





Abstract
3-hydroxy-3-methylglutaryl coenzyme-A (HMG Co-A) reductase inhibitors (statins) are standard treatment for hyperlipidaemia. In addition to lipid-lowering abilities, statins exhibit multiple anti-inflammatory effects. The objectives of this study were to determine whether treatment of patients with RA with lovastatin decreased CRP or reduced disease activity.
We conducted a randomized double-blind placebo-controlled 12 week trial of lovastatin vs placebo in 64 RA patients with mild clinical disease activity but an elevated CRP. The primary efficacy end point was the reduction in mean log CRP. Secondary end points included disease activity, RF and anti–CCP antibody titres. Mechanistic end points included levels of serum cytokines. Safety was assessed; hepatic and muscle toxicities were of particular interest.
Baseline features were similar between groups. No significant difference in mean log CRP reduction between the two groups was observed, and disease activity did not change from baseline in either treatment group. Mechanistic analyses did not reveal significant changes in any biomarkers. A post hoc analysis of subjects not using biologic therapy demonstrated a significantly greater proportion achieving ⩾20% reduction in CRP from baseline in the lovastatin group compared with placebo (P-value = 0.007). No difference was observed in subjects receiving biologics. Lovastatin was well tolerated with no serious safety concerns.
This study showed no anti-inflammatory or clinical effects on RA disease activity after 12 weeks of treatment with lovastatin. Lovastatin had a modest effect on CRP in subjects not using biologics, suggesting statins may be anti-inflammatory in selected patients.
ClinicalTrials.gov, http://clinicaltrials.gov, NCT00302952.
Statins (HMG Co-Reductase Inhibitors) have multiple properties beyond cholesterol reduction, which include anti-inflammatory actions.
12 weeks of lovastatin in patients with mildly active RA had no adjunctive therapeutic effects.
RA patients who were not receiving biologic therapies achieved ⩾20% reduction in CRP with lovastatin.
Introduction
3-hydroxy-3-methylglutaryl coenzyme-A (HMG Co-A) reductase inhibitors (statins) lower lipid levels, reduce cardiovascular events and mortality, and have numerous anti-inflammatory and immunomodulatory properties. Statins decrease production of inflammatory chemokines and cytokines by T cells and macrophages, decrease the viability of plasma cells [1–4], and inhibit endothelial cell activation and angiogenesis [5–7]. In individuals with normal lipid profiles, statins reduce inflammatory markers and improve cardiovascular outcomes [8, 9]. These anti-inflammatory effects are attributed to reductions in mevalonate, a cholesterol precursor. Mevalonate is also a precursor for isoprenoid intermediates required for the functioning of guanosine triphosphatases (GTPases), Ras, Rho and Rab, which control cell behaviour through signal transduction pathways. In addition, statins, particularly lovastatin, may sterically inhibit the interaction of lymphocyte function-associated antigen (LFA-1) with its ligand intracellular adhesion molecule (ICAM-1) in a mevalonate-independent manner [10].
Given these properties, we conducted a prospective trial evaluating the anti-inflammatory effect and efficacy of lovastatin, a statin with potent in vivo and in vitro anti-inflammatory properties, as a non-toxic adjunct therapy in RA patients with mild clinical disease activity. This study examined the short-term effects of exposure to lovastatin on serum CRP, disease activity and a number of RA-related biologic markers.
Methods
In this multicentre, double-blind, Phase II trial (ClinicalTrials.gov Identifier NCT00302952), subjects with RA, with mild clinical disease activity, were randomized (1:1) to receive placebo or 80 mg lovastatin daily for 12 weeks. A dose of 80 mg/day of lovastatin was based upon drug concentrations used in in vitro studies. The primary objective was to examine the effect of lovastatin on CRP. Secondary objectives included evaluating the effects of lovastatin on disease activity, as well as assessing safety and tolerability. Disease activity was measured using DAS28-CRP, and clinical response was determined by ACR20 response and DAS28-CRP EULAR (European League Against Rheumatism) response indices [11, 12])]. Mechanistic objectives included exploring effects of lovastatin on RF and anti-CCP autoantibody titres, inflammatory mediators and pathways, and autoreactive B cells.
Subjects meeting 1987 ACR classification criteria for RA with mildly active clinical disease, defined by joint counts (2–8 tender joints and 1–6 swollen joints) and an elevated CRP (>5 mg/l) were recruited. DMARD and/or biologic therapy and/or stable prednisone ⩽10 mg/day were permitted; however, the addition or increase of medications for RA disease activity during the study was prohibited. Exclusion criteria included statin use, infection, myositis, treatment with medications metabolized using the cytochrome P3A4 pathway, elevated creatinine phosphokinase, serum alanine aminotransaminase, aspartate aminotransaminase, or serum creatinine, pregnancy or ACR Functional Status Class IV. Treatment with infliximab within 3 months of screening or prior treatment with rituximab were also exclusions due to concerns about a loss of drug effect and a subsequent rebound of disease activity occurring during a subject’s participation in the clinical trial. Institutional Review Boards approved the study at each site and the NIAID Autoimmune Data and Safety Monitoring Board provided study oversight. All participants provided informed consent prior to initiation of study procedures.
Eligible subjects were randomized using an adaptive randomization scheme to ensure balance on key baseline characteristics [13]: DAS28-CRP, race, MTX use, anti-TNF use, and disease duration.
Per protocol, temporary discontinuation of study treatment or dose adjustments were allowed for elevations in transaminase or creatinine phosphokinase levels, study drug intolerance, development of a condition that increased the risk for statin-related myopathy, or an adverse event (AE). Upon resolution, the study drug could be resumed and continued at 40 mg/day.
Laboratory assessments
Local laboratories performed screening CRP assessments. Sera for subsequent CRP measurements were batched and assessed centrally. At baseline and end-of-study, lipid levels, RF, anti-CCP antibodies, and a panel of 18 potential RA biomarkers were evaluated centrally. RF was measured by an ELISA using human IgG fragment crystallizable (IgG Fc) (Southern Biotech, Birmingham, AL) [14]. Anti-CCP was measured using the QUANTA Lite CCP3 IgG assay (QUANTA Lite, Inova Diagnostics Inc, Davis CA). Analytes on the biomarker panel (MIP1α, G-CSF, IFNγ, IL1β, ICAM-1, IL6, OPG, VCAM-1, IL12p70, IL10, IL17A, RANTES, TNF, RANKL, MCP-1, IL1RA, E-selectin, and BAFF) were measured using a magnetic bead multiplexed assay (Affymetrix, San Diego CA).
IgM-secreting B cells were enumerated in Dr Davidson’s laboratory at the Feinstein Institute by ELISpot (Enzyme-Linked ImmunoSpot) on a subset of 9 subjects (6 placebo, 3 lovastatin) enrolled at the Feinstein Institute, using fresh peripheral blood mononuclear cells (PBMCs) [15]. Statins inhibit the release of monocyte chemoattractant protein-1 (MCP-1) from PBMCs following mitogen stimulation in a mevalonate-dependent manner [16]. We conducted additional mechanistic studies to evaluate this anticipated effect. PBMCs obtained from the same subset of 9 subjects on days 0 and 84 were treated with lipopolysaccharide (LPS) 100 ng/ml (Sigma Aldrich, St Louis, MO) in the presence of lovastatin10 uM (Teva Pharmaceuticals, Parsippany, NJ), mevalonate 100 µM (Sigma Aldrich, St Louis, MO) or lovastatin + mevalonate. Control wells received no stimulation. Supernatants were harvested after 24 hours and tested for MCP-1 by ELISA (eBioscience, San Diego CA).
Sample size and statistical analyses
The primary efficacy outcome was the change in mean log CRP from baseline to day 84. A 50% reduction in CRP was observed in a study of atorvastatin in RA (TARA). In TARA, the mean (standard deviation) of log CRP was 2.5 (1.2) log mg/l at baseline [17]. After 6 months of treatment, the difference in mean change in log CRP between groups was −0.58. Assuming a consistent standard deviation over time (1.2 log mg/l) and a correlation of 0.75 between baseline and follow-up, 34 subjects per arm gives 80% power to detect the identical group difference using a 2-sided test with α = 0.05. The planned enrolment was 40 per arm to account for potential dropouts.
Pre-specified secondary end points included DAS28-CRP, DAS28-CRP EULAR response, ACR20 response, RF, anti-CCP antibody titres, and serum concentrations of biomarkers. In addition, achievement of a 15% reduction of CRP was pre-specified for this trial, as a reduction of this magnitude was previously observed in a trial of lovastatin for primary prevention of coronary events [9]. A 20% reduction of CRP was added post hoc given its role in contributing to the achievement of an ACR20 response [11].
A modified intent-to-treat (mITT) population was defined as randomized participants receiving at least one dose of study drug and possessing a baseline CRP. The primary efficacy analysis included mITT subjects whose visit at day 84 (14 days) was within 7 days of their last dosing day. Secondary analyses included mITT subjects with available data. Analysis of adverse events used the safety population, consisting of all participants receiving at least 1 dose of study drug.
For the primary end point, an analysis of covariance model was used to compare treatment groups after adjusting for baseline log CRP, baseline DAS28-CRP, race, MTX use, anti-TNF use, and disease duration. Summary statistics for CRP are presented on the untransformed scale. Continuous secondary efficacy end points were analysed in a similar fashion except that only covariates significant at the 0.05 level were included in the final models. Categorical end points were evaluated using Pearson’s χ2 test or a Fisher’s exact test. Changes in lipid profiles from baseline were evaluated using t-tests. Post hoc analyses included comparisons between treatment arms in subjects using and not using biologic agents.
The primary hypothesis was evaluated at the 0.05 level of significance. Secondary analyses were considered exploratory; P-values are presented without adjustment for multiple comparisons.
Results
A total of 132 subjects were screened at 15 centres, and 64 were randomized (Fig. 1). The study was prematurely terminated because of slow enrolment and study drug expiration; patients meeting the definition of mildly active clinical disease and sufficiently elevated CRP proved difficult to identify. The safety population included all 64 randomized subjects (34 lovastatin, 30 placebo). The mITT population comprised 63 participants (34 lovastatin, 29 placebo); 55 (30 lovastatin, 25 placebo) of which met requirements for inclusion in the primary analysis. Thirty (88.2%) lovastatin and 24 (82.8%) placebo subjects received ⩾80% of expected doses.
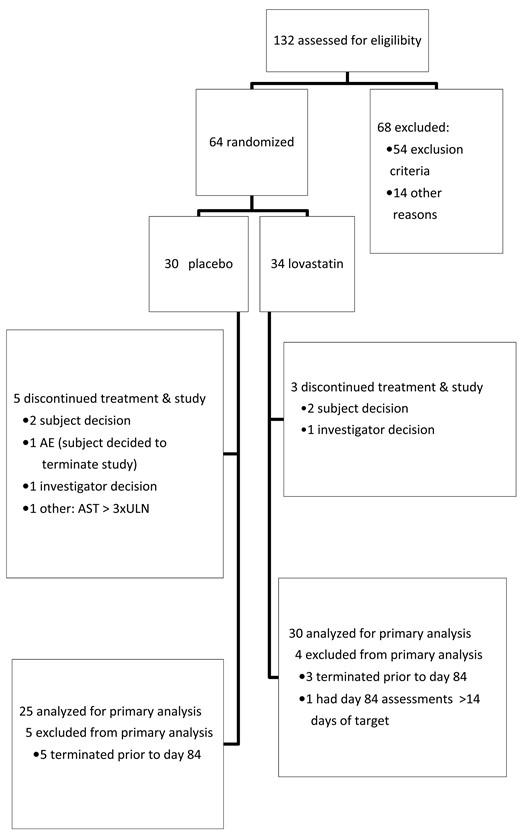
Flow chart showing disposition of the study subjects
AE: adverse event; AST: serum aspartate transaminase; ULN: upper limit of normal.
Baseline characteristics
Baseline characteristics of subjects were consistent with expectations for a RA population with mild clinical activity (Table 1). Study participants had mildly active clinical disease with a mean of 4.6 tender joints and 3.6 swollen joints. The mean (s.d.) baseline CRP levels were 12.2 (11.42) and 12.6 (16.43) mg/l for lovastatin and placebo groups, respectively. DAS-28 scores were similar in the lovastatin and placebo groups (3.9 and 4.2 respectively). As DAS-28 incorporates measurement of an acute phase reactant (i.e. CRP), which was required to be elevated for study entry, patients in this study had mild to moderate DAS-defined RA disease activity. The treatment groups were similar except for biologic use, which was numerically higher in the lovastatin arm (58.8%) than in the placebo arm (37.9%).
Demographic and baseline characteristics by treatment group (mITT)
| Parameter | Lovastatin | Placebo |
|---|---|---|
| (N = 34) | (N = 29) | |
| Agea | 55.8 (7.1) | 52.6 (10.4) |
| Female (%) | 32 (94.1) | 28 (96.6) |
| Hispanic (%) | 6 (17.6) | 8 (27.6) |
| Race, White (%) | 26 (76.5) | 20 (69.0) |
| Black (%) | 6 (17.6) | 5 (17.2) |
| Other (%) | 2 (5.9) | 4 (13.8) |
| Disease duration (years)a | 12.5 (9.6) | 11.9 (12.6) |
| Current biologic use (%) | 20 (58.8) | 11 (37.9) |
| CRP (mg/l)a | 12.2 (11.4) | 12.6 (16.4) |
| Tender joint counta | 4.4 (2.5) | 4.8 (2.2) |
| Swollen joint counta | 3.5 (1.4) | 3.8 (1.6) |
| Patient Global Disease Activitya | 3.5 (2.3) | 4.5 (1.6) |
| DAS28a | 3.9(0.7) | 4.2 (0.4) |
| ACR functional class, I (%) | 20 (58.8) | 15 (51.7) |
| II (%) | 12 (35.3) | 9 (31.0) |
| III (%) | 2 (5.9) | 5 (17.2) |
| Cholesterola | 190 (39.2) | 196 (31.0) |
| LDL-cholesterola | 117 (31.0) | 120 (26.9) |
| HDL-cholesterola | 48 (14.4) | 51.4 (14.3) |
| Triglyceridea | 125 (73.1) | 124.7 (60.0) |
| Parameter | Lovastatin | Placebo |
|---|---|---|
| (N = 34) | (N = 29) | |
| Agea | 55.8 (7.1) | 52.6 (10.4) |
| Female (%) | 32 (94.1) | 28 (96.6) |
| Hispanic (%) | 6 (17.6) | 8 (27.6) |
| Race, White (%) | 26 (76.5) | 20 (69.0) |
| Black (%) | 6 (17.6) | 5 (17.2) |
| Other (%) | 2 (5.9) | 4 (13.8) |
| Disease duration (years)a | 12.5 (9.6) | 11.9 (12.6) |
| Current biologic use (%) | 20 (58.8) | 11 (37.9) |
| CRP (mg/l)a | 12.2 (11.4) | 12.6 (16.4) |
| Tender joint counta | 4.4 (2.5) | 4.8 (2.2) |
| Swollen joint counta | 3.5 (1.4) | 3.8 (1.6) |
| Patient Global Disease Activitya | 3.5 (2.3) | 4.5 (1.6) |
| DAS28a | 3.9(0.7) | 4.2 (0.4) |
| ACR functional class, I (%) | 20 (58.8) | 15 (51.7) |
| II (%) | 12 (35.3) | 9 (31.0) |
| III (%) | 2 (5.9) | 5 (17.2) |
| Cholesterola | 190 (39.2) | 196 (31.0) |
| LDL-cholesterola | 117 (31.0) | 120 (26.9) |
| HDL-cholesterola | 48 (14.4) | 51.4 (14.3) |
| Triglyceridea | 125 (73.1) | 124.7 (60.0) |
Mean (s.d.).
Note: The biologic agents used by patients in this trial were: abatacept, adalimumab, betaseron, certolizumab pegol, etanercept, and ustekinumab. Betaseron was used in a patient with concomitant multiple sclerosis, and ustekinumab was used in a patient with psoriasis with no evidence of PsA.
LDL: low-density lipoprotein; HDL: high-density lipoprotein; mITT: modified intent-to-treat.
Demographic and baseline characteristics by treatment group (mITT)
| Parameter | Lovastatin | Placebo |
|---|---|---|
| (N = 34) | (N = 29) | |
| Agea | 55.8 (7.1) | 52.6 (10.4) |
| Female (%) | 32 (94.1) | 28 (96.6) |
| Hispanic (%) | 6 (17.6) | 8 (27.6) |
| Race, White (%) | 26 (76.5) | 20 (69.0) |
| Black (%) | 6 (17.6) | 5 (17.2) |
| Other (%) | 2 (5.9) | 4 (13.8) |
| Disease duration (years)a | 12.5 (9.6) | 11.9 (12.6) |
| Current biologic use (%) | 20 (58.8) | 11 (37.9) |
| CRP (mg/l)a | 12.2 (11.4) | 12.6 (16.4) |
| Tender joint counta | 4.4 (2.5) | 4.8 (2.2) |
| Swollen joint counta | 3.5 (1.4) | 3.8 (1.6) |
| Patient Global Disease Activitya | 3.5 (2.3) | 4.5 (1.6) |
| DAS28a | 3.9(0.7) | 4.2 (0.4) |
| ACR functional class, I (%) | 20 (58.8) | 15 (51.7) |
| II (%) | 12 (35.3) | 9 (31.0) |
| III (%) | 2 (5.9) | 5 (17.2) |
| Cholesterola | 190 (39.2) | 196 (31.0) |
| LDL-cholesterola | 117 (31.0) | 120 (26.9) |
| HDL-cholesterola | 48 (14.4) | 51.4 (14.3) |
| Triglyceridea | 125 (73.1) | 124.7 (60.0) |
| Parameter | Lovastatin | Placebo |
|---|---|---|
| (N = 34) | (N = 29) | |
| Agea | 55.8 (7.1) | 52.6 (10.4) |
| Female (%) | 32 (94.1) | 28 (96.6) |
| Hispanic (%) | 6 (17.6) | 8 (27.6) |
| Race, White (%) | 26 (76.5) | 20 (69.0) |
| Black (%) | 6 (17.6) | 5 (17.2) |
| Other (%) | 2 (5.9) | 4 (13.8) |
| Disease duration (years)a | 12.5 (9.6) | 11.9 (12.6) |
| Current biologic use (%) | 20 (58.8) | 11 (37.9) |
| CRP (mg/l)a | 12.2 (11.4) | 12.6 (16.4) |
| Tender joint counta | 4.4 (2.5) | 4.8 (2.2) |
| Swollen joint counta | 3.5 (1.4) | 3.8 (1.6) |
| Patient Global Disease Activitya | 3.5 (2.3) | 4.5 (1.6) |
| DAS28a | 3.9(0.7) | 4.2 (0.4) |
| ACR functional class, I (%) | 20 (58.8) | 15 (51.7) |
| II (%) | 12 (35.3) | 9 (31.0) |
| III (%) | 2 (5.9) | 5 (17.2) |
| Cholesterola | 190 (39.2) | 196 (31.0) |
| LDL-cholesterola | 117 (31.0) | 120 (26.9) |
| HDL-cholesterola | 48 (14.4) | 51.4 (14.3) |
| Triglyceridea | 125 (73.1) | 124.7 (60.0) |
Mean (s.d.).
Note: The biologic agents used by patients in this trial were: abatacept, adalimumab, betaseron, certolizumab pegol, etanercept, and ustekinumab. Betaseron was used in a patient with concomitant multiple sclerosis, and ustekinumab was used in a patient with psoriasis with no evidence of PsA.
LDL: low-density lipoprotein; HDL: high-density lipoprotein; mITT: modified intent-to-treat.
Overall efficacy
CRP levels did not change appreciably from baseline to day 84 [mean (s.e.) for change in CRP (mg/l): −2.0 (1.2) lovastatin; −2.2 (1.0) placebo (Table 2)]. Treatment groups did not differ significantly after adjustment for baseline CRP and pre-specified covariates (P-value = 0.8, primary analysis). Subjects with higher baseline disease activity DAS or log CRP did tend to have a greater decline in log CRP over the 84 days of study after accounting for other factors (P-value = 0.017, P-value = 0.272, ANCOVA). Since infection can raise CRP values, an analysis was performed excluding the 6 subjects, 3 lovastatin and 3 placebo treated, with an infection at day 84; the differences between treatment groups remained non-significant. Additional sensitivity analyses such as excluding outliers and imputing missing data, did not change the result of this analysis.
CRP (mg/l) by treatment group and biologic use (mITT with available data)
| Lovastatin | Placebo | P-value | ||
|---|---|---|---|---|
| Primary analysisa | N = 30 | N = 25 | ||
| Day 0 | Mean (s.e.) | 10.8 (2.1) | 12.9 (3.5) | |
| Day 84 | Mean (s.e.) | 8.8 (1.6) | 10.7 (3.0) | |
| Change | Mean (s.e.) | −2.0 (1.2) | −2.2 (1.0) | 0.8* |
| Median (range) | −1.1 (−22.1, 18.0) | −0.6 (−18.0, 3.6) | ||
| No biologic useb | N = 12 | N = 15 | ||
| Day 0 | Mean (s.e.) | 16.6 (4.7) | 16.2 (5.8) | |
| Day 84 | Mean (s.e.) | 11.8 (3.5) | 14.2 (4.7) | |
| Change | Mean (s.e.) | −4.8 (3.0) | −2.0 (1.7) | 0.1** |
| Median range) | −3.4 (−22.1, 18.0) | 1.3 (−18.0, 3.6) | ||
| Biologic useb | N = 19 | N = 10 | ||
| Day 0 | Mean (s.e.) | 7.9 (1.1) | 7.9 (0.9) | |
| Day 84 | Mean (s.e.) | 6.7 (1.0) | 5.5 (0.9) | |
| Change | Mean (s.e.) | −1.2 (1.0) | −2.4 (0.9) | 0.1** |
| Median (range) | −0.5 (−15.1, 3.7) | −2.0 (−6.7, 2.5) |
| Lovastatin | Placebo | P-value | ||
|---|---|---|---|---|
| Primary analysisa | N = 30 | N = 25 | ||
| Day 0 | Mean (s.e.) | 10.8 (2.1) | 12.9 (3.5) | |
| Day 84 | Mean (s.e.) | 8.8 (1.6) | 10.7 (3.0) | |
| Change | Mean (s.e.) | −2.0 (1.2) | −2.2 (1.0) | 0.8* |
| Median (range) | −1.1 (−22.1, 18.0) | −0.6 (−18.0, 3.6) | ||
| No biologic useb | N = 12 | N = 15 | ||
| Day 0 | Mean (s.e.) | 16.6 (4.7) | 16.2 (5.8) | |
| Day 84 | Mean (s.e.) | 11.8 (3.5) | 14.2 (4.7) | |
| Change | Mean (s.e.) | −4.8 (3.0) | −2.0 (1.7) | 0.1** |
| Median range) | −3.4 (−22.1, 18.0) | 1.3 (−18.0, 3.6) | ||
| Biologic useb | N = 19 | N = 10 | ||
| Day 0 | Mean (s.e.) | 7.9 (1.1) | 7.9 (0.9) | |
| Day 84 | Mean (s.e.) | 6.7 (1.0) | 5.5 (0.9) | |
| Change | Mean (s.e.) | −1.2 (1.0) | −2.4 (0.9) | 0.1** |
| Median (range) | −0.5 (−15.1, 3.7) | −2.0 (−6.7, 2.5) |
Note: Summary statistics for CRP are presented on the untransformed scale; P-values are based on the log CRP change from baseline.
Only subjects with the day 84 visit within 14 days of the day 84 target date and within 7 days of their last dosing day were included in the primary efficacy analysis.
Secondary analyses were based on the mITT population; subjects with the day 84 visit >14 days from the day 84 target date were included.
*P-value tested for treatment effect for the change from baseline in log CRP using an analysis of covariance with adjustments for baseline log CRP, baseline DAS28-CRP score, race, MTX use, anti-TNF use, and disease duration.
**P-value for treatment effect for the change from baseline in log CRP in biologic users vs non-users using a Wilcoxon rank sum test.
mITT: modified intent-to-treat.
CRP (mg/l) by treatment group and biologic use (mITT with available data)
| Lovastatin | Placebo | P-value | ||
|---|---|---|---|---|
| Primary analysisa | N = 30 | N = 25 | ||
| Day 0 | Mean (s.e.) | 10.8 (2.1) | 12.9 (3.5) | |
| Day 84 | Mean (s.e.) | 8.8 (1.6) | 10.7 (3.0) | |
| Change | Mean (s.e.) | −2.0 (1.2) | −2.2 (1.0) | 0.8* |
| Median (range) | −1.1 (−22.1, 18.0) | −0.6 (−18.0, 3.6) | ||
| No biologic useb | N = 12 | N = 15 | ||
| Day 0 | Mean (s.e.) | 16.6 (4.7) | 16.2 (5.8) | |
| Day 84 | Mean (s.e.) | 11.8 (3.5) | 14.2 (4.7) | |
| Change | Mean (s.e.) | −4.8 (3.0) | −2.0 (1.7) | 0.1** |
| Median range) | −3.4 (−22.1, 18.0) | 1.3 (−18.0, 3.6) | ||
| Biologic useb | N = 19 | N = 10 | ||
| Day 0 | Mean (s.e.) | 7.9 (1.1) | 7.9 (0.9) | |
| Day 84 | Mean (s.e.) | 6.7 (1.0) | 5.5 (0.9) | |
| Change | Mean (s.e.) | −1.2 (1.0) | −2.4 (0.9) | 0.1** |
| Median (range) | −0.5 (−15.1, 3.7) | −2.0 (−6.7, 2.5) |
| Lovastatin | Placebo | P-value | ||
|---|---|---|---|---|
| Primary analysisa | N = 30 | N = 25 | ||
| Day 0 | Mean (s.e.) | 10.8 (2.1) | 12.9 (3.5) | |
| Day 84 | Mean (s.e.) | 8.8 (1.6) | 10.7 (3.0) | |
| Change | Mean (s.e.) | −2.0 (1.2) | −2.2 (1.0) | 0.8* |
| Median (range) | −1.1 (−22.1, 18.0) | −0.6 (−18.0, 3.6) | ||
| No biologic useb | N = 12 | N = 15 | ||
| Day 0 | Mean (s.e.) | 16.6 (4.7) | 16.2 (5.8) | |
| Day 84 | Mean (s.e.) | 11.8 (3.5) | 14.2 (4.7) | |
| Change | Mean (s.e.) | −4.8 (3.0) | −2.0 (1.7) | 0.1** |
| Median range) | −3.4 (−22.1, 18.0) | 1.3 (−18.0, 3.6) | ||
| Biologic useb | N = 19 | N = 10 | ||
| Day 0 | Mean (s.e.) | 7.9 (1.1) | 7.9 (0.9) | |
| Day 84 | Mean (s.e.) | 6.7 (1.0) | 5.5 (0.9) | |
| Change | Mean (s.e.) | −1.2 (1.0) | −2.4 (0.9) | 0.1** |
| Median (range) | −0.5 (−15.1, 3.7) | −2.0 (−6.7, 2.5) |
Note: Summary statistics for CRP are presented on the untransformed scale; P-values are based on the log CRP change from baseline.
Only subjects with the day 84 visit within 14 days of the day 84 target date and within 7 days of their last dosing day were included in the primary efficacy analysis.
Secondary analyses were based on the mITT population; subjects with the day 84 visit >14 days from the day 84 target date were included.
*P-value tested for treatment effect for the change from baseline in log CRP using an analysis of covariance with adjustments for baseline log CRP, baseline DAS28-CRP score, race, MTX use, anti-TNF use, and disease duration.
**P-value for treatment effect for the change from baseline in log CRP in biologic users vs non-users using a Wilcoxon rank sum test.
mITT: modified intent-to-treat.
A ⩾15% reduction in day 84 CRP was achieved by 61.3% and 44.0% of subjects in the lovastatin and placebo groups, respectively (P-value = 0.3, Fisher’s exact test; Table 3). In addition, a reduction of ⩾20% was observed in 54.8% and 32.0% in the lovastatin and placebo groups, respectively (P-value = 0.1, Fisher’s exact test).
Response rates by treatment group and biologic use (mITT with available data)
| Lovastatin | Placebo | P-value | |
|---|---|---|---|
| ≥ 20% Reduction in CRP at day 84 | |||
| No biologic use | 9/12 (75.0) | 3/15 (20.0) | 0.007* |
| Biologic use | 8/19 (42.1) | 5/10 (50.0) | 0.71* |
| Overall | 17/31 (54.8) | 8/25 (32.0) | 0.11* |
| ≥15% Reduction in CRP at day 84 | |||
| No biologic use | 10/12 (83.3) | 5/15 (33.3) | 0.019* |
| Biologic use | 9/19 (47.4) | 6/10 (60.0) | 0.70* |
| Overall | 19/31 (61.3) | 11/25 (44.0) | 0.28* |
| ACR20 Response at day 84 | |||
| No biologic use | 4/12 (33.3) | 6/15 (40.0) | 0.72** |
| Biologic use | 5/19 (26.3) | 4/10 (40.0) | 0.45** |
| Overall | 9/31 (29.0) | 10/25 (40.0) | 0.39** |
| DAS28-CRP EULAR response at day 84 | |||
| No biologic use | 5/12 (41.7) | 7/15 (46.7) | 0.80** |
| Biologic use | 9/19 (47.4) | 5/10 (50.0) | 0.89** |
| Overall | 14/31 (45.2) | 12/25 (48.0) | 0.83** |
| Lovastatin | Placebo | P-value | |
|---|---|---|---|
| ≥ 20% Reduction in CRP at day 84 | |||
| No biologic use | 9/12 (75.0) | 3/15 (20.0) | 0.007* |
| Biologic use | 8/19 (42.1) | 5/10 (50.0) | 0.71* |
| Overall | 17/31 (54.8) | 8/25 (32.0) | 0.11* |
| ≥15% Reduction in CRP at day 84 | |||
| No biologic use | 10/12 (83.3) | 5/15 (33.3) | 0.019* |
| Biologic use | 9/19 (47.4) | 6/10 (60.0) | 0.70* |
| Overall | 19/31 (61.3) | 11/25 (44.0) | 0.28* |
| ACR20 Response at day 84 | |||
| No biologic use | 4/12 (33.3) | 6/15 (40.0) | 0.72** |
| Biologic use | 5/19 (26.3) | 4/10 (40.0) | 0.45** |
| Overall | 9/31 (29.0) | 10/25 (40.0) | 0.39** |
| DAS28-CRP EULAR response at day 84 | |||
| No biologic use | 5/12 (41.7) | 7/15 (46.7) | 0.80** |
| Biologic use | 9/19 (47.4) | 5/10 (50.0) | 0.89** |
| Overall | 14/31 (45.2) | 12/25 (48.0) | 0.83** |
*P-values are from a Fisher’s exact test.
**P-values are from a χ2 test.
mITT: modified intent-to-treat.
Response rates by treatment group and biologic use (mITT with available data)
| Lovastatin | Placebo | P-value | |
|---|---|---|---|
| ≥ 20% Reduction in CRP at day 84 | |||
| No biologic use | 9/12 (75.0) | 3/15 (20.0) | 0.007* |
| Biologic use | 8/19 (42.1) | 5/10 (50.0) | 0.71* |
| Overall | 17/31 (54.8) | 8/25 (32.0) | 0.11* |
| ≥15% Reduction in CRP at day 84 | |||
| No biologic use | 10/12 (83.3) | 5/15 (33.3) | 0.019* |
| Biologic use | 9/19 (47.4) | 6/10 (60.0) | 0.70* |
| Overall | 19/31 (61.3) | 11/25 (44.0) | 0.28* |
| ACR20 Response at day 84 | |||
| No biologic use | 4/12 (33.3) | 6/15 (40.0) | 0.72** |
| Biologic use | 5/19 (26.3) | 4/10 (40.0) | 0.45** |
| Overall | 9/31 (29.0) | 10/25 (40.0) | 0.39** |
| DAS28-CRP EULAR response at day 84 | |||
| No biologic use | 5/12 (41.7) | 7/15 (46.7) | 0.80** |
| Biologic use | 9/19 (47.4) | 5/10 (50.0) | 0.89** |
| Overall | 14/31 (45.2) | 12/25 (48.0) | 0.83** |
| Lovastatin | Placebo | P-value | |
|---|---|---|---|
| ≥ 20% Reduction in CRP at day 84 | |||
| No biologic use | 9/12 (75.0) | 3/15 (20.0) | 0.007* |
| Biologic use | 8/19 (42.1) | 5/10 (50.0) | 0.71* |
| Overall | 17/31 (54.8) | 8/25 (32.0) | 0.11* |
| ≥15% Reduction in CRP at day 84 | |||
| No biologic use | 10/12 (83.3) | 5/15 (33.3) | 0.019* |
| Biologic use | 9/19 (47.4) | 6/10 (60.0) | 0.70* |
| Overall | 19/31 (61.3) | 11/25 (44.0) | 0.28* |
| ACR20 Response at day 84 | |||
| No biologic use | 4/12 (33.3) | 6/15 (40.0) | 0.72** |
| Biologic use | 5/19 (26.3) | 4/10 (40.0) | 0.45** |
| Overall | 9/31 (29.0) | 10/25 (40.0) | 0.39** |
| DAS28-CRP EULAR response at day 84 | |||
| No biologic use | 5/12 (41.7) | 7/15 (46.7) | 0.80** |
| Biologic use | 9/19 (47.4) | 5/10 (50.0) | 0.89** |
| Overall | 14/31 (45.2) | 12/25 (48.0) | 0.83** |
*P-values are from a Fisher’s exact test.
**P-values are from a χ2 test.
mITT: modified intent-to-treat.
Lovastatin had no significant effect on disease activity. The mean change (s.e.) in DAS28-CRP from baseline to day 84 was −0.4 (0.2) and −0.6 (0.2) in the lovastatin and placebo arms, respectively (P-value = 0.5). Furthermore, lovastatin did not significantly affect either DAS-28 CRP EULAR or ACR20 responder indices at day 84. For the DAS28-CRP EULAR index, 45% and 48% of lovastatin and placebo subjects, respectively, met the response criteria (P-value = 0.8). For ACR20, 29% and 40% of lovastatin and placebo subjects, respectively, met response criteria (P-value = 0.4).
There were no significant changes in autoantibody titres, RF or anti-CCP antibody from baseline to day 84 in either arm, and there were no differences between arms (see Supplementary Figure S1, available at Rheumatology online). In the limited sample of 9 subjects (6 placebo, 3 lovastatin), RF-secreting B cells were detected in all subjects.
Evaluation of efficacy with and without biologic use
Exploratory analyses compared the effects of lovastatin on subjects who did and did not use biologic agents (Table 2). Among those who did not use biologics, CRP levels at baseline were comparable between groups [mean (s.e.) mg/l: 16.6 (4.7) lovastatin, 16.2 (5.8) placebo]. Changes in CRP from baseline to day 84 did not differ significantly between lovastatin- and placebo-treated groups [mean (s.e.) mg/l: −4.8 (3.0) lovastatin; −2.0 (1.7) placebo; P-value = 0.1 Wilcoxon]. In subjects receiving biologics, baseline CRP levels were lower than in those not receiving biologics, but were comparable between arms [mean (s.e.) mg/l: 7.9 (1.1) lovastatin; 7.9 (0.9) placebo]. Changes in CRP from baseline to day 84 were smaller for lovastatin compared with placebo but the difference was not statistically significant [mean (s.e.) mg/l: −1.2 (1.0) lovastatin; −2.4 (0.9) mg/l placebo; P-value = 0.2 Wilcoxon]. Furthermore, irrespective of biologic use, clinical response criteria (DAS28-CRP EULAR or ACR20) did not differ significantly between arms (Table 3).
However, in subjects not using biologic agents, a significant difference was observed in the proportion of subjects reaching a meaningful reduction in CRP (⩾20%) at day 84 compared with baseline in the lovastatin arm (75%) compared with the placebo arm (20%; P-value = 0.007, Fisher’s exact). This difference was not evident in those who used biologics (Table 3). Similar statistically significant results were observed in the 15% reduction in baseline CRP.
Mechanistic analyses
Serum levels of 18 inflammatory markers did not differ between arms on Days 0 or 84, either overall or for subsets based on biologic use. Similar results were seen when comparing individuals who received biologic agents with those who did not. Changes in cytokine levels were uncorrelated with baseline DAS or CRP (see Supplementary Figure S2, available at Rheumatology online).
Statins can block MCP-1 secretion by LPS-stimulated PBMCs in a mevalonate-dependent manner [16]. Among 9 participants, unstimulated PBMCs from day 0 produced variable MCP-1 levels after 24 h of culture, and induction by LPS occurred in only 5 individuals. The effect of exogenous lovastatin on suppression of MCP-1 was also variable; no differences were detected either between days 0 and 84 or between treatment groups (see Supplementary Figure S3, available at Rheumatology online).
Lipid profile
Significant improvements were seen in total cholesterol, low-density lipoprotein and high-density lipoprotein in the lovastatin arm compared with the placebo arm at day 84 [mean (s.e.) change (mg/dl): total −39 (5.3) lovastatin, −6 (3.9) placebo, P-value <0.001; low-density lipoprotein −39 (4.5) lovastatin, −6 (3.5) placebo, P-value < 0.001; high-density lipoprotein 3.9 (1.5) lovastatin, −0.8 (1.6) placebo, P-value 0.04]. The change in triglycerides from baseline to day 84 did not differ between arms [mean (s.e.) change (mg/dl): −21.2 (11.8) lovastatin, 0.7 (9.2) placebo, P-value = 0.2].
Safety
The administration of a high-dose statin was well tolerated without significant safety signals. Fifty-nine participants in the safety population [29 (85%) lovastatin, 30 (100%) placebo] experienced 219 adverse events (AEs, 109 lovastatin, 110 placebo). Most AEs were mild (grade 1) including abnormalities of laboratory values. There were no grade 4 or 5 events. Four subjects (2 lovastatin, 2 placebo) experienced six grade 3 events (2 lovastatin, 4 placebo). Two serious AEs were reported: a subject (lovastatin) was hospitalized for an upper respiratory tract infection that was deemed unrelated to the study therapy; a subject (placebo) was hospitalized for grade 3 haematemesis. Seven subjects (4 lovastatin, 3 placebo) experienced transient elevations in creatinine phosphokinase. Eleven subjects (6 lovastatin, 5 placebo) experienced transaminase abnormalities. Eight subjects (3 lovastatin, 5 placebo) prematurely discontinued study therapy and withdrew from the study (Fig. 1), while 4 subjects (1 lovastatin, 3 placebo) remained in the study but underwent a reduction of their dose of the study drug or had a temporary discontinuation but then resumed at 40 mg/day, per protocol. There were no cases of myositis. Among the 36 musculoskeletal AEs, 6 subjects (4 lovastatin, 2 placebo) experienced new or worsening arthralgia, and 2 subjects (1 per arm) experienced myalgia.
Discussion
Because of its numerous anti-inflammatory properties, we evaluated lovastatin as a potential candidate for a safe adjunct to existing RA therapies. The anti-inflammatory effect of lovastatin on CRP, a marker of inflammation was anticipated to occur relatively quickly after statin exposure. However, a 12 week course of lovastatin in RA patients with mild clinical disease activity did not significantly reduce CRP levels compared with placebo. Neither mean changes in CRP nor proportions achieving meaningful reductions of CRP (15 or 20%) differed significantly between treatment arms. Although disease activity (DAS28-CRP) did not decline, a considerable proportion of both lovastatin- and placebo-treated subjects met DAS28-CRP EULAR or ACR20 response criteria. In the placebo group, response rates were higher than expected (ACR20: 40%, DAS28-CRP EULAR response: 48%), and no difference was observed between treated and placebo groups. The placebo response may be attributable to the short study duration, and it may not have been sustained in a longer study. Additionally, this study targeted subjects without severe disease activity, who may have been more susceptible to the placebo response.
In addition to assessing the effect of lovastatin on disease activity and CRP, we studied a number of mechanistic end points, including autoantibody titres and a panel of 18 potential protein biomarkers. Serum titres of autoantibodies and biomarkers correlate with changes in disease activity and may predict subsequent clinical response [18]. Serum biomarkers may also respond to successful therapeutic intervention, reflecting effects of therapy on pathophysiologic pathways. In this study, IL-6 and VCAM-1, robust markers of change in disease activity, remained stable in both lovastatin and placebo groups. Additionally, although statins have known effects on MCP-1, in vivo, these levels did not change after lovastatin exposure. However, since these studies were performed in a limited number of subjects, results are inconclusive. Overall, the mechanistic findings are consistent with our observation that lovastatin had no effect on either disease activity or on CRP.
Initial studies of statins in RA demonstrated a beneficial effect on RA disease activity and CRP. In 2004, the TARA study reported a significant improvement in DAS 28 and CRP after 6 months of treatment with atorvastatin vs placebo [17]. These subjects entered the study with quite active disease. Many, but not all, subsequent studies of statins in RA have also demonstrated significant improvement in disease activity. Differences in trial duration, disease activity at enrolment, the statin studied and background concomitant RA medications likely contributed to the response heterogeneity observed among these studies [19–29]. Our 12 week trial studied subjects with mild clinical disease activity with a mean entry DAS28 of 4.0. In addition, our study population included subjects using biologic agents, whereas in the TARA trial, no subjects received biologic therapy. These differences may account for the differences observed between the two trials.
Background medication may modulate the effect of statins on CRP. Although randomization of subjects was stratified by use of TNF inhibition (the predominant class of biologic at the time this study was conducted), a greater number of subjects randomized to lovastatin were receiving biologic agents than subjects receiving placebo. Previous studies of statins in RA of subjects receiving only DMARD therapy, without inclusion of subjects on concomitant biologic agents, have demonstrated a significant reduction in CRP [17, 20]. Only two studies evaluating patients receiving DMARDs only have failed to show a significant reduction in CRP [24, 26]. One of these studies was in patients receiving ‘triple therapy’, an approach considered equivalent to biologic treatment [26]. Furthermore, all RA studies of statins that have included subjects using biologic therapy or JAK inhibition [with the exception of one (Tang et al..)] have failed to observe a significant reduction in CRP [19, 25, 27–29]. Thus, our study, which included subjects treated with DMARDs and biologic therapies, is consistent with these previous observations. It is of interest that within the small subgroup of subjects who were not taking biologics, we observed trends suggesting that lovastatin reduced serum CRP, and was associated with increased percentages of subjects achieving significant reductions in CRP from baseline (P-value = 0.007 and 0.019 for 20% and 15%, respectively). One can postulate that subjects in this study who were receiving treatment with biologic agents are inherently different from biologic-naïve patients, and that ‘biologic treatment’ is a confounder for a subset of patients with more severe and resistant disease who are less likely to respond to adjunctive therapy with a statin. Alternatively, the anti-inflammatory effect of a statin in the context of treatment with a biologic may be minimal.
This study had several limitations. Because the study did not fully enrol, failure to observe a difference between arms could be a type II error. This possibility, however, seems unlikely, as no suggestion of a trend was detected with 55 subjects, even after performing various sensitivity analyses. The difficulties in recruitment suggest that subjects with mild clinical disease activity and an elevated CRP may not be representative of the majority of patients with RA. Our observation of a potential beneficial effect of statins in subjects not taking biologic therapy is of interest, particularly given observations of recent studies. However, this was observed in a post hoc analysis, was not associated with clinical benefit and must be viewed with caution. Furthermore, the numbers of patients receiving a biologic agent targeting the same pathway (e.g. TNF) were insufficient to develop a mechanistic hypothesis.
Factors with the potential to influence the inflammatory response are not well defined. Concomitant therapy with a potent biologic agent that lowers baseline CRP values may mask or impair our ability to detect an adjunctive benefit of statins. In contrast, patients with high disease activity may be more susceptible to the anti-inflammatory effects of statins. Furthermore, genetic polymorphisms may affect CRP levels in both normal individuals and RA patients. CRP polymorphisms may be associated with higher (or lower) levels of CRP, but their contribution to the responsiveness of CRP to changes in the inflammatory milieu is unclear [30]. Another possibility is that the factors contributing to elevated acute phase reactants in mild or moderate disease differ from those associated with more active disease and are less responsive to intervention. In any case, the stability of autoantibody titres, inflammatory markers, and disease activity during the trial suggest that treatment with lovastatin was not efficacious in the population studied.
Although we observed no anti-inflammatory or clinical effects of lovastatin, statins may still benefit RA patients because accelerated atherosclerosis and cardiovascular events are increased in RA. Control of modifiable cardiovascular risk factors such as hyperlipidaemia is an important component of the care of these patients. Interestingly, although reduced lipids (i.e. low-density lipoprotein) are associated with an increased risk of cardiovascular disease in RA, treatment with DMARDs may result in an increase in serum lipids yet a reduction in cardiovascular disease, the ‘lipid paradox’ (reviewed in 31). Nevertheless, initiation of statins in RA patients has been shown to be associated with decreased mortality [31]. Identification of subpopulations of RA patients who may have an anti-inflammatory response to statins remains to be demonstrated in future studies [32].
Funding: This study was funded by the Autoimmunity Centers of Excellence (U19 AI-056363 and U19 AI-0563662), a consortium funded by the National Institute of Allergy and Infectious Disease (NIAID). Research reported in this publication was additionally supported by the Autoimmune Disease Clinical Trials – Statistical and Clinical Coordinating Center funded by NIAID (1UMZAI117870 and contract: HHSN272200900057C). Lovastatin and placebo were provided by Teva Pharmaceutical Industries, which had no role in the study design or collection, analysis and interpretation of the data, the writing of the report, nor in the decision to submit the paper for publication.
Disclosure statement: The authors have declared no conflicts of interest.

{kind=link}
Comments