





The imaging features of multicentric Castleman disease and IgG4-related kidney disease can be very similar.
DEAR EDITOR, Human herpesvirus-8 (HHV-8)-negative/idiopathic multicentric Castleman disease (iMCD) is a systemic inflammatory disorder showing multicentric lymphadenopathy with characteristic histopathology and multiple organ dysfunction due to a cytokine storm often including IL-6 [1]. iMCD encompasses hyaline vascular/hypervascular to plasmacytic histopathologic subtypes. Elevated serum immunoglobulin G4 (IgG4) levels and tissue infiltrating IgG4-positive (IgG4+) plasma cells (PCs) are sometimes found in iMCD [2, 3], making it difficult to distinguish it from IgG4-related disease (IgG4-RD), even after publication of the 2019 ACR/EULAR classification criteria for IgG4-RD [4, 5]. IgG4-RD generally has lower IgA, IL-6 and CRP, and decreased serum albumin and haemoglobin levels are important features of iMCD while these are usually normal in IgG4-RD [3]. Although MCD is included in the exclusion criteria of the new classification criteria [4, 5], it is sometimes difficult to differentiate these diseases in daily clinical practice because neither has a highly specific diagnostic biomarker. Herein, we describe two cases of MCD with imaging features very similar to those of IgG4-related kidney disease.
Patient 1 was a 53-year-old woman admitted due to renal insufficiency. Six years earlier, 18F-fluorodeoxyglucose positron emission tomography (FDG-PET)-positive bilateral cervical lymphadenopathy was noted. Lymph node (LN) biopsy showed regressed germinal centres (GCs) with sheet-like interfollicular plasmacytosis, consistent with the plasmacytic histopathologic subtype of iMCD [1], and many IgG4+PCs (IgG4+PC 155/hpf; IgG4/IgG 30%) (Fig. 1C and E). Two years following the iMCD diagnosis, proteinuria appeared and renal function gradually decreased. She had moderate anaemia (Hb 7.6 g/dl) with hypoalbuminemia (Alb 3.1 g/dl). Her serum IgG on admission was 8224 mg/dl (normal: 870–1700), IgA 949 mg/dl (110–410), IgG4 625 mg/dl (<135), IL-6 15.9 pg/ml (<4.0) and CRP 5.4 mg/dl (<0.3). Contrast-enhanced (CE) CT showed multicentric lymphadenopathy with bilateral swollen kidneys and left renal pelvic wall thickening (Fig. 1A). Renal biopsy revealed copious lymphoplasmacytic infiltration, predominantly IgG4+PCs (IgG4+PC 50/hpf; IgG4/IgG 32%) but no fibrosis. Prednisolone at 25 mg daily was not effective, so tocilizumab was added, inducing complete symptomatic improvement and partial improvement of renal function; the serum IL-6 level remained very high (830 pg/ml).
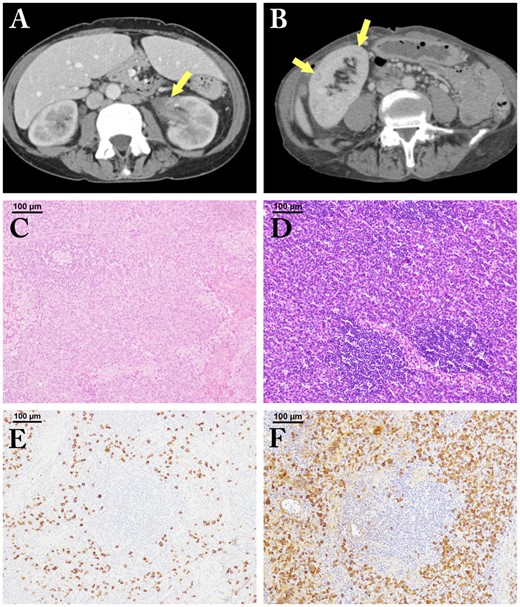
The imaging findings of kidney lesions and histopathological findings of lymph node lesions
Patient 1 (A, C, E). Patient 2 (B, D, F). A: left renal pelvic wall thickening is seen (arrow); B: multiple low-density lesions in the right kidney are noted (arrows); C: regressed germinal centres (GCs) with sheet-like plasmacytosis (hematoxylin and eosin staining; 100μm); D: burned out GCs with sheet-like plasmacytosis (hematoxylin and eosin staining; 100μm); E, F: many IgG4-positive plasma cell infiltrates are seen (IgG4-immunostaining; 100μm).
Patient 2 was a 67-year-old woman admitted because of respiratory failure. Twelve years earlier, a lung biopsy had revealed plasmacytosis. Treatment with 30 mg prednisolone daily was started, but discontinued shortly thereafter. Four years later, steroids were re-started when autoimmune haemolytic anaemia appeared. She had moderate anaemia (Hb 7.8 g/dl) with hypoalbuminemia (Alb 3.1 g/dl). Her serum IgG on admission was 5669 mg/dl, IgA 777 mg/dl, IgG4 738 mg/dl, CRP 5.1 mg/dl and eGFR 42.4 ml/min/1.73m2. CE-CT showed multicentric lymphadenopathy. In the lung, multiple patchy shadows accompanied by ground-glass opacities, thickening of interlobular septa, multiple cysts and bronchiectasis were seen. Multiple low-density lesions were present in the bilateral kidneys (Fig. 1B). On LN biopsy, GCs showed regressive changes with sheet-like interfollicular plasmacytosis, consistent with the plasmacytic sub-type of iMCD (Fig. 1D). However, most infiltrating PCs were IgG4-positive (IgG4+PC 323/hpf; IgG4/CD138 67%) (Fig. 1F). Renal biopsy revealed marked lymphoplasmacytic infiltration (IgG4+PC 20/hpf; IgG4/IgG 25%) with atrophic tubuli. We tentatively diagnosed IgG4-RD and 40 mg prednisolone daily was started. However, the response was only partial and anaemia and severe fatigue developed. Therefore, we re-diagnosed the patient with iMCD. We added tocilizumab to 13 mg prednisolone daily, resulting in complete control of inflammation, anaemia and fatigue, but the serum IL-6 level remained very high (630 pg/ml).
Distinguishing IgG4-RD and iMCD is extremely important as their clinical courses and treatments differ greatly; iMCD often requires anti-IL-6 and/or more potent immunosuppressive/immunoablative agents [6]. However, distinguishing these diseases is often difficult, particularly when iMCD has many IgG4+PC infiltrates histopathologically. In our cases, we noted iMCD LN histopathology and overlapping abnormalities, including copious IgG4+PC infiltrates in kidneys and LNs and elevated serum IgG4 levels. Moreover, these patients have very similar imaging features with IgG4-related kidney disease; i.e., renal pelvic thickening/soft tissue (numerical weight +8 in the classification criteria [4, 5]) and bilateral renal cortex low-density areas (+10) respectively. In contrast, some of the pulmonary imaging findings in case 2 were partially very similar to those of IgG4-RD, i.e. thickening of interlobular septa, whereas multiple lung cysts are the distinguishing feature in iMCD. Storiform fibrosis, a hallmark of IgG4-RD, was not observed. Considering the classification criteria and available data, we consider these cases iMCD with features of IgG4-RD. Glucocorticoid resistance and persistently elevated CRP levels strongly support this distinction.
IL-6 levels continued to increase after symptomatic improvement on tocilizumab, surpassing 100-fold the upper limit of normal (<4 pg/ml) at follow-up. Persistent IL-6 elevation after complete response on tocilizumab has been reported in iMCD [7], whereas IL-6 levels return to normal in rheumatoid arthritis with tocilizumab-induced remission [8] and have not been reported elevated in IgG4-RD. This may further support that these are iMCD cases rather than IgG4-RD and may point to a difference in IL-6’s role in them.
These cases highlight the challenge of distinguishing iMCD and IgG4-RD and the need for further research into optimal diagnostic biomarkers and treatment approaches.
Funding: No specific funding was received from any funding bodies in the public, commercial or not-for-profit sectors to carry out the work described in this manuscript.
Disclosure statement: D.C.F. has received research funding from Janssen Pharmaceuticals and EUSA Pharma for the ACCELERATE natural history registry. The remaining authors have declared no conflicts of interest.
Acknowledgement
We thank John Gelblum for his critical reading of the manuscript.

{kind=link}
Comments