = 0.11). Changes in the HAQ-DI summary score were the same in both treatment arms, but numerical differences in favour of the rituximab arm were observed for some dimensions, including activities and grip.

Dimensions of SF-36, HAQ-DI and FACIT scores at baseline and week 24
Higher scores are better for SF-36 and FACIT (dimensions F7 and F8 were rescaled so that higher levels were better for all dimensions) and worse for HAQ-DI. *P-value ≤0.05 for rituximab + LEF vs placebo + LEF. Data for mean change from baseline to week 24 are presented in supplementary Table S1, available at Rheumatology online. FACIT: Functional Assessment of Chronic Illness-Therapy fatigue scale; HAQ-DI: HAQ Disability Index; SF-36: Short Form-36.
Safety
Two hundred treatment-emergent AEs were reported in 66 (71%) subjects in the rituximab + LEF arm and 113 in 33 (70%) subjects in the placebo + LEF arm during the 24-week study (Table 2). The most common AEs by system organ class were infections and infestations and musculoskeletal and connective tissue disorders, primarily RA complications or flares. AE rates were generally similar between groups. AEs were equally distributed throughout the study and there was no obvious pattern with respect to duration of treatment.
Treatment-emergent adverse events over 24 weeksa
| Treatment-emergent AE | RTX + LEF (n = 93) | PL + LEF (n = 47) |
|---|---|---|
| Any AE | 66 (71.0) | 33 (70.2) |
| AEs occurring in ≥5% of subjects in either group | ||
| Infections and infestations | 37 (39.8) | 16 (34.0) |
| Nasopharyngitis | 11 (11.8) | 6 (12.8) |
| Urinary tract infection | 8 (8.6) | 2 (4.3) |
| Musculoskeletal and connective tissue disorders | 20 (21.5) | 11 (23.4) |
| RA | 6 (6.5) | 2 (4.3) |
| Vascular disorders | 15 (16.1) | 7 (14.9) |
| Hypertension | 11 (11.8) | 5 (10.6) |
| Gastrointestinal disorders | 12 (12.9) | 5 (10.6) |
| Diarrhoea | 7 (5.3) | 5 (10.6) |
| Investigations | 11 (11.8) | 3 (6.4) |
| Nervous system disorders | 6 (6.5) | 8 (17.0) |
| General disorders and administration site conditions | 7 (7.5) | 5 (10.6) |
| Injury, poisoning and procedural complications | 9 (9.7) | 2 (4.3) |
| Skin and s.c. tissue disorders | 7 (7.5) | 4 (8.5) |
| Blood and lymphatic system disorders | 5 (5.4) | 2 (4.3) |
| Metabolism and nutrition disorders | 3 (3.2) | 4 (8.5) |
| Respiratory, thoracic and mediastinal disorders | 5 (5.4) | 2 (4.3) |
| Reproductive system and breast disorders | 1 (1.1) | 3 (6.4) |
| Cardiac disorders | – | 3 (6.4) |
| Any SAE | 19 (20.4) | 1 (2.1) |
| SAE by system organ class | ||
| Infections and infestations | 5 (5.4) | – |
| Musculoskeletal and connective tissue disorders | 5 (5.4) | – |
| Gastrointestinal disorders | 3 (3.2) | 1 (2.1) |
| Investigations | 3 (3.2) | – |
| Surgical and medical procedures | 3 (3.2) | – |
| General disorders and administration site conditions | 2 (2.2) | – |
| Injury, poisoning and procedural complications | 1 (1.1) | – |
| Neoplasms benign, malignant and unspecified | 1 (1.1) | – |
| Nervous system disorders | 1 (1.1) | – |
| Respiratory, thoracic and mediastinal disorders | 1 (1.1) | – |
| Treatment-emergent AE | RTX + LEF (n = 93) | PL + LEF (n = 47) |
|---|---|---|
| Any AE | 66 (71.0) | 33 (70.2) |
| AEs occurring in ≥5% of subjects in either group | ||
| Infections and infestations | 37 (39.8) | 16 (34.0) |
| Nasopharyngitis | 11 (11.8) | 6 (12.8) |
| Urinary tract infection | 8 (8.6) | 2 (4.3) |
| Musculoskeletal and connective tissue disorders | 20 (21.5) | 11 (23.4) |
| RA | 6 (6.5) | 2 (4.3) |
| Vascular disorders | 15 (16.1) | 7 (14.9) |
| Hypertension | 11 (11.8) | 5 (10.6) |
| Gastrointestinal disorders | 12 (12.9) | 5 (10.6) |
| Diarrhoea | 7 (5.3) | 5 (10.6) |
| Investigations | 11 (11.8) | 3 (6.4) |
| Nervous system disorders | 6 (6.5) | 8 (17.0) |
| General disorders and administration site conditions | 7 (7.5) | 5 (10.6) |
| Injury, poisoning and procedural complications | 9 (9.7) | 2 (4.3) |
| Skin and s.c. tissue disorders | 7 (7.5) | 4 (8.5) |
| Blood and lymphatic system disorders | 5 (5.4) | 2 (4.3) |
| Metabolism and nutrition disorders | 3 (3.2) | 4 (8.5) |
| Respiratory, thoracic and mediastinal disorders | 5 (5.4) | 2 (4.3) |
| Reproductive system and breast disorders | 1 (1.1) | 3 (6.4) |
| Cardiac disorders | – | 3 (6.4) |
| Any SAE | 19 (20.4) | 1 (2.1) |
| SAE by system organ class | ||
| Infections and infestations | 5 (5.4) | – |
| Musculoskeletal and connective tissue disorders | 5 (5.4) | – |
| Gastrointestinal disorders | 3 (3.2) | 1 (2.1) |
| Investigations | 3 (3.2) | – |
| Surgical and medical procedures | 3 (3.2) | – |
| General disorders and administration site conditions | 2 (2.2) | – |
| Injury, poisoning and procedural complications | 1 (1.1) | – |
| Neoplasms benign, malignant and unspecified | 1 (1.1) | – |
| Nervous system disorders | 1 (1.1) | – |
| Respiratory, thoracic and mediastinal disorders | 1 (1.1) | – |
Data are presented as n (%). aSubjects could have more than one AE or SAE. AE: adverse event; PL: placebo; RTX: rituximab; SAE: serious adverse event.
Treatment-emergent adverse events over 24 weeksa
| Treatment-emergent AE | RTX + LEF (n = 93) | PL + LEF (n = 47) |
|---|---|---|
| Any AE | 66 (71.0) | 33 (70.2) |
| AEs occurring in ≥5% of subjects in either group | ||
| Infections and infestations | 37 (39.8) | 16 (34.0) |
| Nasopharyngitis | 11 (11.8) | 6 (12.8) |
| Urinary tract infection | 8 (8.6) | 2 (4.3) |
| Musculoskeletal and connective tissue disorders | 20 (21.5) | 11 (23.4) |
| RA | 6 (6.5) | 2 (4.3) |
| Vascular disorders | 15 (16.1) | 7 (14.9) |
| Hypertension | 11 (11.8) | 5 (10.6) |
| Gastrointestinal disorders | 12 (12.9) | 5 (10.6) |
| Diarrhoea | 7 (5.3) | 5 (10.6) |
| Investigations | 11 (11.8) | 3 (6.4) |
| Nervous system disorders | 6 (6.5) | 8 (17.0) |
| General disorders and administration site conditions | 7 (7.5) | 5 (10.6) |
| Injury, poisoning and procedural complications | 9 (9.7) | 2 (4.3) |
| Skin and s.c. tissue disorders | 7 (7.5) | 4 (8.5) |
| Blood and lymphatic system disorders | 5 (5.4) | 2 (4.3) |
| Metabolism and nutrition disorders | 3 (3.2) | 4 (8.5) |
| Respiratory, thoracic and mediastinal disorders | 5 (5.4) | 2 (4.3) |
| Reproductive system and breast disorders | 1 (1.1) | 3 (6.4) |
| Cardiac disorders | – | 3 (6.4) |
| Any SAE | 19 (20.4) | 1 (2.1) |
| SAE by system organ class | ||
| Infections and infestations | 5 (5.4) | – |
| Musculoskeletal and connective tissue disorders | 5 (5.4) | – |
| Gastrointestinal disorders | 3 (3.2) | 1 (2.1) |
| Investigations | 3 (3.2) | – |
| Surgical and medical procedures | 3 (3.2) | – |
| General disorders and administration site conditions | 2 (2.2) | – |
| Injury, poisoning and procedural complications | 1 (1.1) | – |
| Neoplasms benign, malignant and unspecified | 1 (1.1) | – |
| Nervous system disorders | 1 (1.1) | – |
| Respiratory, thoracic and mediastinal disorders | 1 (1.1) | – |
| Treatment-emergent AE | RTX + LEF (n = 93) | PL + LEF (n = 47) |
|---|---|---|
| Any AE | 66 (71.0) | 33 (70.2) |
| AEs occurring in ≥5% of subjects in either group | ||
| Infections and infestations | 37 (39.8) | 16 (34.0) |
| Nasopharyngitis | 11 (11.8) | 6 (12.8) |
| Urinary tract infection | 8 (8.6) | 2 (4.3) |
| Musculoskeletal and connective tissue disorders | 20 (21.5) | 11 (23.4) |
| RA | 6 (6.5) | 2 (4.3) |
| Vascular disorders | 15 (16.1) | 7 (14.9) |
| Hypertension | 11 (11.8) | 5 (10.6) |
| Gastrointestinal disorders | 12 (12.9) | 5 (10.6) |
| Diarrhoea | 7 (5.3) | 5 (10.6) |
| Investigations | 11 (11.8) | 3 (6.4) |
| Nervous system disorders | 6 (6.5) | 8 (17.0) |
| General disorders and administration site conditions | 7 (7.5) | 5 (10.6) |
| Injury, poisoning and procedural complications | 9 (9.7) | 2 (4.3) |
| Skin and s.c. tissue disorders | 7 (7.5) | 4 (8.5) |
| Blood and lymphatic system disorders | 5 (5.4) | 2 (4.3) |
| Metabolism and nutrition disorders | 3 (3.2) | 4 (8.5) |
| Respiratory, thoracic and mediastinal disorders | 5 (5.4) | 2 (4.3) |
| Reproductive system and breast disorders | 1 (1.1) | 3 (6.4) |
| Cardiac disorders | – | 3 (6.4) |
| Any SAE | 19 (20.4) | 1 (2.1) |
| SAE by system organ class | ||
| Infections and infestations | 5 (5.4) | – |
| Musculoskeletal and connective tissue disorders | 5 (5.4) | – |
| Gastrointestinal disorders | 3 (3.2) | 1 (2.1) |
| Investigations | 3 (3.2) | – |
| Surgical and medical procedures | 3 (3.2) | – |
| General disorders and administration site conditions | 2 (2.2) | – |
| Injury, poisoning and procedural complications | 1 (1.1) | – |
| Neoplasms benign, malignant and unspecified | 1 (1.1) | – |
| Nervous system disorders | 1 (1.1) | – |
| Respiratory, thoracic and mediastinal disorders | 1 (1.1) | – |
Data are presented as n (%). aSubjects could have more than one AE or SAE. AE: adverse event; PL: placebo; RTX: rituximab; SAE: serious adverse event.
Twenty-three SAEs were reported in the rituximab + LEF arm over a cumulative treatment duration of 37.5 patient-years (61.3 SAEs/100 patient-years), and 5 additional SAEs occurred after treatment discontinuation/during rescue treatment for a total of 28 SAEs in 19 (20%) subjects (Table 2; supplementary Fig. S2, available at Rheumatology online). In the placebo + LEF arm, two SAEs occurred over 18.4 patient-years (10.9 SAEs/100 patient-years; 2% of subjects). The most common SAEs in rituximab + LEF subjects by system organ class were infections and infestations [five events in five subjects (5.4%): urosepsis, gastroenteritis, respiratory tract infection, erysipelas and otitis media] and musculoskeletal and connective tissue disorders [six events in five subjects (5.4%): intervertebral disc protrusion, muscular weakness, osteochondrosis, rotator cuff syndrome, tendon calcification, and arthropathy/OA). Of the six patients who fulfilled SAE criteria due to underlying infection, one was treated with oral antibiotics, four were treated with i.v. antibiotics after hospitalization and one was hospitalized without need for antibiotic treatment. None of the patients developed septicaemia. SAEs were considered treatment-related in eight rituximab + LEF subjects. Four of the infections/infestations and none of the musculoskeletal and connective tissue disorders were considered treatment-related. There was no obvious pattern of SAEs by preferred term, and none was reported in more than one subject. One case of neoplasm (gastric neoplasm) occurred in the rituximab + LEF arm. The lesion was successfully resected and no signs of lymph node metastasis or tissue infiltration were observed. No deaths occurred during the study period. One subject in the rituximab + LEF arm died 15 weeks after the last study visit and 37 weeks after the last administration of rituximab. The death was declared to be due to natural causes and no post-mortem examination was performed.
As expected, the rituximab + LEF arm showed rapid and marked B cell depletion as indicated by a decrease in peripheral CD19+/CD20+ cells from baseline levels of 10 cells/µl to <1 cell/µl from weeks 2 through 24 (data not shown). The development of hypogammaglobulinaemia was monitored at baseline, before re-treatment and at study discontinuation. One patient showed a slightly reduced IgG level of 6.1 g/l (normal range 7–16 g/l), but this was not considered clinically significant.
Discussion
Rituximab is among the most frequently used biologic DMARDs in Germany [21], so information on possible concomitant therapies is highly relevant to everyday treatment of patients with RA. Although the combination of rituximab and LEF is used in clinical practice [9–13], there is insufficient information on this combination to guide rheumatologists. In this study, we conducted a randomized, investigator-initiated, multicentre, placebo-controlled clinical trial to obtain data on the efficacy and safety of combination therapy with rituximab + LEF in patients with RA; this is the first randomized trial to evaluate objective and subjective outcomes associated with this treatment regimen. The combination of rituximab + LEF failed to meet the primary outcome. However, other endpoints suggested a beneficial effect for rituximab added to ongoing LEF, including significant improvements in ACR20 response rates, the primary endpoint in some rituximab trials [22, 23], at weeks 12 through 24, ACR50 response rates at week 16, and ACR70 response rates at weeks 8 and 16. An improved therapeutic response in the rituximab + LEF arm was also supported by significant improvements in DAS28 and in the SF-36 dimensions of bodily pain and emotional role functioning.
On the basis of earlier reports of increased rituximab response rates in seropositive RA [24, 25], we evaluated the effect of anti-CCP seropositivity on ACR response rates. In the rituximab + LEF arm, ACR response rates were consistently higher in anti-CCP-seropositive patients compared with anti-CCP-seronegative patients, with statistically significant differences for most evaluations. A similar effect was not observed in the placebo + LEF arm. However, multivariable regression analysis did not identify anti-CCP seropositivity as a significant variable for the primary outcome.
The major limitation of this study is that it was likely underpowered for the predefined primary outcome of ACR50 response at week 24. ACR50 was chosen as the primary endpoint based on a previous RA study in which rituximab + MTX resulted in a significantly higher rate of ACR50 responses at week 24 compared with MTX monotherapy [2], along with findings from noninterventional studies in which a higher level of effectiveness was observed with rituximab + LEF compared with rituximab + MTX or rituximab monotherapy [10, 26]. However, enrolment in this trial was lower than expected, and dropouts were higher (>30% rather than the estimated 10%), leading to a need to readjust the statistical power to 80%. Difficulties in enrolling and the high dropout rate were due in part to publication of a report on an association between LEF and pancreatic cancer [19], which was not confirmed in a subsequent analysis [20], and to the rituximab manufacturer’s decision not to apply for a first-line biologic DMARD RA indication during the study. As a result of this decision, the European Medicines Agency indication for rituximab required patients to first fail therapy with a TNF inhibitor, a requirement met by only 11% of patients enrolled in this trial. Biologic-naïve patients who enrolled in the study were not eligible to receive post-study reimbursement for rituximab under most healthcare plans, which likely discouraged potential participants from enrolling. The high discontinuation rate potentially affected the statistical power and reliability of our findings. We estimated discontinuation rates based on older (2004 and 2006) studies in RA. More recent studies show higher rates of discontinuation (30% for LEF or MTX + TNF inhibitors at 24 weeks [8]; 26% for rituximab, 52% for TNF inhibitors and 49% for abatacept at 1 year [27]) that are more in line with the rates reported here. The ACR50 response rate at week 24 in the rituximab + LEF arm was lower than expected (27 vs 43% in a study of rituximab + MTX) [2], but similar to the ACR50 rate for rituximab + MTX in patients previously treated with anti-TNF agents (27%) [18]. The study with the 43% ACR50 response rate involved 40 patients with higher disease activity than our patient cohort (mean DAS28 of 6.8 vs 5.6) [2]. Because patients in the previous study had more room for improvement, they may have had an increased likelihood of achieving an ACR50 response, as has been observed in other studies [28]. Our data are more likely to reflect outcomes representing daily practice during an era when multiple effective RA therapies are available and treat-to-target strategies are frequently employed.
It is also possible that our choice of a primary outcome contributed to the null findings [28]. Although ACR improvement criteria are frequently used as an efficacy endpoint in RA trials, simulation models have shown that dichotomized responses such as responder analyses require a large population of patients in order to detect a therapeutic difference [29]; >200 patients are required to detect a therapeutic difference in ACR20 in RA patients with late disease [30]. The likelihood of finding a positive effect was made more difficult by our choice of the stringent criteria of ACR50, as well as a week 24 timepoint, the time at which the rituximab prescribing information suggests considering a further course of treatment. Our data suggest that the efficacy of rituximab + LEF reaches its highest level at week 16, after which the effect may plateau or wane. These combined challenges likely resulted in a patient cohort that was insufficient in size to show a significant difference in ACR50 response rate at week 24. However, the lack of a positive finding for the primary objective does not negate the clinical benefit observed in our study. Similar to other studies in rheumatology that failed to meet the primary endpoint [31], our data support clinical consideration of the treatment regimen and additional studies to verify its safety and efficacy.
The AE profiles of the two treatment arms were generally similar and no unexpected AEs were observed. However, the SAE rate for rituximab + LEF was higher than expected (20 vs 2% for placebo), even when calculated as events per 100 patient-years to take into account longer study continuation in the rituximab arm (61.3 vs 10.9). For comparison, studies of rituximab + MTX reported SAE rates of 5–7% [18, 22]. The majority of SAEs observed with rituximab + LEF were not considered treatment-related. The higher SAE rate in the rituximab + LEF arm was driven in part by serious infections, which occurred in 5.4% of patients vs 0% for placebo. This serious infection rate is similar to the rate for rituximab in a study from the British Society for Rheumatology Biologics Register for RA, which found comparable serious infection rates for rituximab (5.8%) and TNF inhibitors (4.8%) [32]. Additional studies are required to evaluate AEs occurring in patients treated with rituximab + LEF. Until more information becomes available, patients treated with rituximab + LEF should be counselled about the potential for SAEs, particularly infections, and monitored carefully.
In summary, combination therapy with rituximab + LEF did not achieve the primary efficacy outcome. However, key secondary outcomes suggest this may be an effective treatment regimen for some patients with active RA and an inadequate response to LEF alone. In particular, this regimen may provide a valuable treatment option for RA patients who are likely to benefit from rituximab, but are intolerant to or have contraindications against MTX. Given the unexplained increase in SAEs in patients treated with this combination, we recommend close monitoring of treated patients.
Acknowledgements
We wish to thank the participating patients and study centre personnel. Medical writing support was provided by Sharon L. Cross, PhD, in consultation with the authors on behalf of and funded by Fraunhofer IME, Translational Medicine & Pharmacology TMP, Frankfurt am Main, Germany. These data were presented in part at the 80th Annual Scientific Meeting of the ACR in Washington, DC, November 2016, the Annual European Congress of Rheumatology in London, UK, June 2016, and the Annual European Congress of Rheumatology in Amsterdam, the Netherlands, June 2018. F.B., M.K., T.R., M.A., G.R.B., E.F., A.R.-R., H.-P.T., S.W. and H.B. designed, the study. F.B., M.K., T.R., R.A, M.A., M.B., G.R.B, E.F., H.K., K.K., U.M.-L., A.R.-R., H.-P.T., S.W. and H.B. acquired the data. E.H. and A.L. led the statistical analyses. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
Funding: This work was supported by Roche Pharma, Germany; they had no influence on the design or conduct of this investigator-initiated trial and were not involved in interpretation of the data. Medical writing services were supported by Fraunhofer IME, Translational Medicine & Pharmacology TMP, Frankfurt am Main, Germany, a nonprofit organization. The clinical research group Frankfurt is supported by the LOEWE-Center TMP of the state of Hesse (Germany), the ArthroMark Consortium, funded by German Federal Ministry of Education and Research (BMBF 01EC1401C Project 4) and the Fraunhofer Cluster of Excellence for Immune-Mediated Diseases CIMD.
Disclosure statement: F.B., M.K., T.R. and H.B. are employees of Fraunhofer Institute IME, which performed the trial in cooperation with Klinisches Studienzentrum Rhein-Main at University Hospital Frankfurt. F.B., M.K. and T.R. received research grants from Roche. R.A. received personal fees from Abbvie, Bristol Myers Squibb (BMS), Celltrion, Gilead, Lilly and Pfizer. M.A. and G.R.B. received personal fees from Roche. E.F. received speakers and consulting fees from Abbvie, Pfizer, Roche/Chugai, Union Chimique Belge (UCB), BMS, Celgene, Merck Sharp & Dohme (MSD), Novartis, Sobi, Sanofi and Lilly. U.M.-L. served as a speaker/advisor for Roche and Medac. A.R.-R. received speakers and consulting fees from Roche, Sanofi, Novartis, Abbvie, Lilly, MSD and UCB. H.-P.T. received speakers and consulting fees from Abbvie, Chugai, Janssen Cilag, Lilly, Novartis, BMS, Gilead, Roche and Sandoz Hexal. S.W. received speakers and consulting fees from Abbvie, Amgen, BMS, Celgene, Gilead, Lilly, Hexal, MSD, Nichi-Iko, Pfizer and Sanofi. H.B. received consultancy fees from Roche. E.H., H.K., K.K., A.L. and M.B. have no competing interests to declare.
Data availability statement
Data from this study are available to researchers who provide a methodologically sound collaborative proposal. Proposals should be directed to the corresponding author.
Supplementary data
Supplementary data are available at Rheumatology online.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments