





Abstract
Lamiales is an order of core eudicots with abundant diversity, and many Lamiales plants have important medicinal and ornamental values. Here, we comparatively reanalyzed 11 Lamiales species with well-assembled genome sequences and found evidence that Lamiales plants, in addition to a hexaploidization or whole-genome triplication (WGT) shared by core eudicots, experienced further polyploidization events, establishing new groups in the order. Notably, we identified a whole-genome duplication (WGD) occurred just before the split of Scrophulariaceae from the other Lamiales families, such as Acanthaceae, Bignoniaceae, and Lamiaceae, suggesting its likely being the causal reason for the establishment and fast divergence of these families. We also found that a WGT occurred ∼68 to 78 million years ago (Mya), near the split of Oleaceae from the other Lamiales families, implying that it may have caused their fast divergence and the establishment of the Oleaceae family. Then, by exploring and distinguishing intra- and intergenomic chromosomal homology due to recursive polyploidization and speciation, respectively, we inferred that the Lamiales ancestral cell karyotype had 11 proto-chromosomes. We reconstructed the evolutionary trajectories from these proto-chromosomes to form the extant chromosomes in each Lamiales plant under study. We must note that most of the inferred 11 proto-chromosomes, duplicated during a WGD thereafter, have been well preserved in jacaranda (Jacaranda mimosifolia) genome, showing the credibility of the present inference implementing a telomere-centric chromosome repatterning model. These efforts are important to understand genome repatterning after recursive polyploidization, especially shedding light on the origin of new plant groups and angiosperm cell karyotype evolution.
Introduction
Lamiales plants, forming an order of angiosperms, play important roles in our daily life, health preservation, medicinal use, ecological balance, and biodiversity conservation. According to the APG IV system (THE ANGIOSPERM PHYLOGENY Group 2016), it includes thousands of plants divided into 24 families, such as Lamiaceae, Gesneriaceae, Plantaginaceae, and Scrophulariaceae. As the largest family in Lamiales, Lamiaceae includes more than 240 genera and more than 7,800 plants, which are widely distributed around the world, with 99 genera and 808 species found on the Chinese mainland (Zhao et al. 2021). Many plants in Lamiaceae have medicinal values, such as basil and perilla (Dudai and Belanger 2016; Ahmed 2019). Plants in Gesneriaceae are widely used in horticulture and green construction (Moller et al. 2019). Plantaginaceae plants have medicinal values such as clearing heat, detoxifying, diuresis, and reducing swelling (Mutinda et al. 2023). Most plants in Scrophulariaceae have certain economic and medicinal values (Cock et al. 2022). To date, there has been considerable progress in sequencing Lamiales plant genomes. According to the PlabiPD database (http://plabipd.de/index.ep), 80 Lamiales plants have been deciphered of their genomes, distributed in 13 families and 52 genera, including 27 genome sequences assembled to the chromosome level, providing valuable data to understand their biology and evolution.
Polyploidy plays important roles during the evolution of plants (Bowers et al. 2003; Jiao et al. 2011; Barker et al. 2016). Through polyploidization, plants can increase their genotype and phenotype diversity and improve their adaptability to the environment due mainly to the production of thousands of simultaneously duplicated genes, enhancing the evolutionary potentials of plants (Soltis and Soltis 2016; Van de Peer et al. 2017). After polyploidization, a genome often becomes unstable, resulting in phenomena such as chromosome breakages, abnormal recombination, and widespread gene losses. Genome instability provides nearly infinite possibilities for plant evolution and divergence (Leitch and Leitch 2008; Wendel 2015). As one of the most diverse orders in eudicots, Lamiales experienced the whole-genome triplication event common to major core eudicots (γ WGT), having occurred ∼115 to 130 million years ago (Mya; Jaillon et al. 2007). Previously, it was inferred that Oleaceae, a Lamiales family, experienced an extra WGT (named ω WGT) in the late Cretaceous about 66 Mya, likely contributing to the split of Oleaceae and the other Lamiales plants, and the fast diversification of Oleaceae plants (Xu et al. 2022). Further analysis of olive (Olea europaea) and green ash (Fraxinus pennsylvanica), both from the family of Oleaceae, suggested that they experienced an extra whole-genome duplication (named χ WGD) after the ω WGT (Rao et al. 2021; Huff et al. 2022). Extra polyploidization may have occurred in other Lamiales families. For example, it was inferred that rosemary (Salvia rosmarinus), a Lamiaceae plant, underwent 3 polyploidization events, including the γ WGT, an intermediate one having occurred ∼48 Mya, and a recent one ∼15 Mya (Han et al. 2023).
Chromosomes double during a WGD, and after a period of time, chromosome number often decreases mainly due to fusions of chromosomes, accompanied by different types of DNA rearrangement (Schubert and Lysak 2011). By performing pan-family or cross-family analysis of extant genomes, researchers attempted to reconstruct the ancestral cell karyotypes (Salse et al. 2009a, 2009b; Murat et al. 2012). A telomere-centric model was raised to reconstruct not only ancestral cell karyotypes but also the evolutionary trajectories of chromosomes (Wang et al. 2015a, 2015b, 2015c). The model considers the property of telomeres to protect the integrity of chromosomes, and their removal, by crossing-over between telomeres’ proximal regions, would likely facilitate their reorganization. The model explained how chromosome numbers decreased during evolution, especially after polyploidization. Three main types of chromosome reorganization were proposed, namely nested chromosome fusion (NCF), chromosome end–end joining (EEJ), and reciprocal exchanges of chromosomal arms (RTA). The occurrence of NCF is inferred to involve crossing-over between the proximal regions of 2 telomeres from the same chromosome to form a circular chromosome, and the resolution of the crossing-over could result in a free-end chromosome and a satellite chromosome to contain mainly the telomeric DNA. This NCF model explained how recurrent fusions of different chromosomes form extant chromosomes in Brachypodium (Vogel et al. 2010). The free-end chromosome may invade another chromosome and break the latter, eventually merging into it. The occurrence of EEJ is inferred to occur between different chromosomes; that is, a crossing-over may occur between the proximal regions of telomeres of the 2 chromosomes. The resolution of the crossing-over could result in a fused chromosome and a satellite chromosome. The model predicts that the occurrence of EEJ and NCF can form the satellite chromosomes, often called B chromosomes previously, which will be lost during evolution, eventually resulting in chromosome number reduction. The model has been applied to the study of karyotype evolution in multiple plant groups, including Cucurbitales, Leguminosae, Apiales, Poales, and other monocot orders (Wang et al. 2015a, 2015b, 2015c, 2019; Zhuang et al. 2019; Song et al. 2021; Wang et al. 2022). Until now, there has been no research on the ancestral cell karyotypes and Lamiales chromosome evolution.
Here, we reanalyzed the genome sequences of 11 representative Lamiales plants, assembled to the chromosome level (Hamilton et al. 2020; Xu et al. 2020a, 2020b; Friis et al. 2021; Jia et al. 2021; Ma et al. 2021; Rao et al. 2021; Wang et al. 2021a, 2021b; Huff et al. 2022; Xu et al. 2022), by referring to grape (Vitis vinifera), which has a conserved genome and therefore is often used as a reference to understand other eudicot genomes’ structure and polyploidization. We firstly determined whether the abovementioned polyploidization events were shared by Lamiales plants or not, secondly inferred the cell karyotypes of the ancestors of Lamiales at key evolutionary nodes, especially Lamiales proto-chromosomes, and thirdly reconstructed the evolutionary trajectories from those proto-chromosomes to form the extant chromosomes in Lamiales plants under consideration.
Results
Inference of gene collinearity
In order to clarify the polyploidization events experienced by Lamiales plants, we conducted a comparative analysis of the genome structures of 11 representative species in Lamiaceae, Acanthaceae, Bignoniaceae, Scrophulariaceae, and Oleaceae (Supplementary Table S1). Firstly, a phylogenetic tree was constructed with V. vinifera as the outgroup (Fig. 1A). Secondly, using WGDI (Sun et al. 2022), a gene collinearity inference toolkit, we extracted the collinear gene pairs within each genome and between the genomes of the studied species (Supplementary Table S2). Compared to the other species’ genomes under consideration, the genome of F. pennsylvanica contains much more collinear genes, with 31,189 homologous gene pairs residing in 2,126 homologous genomic blocks. In contrast, we revealed only 7,482 pairs in jasmine (Jasminum sambac). In addition, jacaranda (Jacaranda mimosifolia) from Bignoniaceae has more intergenomic collinear genes with the other plants (22,245 to 38,551), located in 1,074 to 1,999 blocks. Thirdly, we estimated the synonymous nucleotide substitution rate (Ks) between collinear gene pairs within a genome or between different genomes, which helped date polyploidization events and speciation events (Fig. 2; Supplementary Table S3).
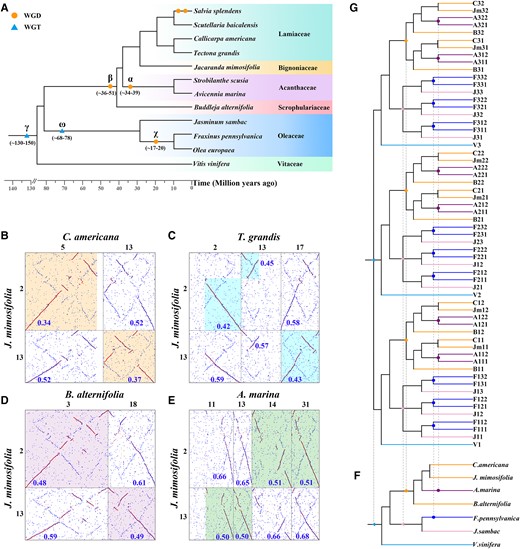
Phylogenetic trees and examples of local homologous gene dot plots of Lamiales plants. A) The phylogenetic relationship between the 5 families of Lamiales plants and the outgroup plant, V. vinifera; circle represents WGD; the light blue triangle represents WGT. B to E) Local homologous gene dot plots, where the numbers represent the Ks values at the corresponding positions. B) The dot plot between J. mimosifolia and C. americana, and the orange boxed segment represents 1 segment of J. mimosifolia to have 1 best-matched or orthologous segment of C. americana. C) The dot plot between J. mimosifolia and T. grandis, and the blue boxed segment represents 1 segment in J. mimosifolia to have 1 best-matched or orthologous segment in T. grandis. D) The dot plot between J. mimosifolia and B. alternifolia, and the purple boxed segment represents 1 segment in J. mimosifolia to have 1 best orthologous segment in B. alternifolia. E) The dot plot between J. mimosifolia and A. marina, and the green boxed segment represents 1 segment in J. mimosifolia to have the 2 best orthologous segments in A. marina. F) Species phylogenetic trees for the V. vinifera, J. sambac, F. pennsylvanica, B. alternifolia, A. marina, J. mimosifolia, and C. americana genomes. The light blue triangle represents the core eudicot common hexaploidization (ECH or γ) event, while the pink triangle, blue circle, orange circle, and purple circle represent the polyploidization events of in Lamiales, respectively. G) Corresponding gene tree. In V. vinifera (V), there were 3 paralogous genes generated by the ECH, namely V1, V2, and V3, if no gene losses had occurred after the ECH, and each grape gene was expected to have 3 orthologous genes in J. sambac (J) genome, 6 orthologs in F. pennsylvanica (F) genome, 2 orthologs in B. alternifolia (B), J. mimosifolia (Jm), and C. americana (C) genomes, and 4 orthologs in A. marina (A) genome.
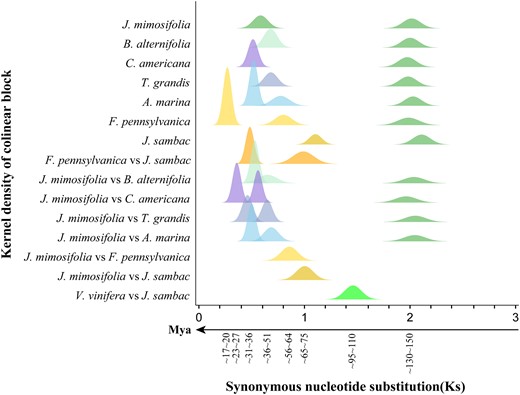
Distribution of Ks among collinearity genes in or between compared genomes. A timeline is provided to estimate dating. Mya, million years ago.
Inference of homology ratios
Ratios of orthology between genomes or paralogy within a genome are key to understanding whether or not polyploidization event(s) are shared and the ploidy levels of a specific polyploidization event. Here, by integrating the information about gene collinearity inferred above, we used Blast-supported putative homologous genes to draw homologous gene dot plots within a genome and between different genomes to find homology ratios (Tang et al. 2011; Wang et al. 2022). The intraspecific homologous gene dot plot of each of these plants, callicarpa (Callicarpa americana; a Lamiaceae plant), teak (Tectona grandis; a Lamiaceae plant), J. mimosifolia (a Bignoniaceae plant), and butterfly bush (Buddleja alternifolia; a Scrophulariaceae plant), indicated that the paralogy ratio, produced by the best-matched genes between their chromosomes, is 1:1. This indicates that each of these plants has experienced a WGD (Supplementary Figs. S1 to S4). Moreover, the interspecific homologous gene dot plots between J. mimosifolia and each of these 3 plants, C. americana, T. grandis, and B. alternifolia, showed that orthology ratios are 1:1, indicating that these plants shared a WGD before the split of Lamiaceae, Bignoniaceae, Acanthaceae, and Scrophulariaceae. That is, there has been no WGD specific to any of them. In comparison, the orthology ratio between J. mimosifolia and Avicennia marina (an Acanthaceae plant) is 1:2 (Fig. 1, B to E; Supplementary Figs. S5 to S7), which indicates that A. marina was affected by an extra duplication event after their split.
To find the homology ratio between J. mimosifolia and B. alternifolia, illustrated in the homologous gene dot plot (Supplementary Fig. S8), we extracted the best-matched genes from the Blast results. Taking the chromosome Jmi2 as an example, the number of best-matched genes in the chromosome Bal3 is 482, and that in Bal18 is 195, showing significant difference (χ2 test, P < 2.004 × 10−5; Supplementary Tables S4 and S5). This shows that Bal3 was likely orthologous to Jmi2, and Bal18 was outparalogous to Jmi2. Here, orthology forms due to speciation and outparalogy due to shared polyploidization. Two genes from different species are orthologous if derived from the same ancestral gene in their latest common ancestor. Two genes from different species are outparalogous if they were derivatives from 2 ancestral genes produced by an ancient gene duplication event before the split of the 2 species. Notably, 2 genes from the same plant can be referred to as outparalogous ones if produced by duplication in the common ancestor of relative species to distinguish paralogous genes produced by duplication in its own lineage (Supplementary Fig. S9). Grossly, a check of orthology and outparalogy across 2 genomes supports an orthology ratio of 1:1, and the orthologous collinear genes produced estimated Ks to be 0.532 ± 0.040. However, the outparalogy pattern was much more complex; a J. mimosifolia chromosome often has multiple outparalogous correspondence. By checking Ks values between outparalogous gene pairs between J. mimosifolia and B. alternifolia, we found that there existed 2 groups of outparalogous genes, with 1 group containing 24,265 gene pairs (313 blocks) and having Ks ∼0.650 ± 0.092 and the other group containing 1,135 gene pairs (118 blocks) and having Ks ∼2.037 ± 0.276 (Supplementary Table S3). The older group obviously resulted from the γ WGT shared by major core eudicots, while the younger group shows that there was a polyploidization shared by the 2 plant families, not revealed before. As the younger polyploidization, an outparalogy ratio of 1:1 can be inferred, showing its tetraploidization nature, or a WGD. Characterization of interspecific Ks values further supports that the WGD is shared among 4 studied Lamiales families, including Acanthaceae, Scrophulariaceae, Bignoniaceae, and Lamiaceae. For convenience, it is named as β WGD.
Similarly, by checking homology ratios and Ks, besides the γ WGT (Ks ∼1.986 ± 0.267), we confirmed that J. sambac, F. pennsylvanica, and O. europaea shared another WGT (Ks ∼0.801 ± 0.144); then, F. pennsylvanica and O. europaea shared a WGD event, namely χ WGD, as to the above context (Ks ∼0.269 ± 0.027; Fig. 2). The orthologous genes between the 2 Oleaceae plants have Ks ∼0.483 ± 0.067, showing that they share the 2 older WGT events, with the oldest being γ WGD and the more recent being ω WGT. Correspondingly, the orthology ratio between V. vinifera and J. sambac is 1:3, the orthology ratio between V. vinifera and F. pennsylvanica is 1:6, and the orthology ratio between O. europaea and F. pennsylvanica is 1:1 (Supplementary Fig. S10). This is consistent with previous reports (Rao et al. 2021; Huff et al. 2022; Xu et al. 2022). As to the homologous gene dot plots between A. marina or Strobilanthes cusia and J. mimosifolia, the orthology ratio between J. mimosifolia and A. marina or S. cusia is 1:2, indicating that the latter 2 species shared an extra WGD event (Supplementary Figs. S11 and S12). A comparison of A. marina and S. cusia revealed 3 polyploidization events, with peaks located at 0.519 ± 0.026, 0.774 ± 0.084, and 2.029 ± 0.386. The oldest one is surely the γ WGT, shared with the other core eudicots, the intermediate one β WGD, and the recent one shared by the 2 plants, here referred to as α WGD. Here, we also provided the 4DTV values, reflecting synonymous nucleotide substitutions involving 4-fold degenerative codons, and the relevant analysis came to the same inference (Supplementary Fig. S13).
The 4DTV values between collinear genes can reflect relative species differentiation events and whole-genome replication events during evolution. Therefore, in order to further validate the polyploidization events revealed earlier, we conducted a 4DTV value analysis on gene pairs within and between collinear segments of the genome (Supplementary Fig. S13). In the J. mimosifolia, B. alternifolia, C. americana, and T. grandis genomes, we can observe 2 peaks, one near 0.2 and the other near 0.4, indicating recent and ancient polyploidization events in all 4 species. One of the ancient events must have been γ WGT, shared by core eudicots; the most recent event is β WGD. In the A. marina genome, we observed 3 peaks located near 0.16, 0.24, and 0.43, corresponding to γ WGT, β WGD, and α WGD, 3 polyploidization events. Similarly, in the F. pennsylvanica and J. sambac genomes, we observed 3 peaks and 2 peaks, respectively, with 2 peaks with higher 4DTV values corresponding to γ WGT and ω WGT; the most recent one is χ WGD. Overall, the results of the 4DTV analysis are consistent with those of the Ks analysis, further confirming the correctness of our previous inference of the polyploidization events.
Supposing the occurrence of γ WGD to be 130 to 150 Mya, the other polyploidization and speciation events were dated (Fig. 2). The ω WGT occurred ∼68 to 78 Mya, near the split of Oleaceae from the other Lamiales families, implying its contribution to their divergence, while β WGD occurred ∼36 to 51 Mya, near the split of Scrophulariaceae from the 3 families, Acanthaceae, Bignoniaceae, and Lamiaceae, implying its contribution to their divergence. Clarifying the polyploidization events affecting the Lamiales plants, we constructed the gene tree reflecting the relationship between homologous collinear genes in these plants (Fig. 1, F and G), which would help study the evolution and functional innovation of genes, especially those simultaneously duplicated during each polyploidization.
Inference of ancestral chromosome karyotypes
The reconstruction of ancestral chromosome karyotypes helps reveal the evolution of Lamiales plants, especially to answer how recursive polyploidization and following genome stability contributed to chromosome rearrangement and number reduction. Here, by checking homologous gene dot plots, we determined 11 Lamiales proto-chromosomes, with approaches as illustrated previously (Zhuang et al. 2019; Sun et al. 2022).
We reconstructed the proto-karyotype of B. alternifolia and C. americana common ancestors. By checking orthology and paralogy ratios shown with the homolog gene dot plots within and between their genomes, we found that the common ancestor of them after the β WGD had n = 2x = 22 chromosomes, or n = x = 11 chromosomes before the β WGD (Fig. 3). Taken as an example, the inference of their ancestral chromosome L1 before β WGD is due to the following fact: B. alternifolia chromosomes Bal3 and Bal18 were paralogous chromosomes, doubled during β WGD, and their corresponding C. americana orthologous chromosomes are Cam5 and Cam13, respectively (Fig. 3A). Ignoring certain local DNA inversion or a few small missing DNA regions, they share homology in their full length. Therefore, a parsimonious explanation is that they were derivatives of an ancestral proto-chromosome and retained the main structure of it, referred to as L1 (Fig. 3A). Taken as another example, as to the inference of ancestral chromosomes L6 and L11, Bal10 and Cam10 are orthologous to one another in their full length and are outparalogous to 1 arm of Bal5 and the corresponding region of Cam3. Meanwhile, Bal17, in its full length, is orthologous to the majority of Cam14 and 1 arm of Cam3. This suggests that Bal10 (Cam10) and Bal17 are likely derivatives of 2 pre-β WGD chromosomes, here referred to as L6 and L11, respectively (Fig. 3B). As to the inference of ancestral chromosome L8, Bal19 in its full length is orthologous to the majority of Cam6 and to the full length of Cam4; meanwhile, 1 arm of Bal1 and Bal15 is orthologous to the corresponding region of Cam4 (Cam6), respectively. Therefore, we infer that they are likely derivatives of ancestral proto-chromosomes and retain their main structure, referred to as L8 (Fig. 3C). As to the inference of ancestor chromosome L9, Bal8 and Cam11 are orthologous to one another in their full length and are orthologous to Cam1in it partly length; at the same time, Bal1 has a corresponding orthologous region in Cam1 and has an orthologous region in Cam11. Ignoring likely gene loss on involved chromosomes, we infer that they likely originated from the same ancestral proto-chromosome, here referred to as L9 (Fig. 3D). As to the inference of ancestor chromosomes L7 and L2, Bal13 in its full length is orthologous to a portion of Cam6, while most of Bal2 is orthologous to regions in Cam8 and Cam9; at the meantime, Bal4 is orthologous to other regions in Cam8 and Cam9, while a portion of Bal11 and Bal16 is directly orthologous to a portion of Cam1. This indicates that the Bal2 portion (part of Cam8 and Cam9) and Bal4 are much likely derivatives of 2 pre-β WGD chromosomes, here referred to as L7 and L2, respectively (Fig. 3E). As to the inference of ancestral chromosome L4, Bal6 and the middle part of Bal1 are paralogous chromosomes resulting from β WGD. Their corresponding C. americana orthologous chromosomes are Cam17 and a part of Cam14. Ignoring certain inversion or a few small missing DNA regions, they share homology in their full length. Therefore, we inferred that they were likely derivatives of an ancestral proto-chromosome and retained the main structure of it, here referred to as L4 (Fig. 3G). Similarly, the ancestral chromosome L10 was inferred based on the homologous regions corresponding to Bal9, Bal12, Bal4 and Cam1, Cam2 (Fig. 3H). The ancestral chromosome L3 was inferred based on the homologous regions corresponding to Bal2, Bal7 and Cam12, Cam15, Cam16 (Fig. 3F). As to the inference of ancestor chromosome L5, based on the correspondence between Bal9, Bal12, and Cam7, it is uncertain whether Cam7 evolved from a complete ancestral chromosome or was formed by the merging of 2 ancestral chromosomes (Fig. 3F). In order to determine the exact number, we used 2 outgroups, F. pennsylvanica and J. sambac, to find the answer. A comparative analysis with 2 outgroups showed that the 2 parts that make up Cam7 are homologous to Fpe5, Fpe8, Fpe17, and Fpe19 at full length and to Jsa3 and Jsa13 in their full length, too, indicating that Cam7 evolved from 1 ancestral chromosome, here referred to as L5 (Fig. 3I). Therefore, the explanation is that an RTA occurred between L5 and L10 to form neo-chromosomes Bal9 and Bal12, as shown in Fig. 3F.
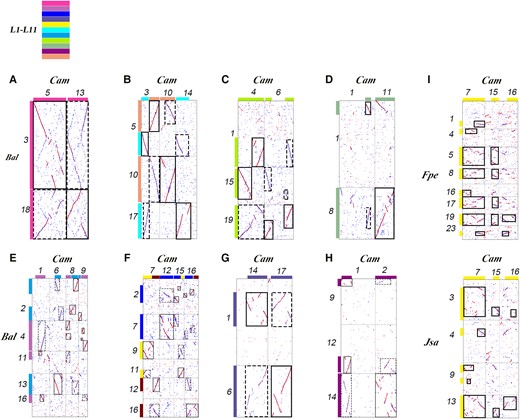
Inference of the ancestral chromosomes of Lamiales. A to H) Inferring ancestral chromosomes based on the homologous gene dot plots between B. alternifolia (Bal) and C. americana (Cam). I) Inferring the proto-chromosome L5 based on comparing C. americana to J. sambac (Jsa) and F. pennsylvanica (Fpe). L1 to L11: distinguishing inferred Lamiales proto-chromosomes using distinct identifiers (Hex codes: f83ba6, be6ab5, 0000ff, 5f52a0, fff100, 00ffff, 00a0e9, b1f005, 8FBC8F, 920783, f29b76).
It is worth noting that the majority of the inferred n = x = 11 proto-chromosomes, duplicated to n = 2x = 22 during the β WGD, have been well preserved in the J. mimosifolia (Jmi) genome. In the evolution after the β WGD, occurring ∼36 to 51 Mya, without considering small chromosomal changes, only 2 EEJs and 2 NCFs occurred to result in chromosome number reduction from 22 to 18 in extant J. mimosifolia plant (Fig. 4A). This means that 4 Jmi chromosomes are composite chromosomes of ancestral chromosomes, and 14 ancestral chromosomes have been preserved (Fig. 4A). Chromosome Jmi1 is a composite chromosome formed by a fusion of a β WGD copy of L4 into a β WGD copy of L5, and the other β WGD copies of L4 and L5 evolved to form Jmi9 and Jmi10, respectively. Chromosome Jmi6 is a composite chromosome formed by a fusion of a β WGD copy of L3 into a β WGD copy of L7, and the other β WGD copies of L3 and L7 evolved to form Jmi8 and Jmi12, respectively. Chromosome Jmi3 is a composite chromosome formed by an EEJ of a β WGD copy of L6 and a copy of L11, and the other β WGD copies evolved to form Jmi11 and Jmi18, respectively. Chromosome Jmi7 is a composite chromosome formed by an EEJ of a β WGD copy of L9 and a copy of L10, and the other β WGD copies evolved to form Jmi15 and Jmi16, respectively.
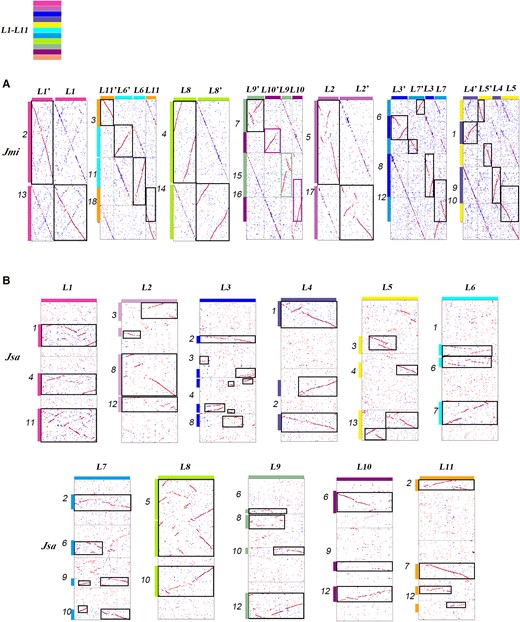
Supporting evidence for ancestral chromosome inference. A and B) Supportive evidence to the correctness of ancestral chromosome inference based on comparing J. mimosifolia (Jmi) chromosomes and homology between J. mimosifolia and J. sambac. L1 to L11: distinguishing inferred Lamiales proto-chromosomes using distinct identifiers (Hex codes: f83ba6, be6ab5, 0000ff, 5f52a0, fff100, 00ffff, 00a0e9, b1f005, 8FBC8F, 920783, f29b76).
In addition, though J. sambac has been affected by a WGT and a WGD after the split from plants in Lamiaceae, Bignoniaceae, Acanthaceae, and Scrophulariaceae, subjected to extensive genome instability and repatterning, 7 of the 11 proto-chromosomes have been preserved as an extant chromosome in J. sambac (Fig. 4B). The other 4 proto-chromosomes also have considerably large patches in extant chromosomes. Therefore, we inferred these 11 proto-chromosomes (L1 to L11) as the common ancestor chromosomes of all Lamiales plants.
We have to note that the findings of well-preserved proto-chromosomes in J. mimosifolia and J. sambac provide supportive evidence that the inference of the 11 proto-chromosomes is most likely credible, in that the inference was based on B. alternifolia and C. americana, being independent of J. mimosifolia and J. sambac.
Reconstruction of chromosome evolutionary trajectories
Then, we inferred karyotype changes from Lamiales ancestors to J. mimosifolia, B. alternifolia, and C. americana (Fig. 5A). As described above, the 22 β WGD duplicated chromosomes underwent 2 EEJs and 2 NCFs to form J. mimosifolia 18 chromosomes (Fig. 5B). The evolutionary trajectories of chromosomes are depicted to show the major changes during the evolution. A similar inference was made with the other genomes. Actually, subjected to 1 EEJ, 2 NCFs, and 3RTAs, B. alternifolia 19 chromosomes formed (Fig. 5C), and subjected to 4 EEJs, 1 NCF, and 2RTAs, C. americana’s 17 chromosomes formed (Fig. 5D).
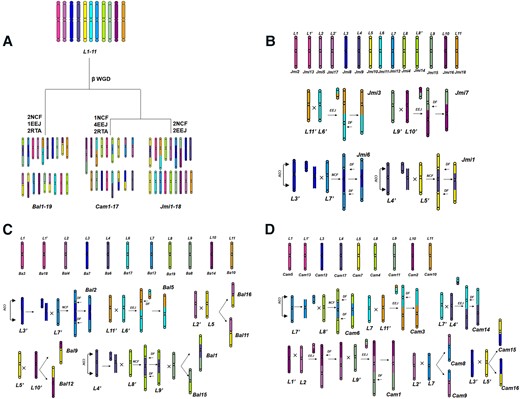
Examples of ancestral karyotypes and evolutionary trajectories from Lamiales common ancestors. A) Reconstructed ancestral karyotypes. L1 to L11: inferred Lamiales proto-chromosomes (each distinguished by a unique identifier, listed in Hex format: f83ba6, be6ab5, 0000ff, 5f52a0, fff100, 00ffff, 00a0e9, b1f005, 8FBC8F, 920783, f29b76). NCF, nested chromosome fusion; EEJ, end–end joining; RTA, reciprocal exchange of chromosomal arms; WGD, whole-genome duplication. B to D) reconstructed evolutionary trajectories to form extant chromosomes in J. mimosifolia (Jmi), B. alternifolia (Bal), and C. americana (Cam). COV and X: crossing-over; DF: the inactivation of extra centromeres; lost: the loss of supposed B chromosomes; L1 to L11 and L1′ to L11′: duplicated copies of L1.
How are the Lamiales proto-chromosomes formed after the split from the other eudicots? Here, we first reconstructed the 11 Lamiales proto-chromosomes or proto-genome using J. mimosifolia extant chromosomes. As to the correspondence between proto-chromosomes and J. mimosifolia chromosomes shown (Fig. 4A), a proto-chromosome was reconstructed by the gene content in the corresponding extant chromosome. In that there are paralogous chromosomes or regions in J. mimosifolia, an alternative one was used, which will not affect the following analysis. Then, by revealing putative homologous genes between the reconstructed proto-chromosomes and V. vinifera, we checked the homology between the Lamiales proto-genome and that of V. vinifera (Fig. 6A). V. vinifera has n = 19 chromosomes, from which revealed the γ WGT and 7 pre-γ WGT proto-chromosomes and 21 post-γ WGT chromosomes common to major core eudicots, making V. vinifera a model plant to understand the structure and evolution of the other eudicot genomes. The 21 post-γ WGT chromosomes were referred to as A1 to A7, B1 to B7, and C1 to C7. We found that 9 EEJs and 1 NCF occurred to form the 11 Lamiales proto-chromosomes (Fig. 6B). L9 and L10 were derivatives of B4 and A3. L5 formed due to an NCF of B2 into B6. L3 formed due to an EEJ between B5 and A2. C4 and C5 formed an intermediate chromosome by an EEJ, which crossed with C6 to form L2 and another intermediate chromosome. The intermediate chromosome then crossed with B1 to form another 2 intermediates, one of which was linked to C1 to form L4 by EEJ. The other Lamiales proto-chromosomes and their evolutionary trajectories were reconstructed similarly.
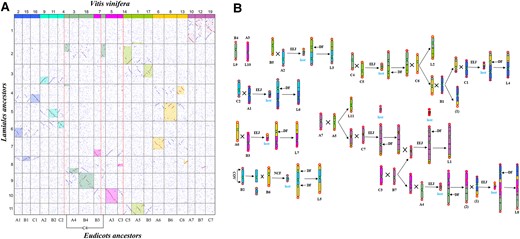
Homologous gene dot plot between the reconstructed Lamiales chromosomes and core eudicot proto-chromosomes inferred by using V. vinifera, and the formation of Lamiales proto-chromosomes. A) Dot plot. Best-matched regions are shown in shadow; each group of 7 ancestor chromosomes is distinguished by distinct identifiers (Hex codes: 3366FF, 00CCFF, FF00FF, 8FBC8F, 99CC00, FFCC00, C46AB5). B) A1 to A7, B1 to B7, and C1 to C7 represent 21 core eudicot ancestral chromosomes; L1 to L11 represent the Lamiales proto-chromosomes. EEJ, end–end joining; NCF, nested chromosome fusion; COV, crossing-over; DF, the inactivation of extra centromeres; lost, the loss of supposed B chromosomes.
Eventually, we inferred ancestral karyotypes at major evolutionary nodes from the major core eudicot proto-chromosomes and reconstructed evolutionary trajectories to extant Lamiales plants under consideration (Fig. 7).
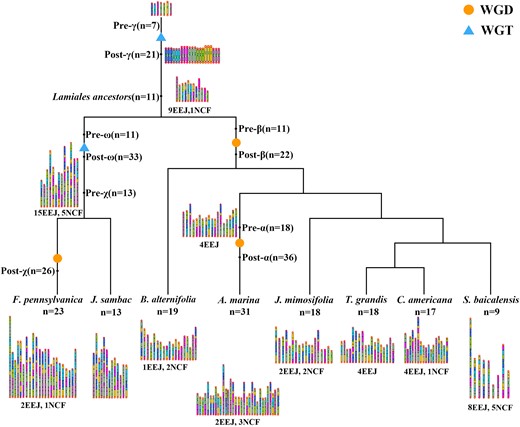
Reconstructing the evolutionary trajectories of chromosomes from core eudicot common ancestor to the extant Lamiales. Orange circles represent WGD (WGD events). Tthe light blue triangles represent WGT (WGT events). ω, χ, β, and α represent the polyploidization events that occur in Lamiales species, respectively. Seven ancestor chromosomes are distinguished by distinct identifiers (Hex codes: 3366FF, 00CCFF, FF00FF, 8FBC8F, 99CC00, FFCC00, C46AB5). The figure shows the number of chromosome fusions (EEJs and NCFs) experienced by each speciation.
Discussion
Polyploidy plays an important role in the evolution and functional innovation of plants (Bowers et al. 2003; Soltis and Soltis 2016). However, due to the complexity of species genomes, polyploid events could be omitted or misidentified (Wang et al. 2018a, 2018b, 2020). A sophisticated comparative genomic analysis showed that a WGD event contributed to the establishment of the Cucurbitaceae family (Wang et al. 2018a, 2018b). This polyploidization event had been overlooked in several previous reports (Huang et al. 2009; Garcia-Mas et al. 2012; Guo et al. 2013). Additionally, a similar reanalysis of Selaginella moellendorffii revealed multiple ancient polyploidization events during the evolution of Lycophytes (Wang et al. 2020), negating the previous inference that Lycophytes did not undergo ancient polyploidization (Banks et al. 2011). This showed that all the vascular plants were affected by the paleopolyploidization. Here, we investigated the polyploid events in Lamiales plants by identifying orthologous and paralogous genes. We found that, in addition to the γ WGT shared by core eudicots, Lamiales plants also experienced certain specific polyploidization events. Apart from the ω WGT and χ WGD reported in Oleaceae species, we found evidence that the β WGD is shared by Lamiaceae, Bignoniaceae, Acanthaceae, and Scrophulariaceae. Notably, we found that the β WGD occurred just before the split of Scrophulariaceae from the 3 families, Acanthaceae, Bignoniaceae, and Lamiaceae, ∼33 to 45 Mya, suggesting its contribution to their divergence of these families. We also found that ω WGT occurred ∼68 to 78 Mya, near the split of Oleaceae from the other Lamiales families ∼65 to 67 Mya, implying its contribution to their divergence. Actually, the link between paleopolyploidization and speciation has been proposed. The fast divergence and establishment of angiosperms and seed plants were related to likely polyploidization (Jiao et al. 2011). There has been genomic evidence that the establishment of the plant families Poaceae (Wang et al. 2015a, 2015b, 2015c), Cucurbitaceae (Wang et al. 2018a, 2018b), Fabaceae (Zhuang et al. 2019), Solanaceae (International Tomato Genome Sequencing Consortium 2012), and Apiaceae (Song et al. 2021) were related to their family-specific polyploidization. However, at least in the case of seed plants, whether they shared a common polyploid ancestor has been still hotly debated (Leebens-Mack et al. 2019; Liu et al. 2021; Han et al. 2022; Wan et al. 2022).
The occurrence of various WGDs or WGTs in Lamiales has been in obscurity. Comparative genomic analysis of C. americana and T. grandis inferred that C. americana underwent 3 ancient WGD events, including 1 WGD detected in the T. grandis lineage but not shared with C. americana (Hamilton et al. 2020). The scarlet sage (Salvia splendens) genome also revealed 3 WGD events after the γ event, with 2 in its own lineage and the other shared by Lamiaceae plants (Jia et al. 2021). The present study provided solid evidence that C. americana and T. grandis shared 2 polyploidization events, including γ WGT shared by major core eudicots and the other one shared by the 4 Lamiales families Scrophulariaceae, Acanthaceae, Bignoniaceae, and Lamiaceae (β WGD). The present reanalysis of S. splendens consolidated the above inference that the β WGD is shared by the abovementioned 4 Lamiales families and 2 additional WGD events experienced by the species alone. In addition, the present reanalysis of 3 Oleaceae plants, J. sambac, F. pennsylvanica, and O. europaea, revealed that the Oleaceae underwent a WGT event, consistent with previous findings (Rao et al. 2021; Huff et al. 2022; Xu et al. 2022). In conclusion, the present research cleared the obscurity over the occurrence of polyploidization during the evolution of Lamiales. Polyploidization, as a type of abrupt evolutionary event, may produce a new species overnight (Ruprecht et al. 2017; Van de Peer et al. 2017). Though the new species has a very slim chance of survival, its parental lines are in a niche that have been most likely adapted to; if it survives, it would have numerous opportunities to have genetic novelties. In a neo-polyploid, the existence of homoeologous chromosomes and thousands of duplicated genes provides enormous chances for mutation (Wang et al. 2015a, 2015b, 2015c; Barker et al. 2016). Multivalent recombination may occur between homologous and homologous chromosomes, likely resulting in chromosome breakages and rearrangements, gene losses and copy number variations, increased evolutionary rates, chromosome number changes (often reduction), etc. (Wendel 2015; Wang et al. 2018a, 2018b; Sun et al. 2022). A fact is though tens of paleopolyploidization events have been inferred as entering the genomic era, and a plant and its ancestral line may have only been affected by 2 to 6 paleopolyploidization events, all plants have been supposed to be derived from an ancestral paleopolyploid (Zhuang et al. 2019; Wang et al. 2021a, 2021b; Sun et al. 2022). The findings here in Lamiales provide further supportive evidence that paleopolyploidization would contribute to fast divergence and establishment of plant groups.
Following polyploidization, a duplicated genome is often unstable, and extensive DNA rearrangements may result in chromosome number changes, often reduction, resulting in cell karyotype variation in derivative plants. Karyotype refers to the characteristics of the number, size, and morphology of chromosomes in a cell. Currently, 2 methods for predicting chromosome karyotypes have been proposed in comparative genomic research. A pure bioinformatic approach was proposed, which uses a cumulative identity percentage (CIP, 60%) and a cumulative alignment length percentage (CALP, 70%) to identify orthologous and paralogous genes, as well as Closeup software to identify blocks (Salse et al. 2009a, 2009b). Then, continuous ancestral regions (CARs) are defined by comparing conserved fragments between 2 genomes (orthologous genes/blocks) and within a single genome (paralogous genes/blocks); we determined the most likely evolution based on CARs. The approach hypothesized that the ancestral genome was formed by replication or rearrangement events of modern species at homologous positions and attempted to explain the evolutionary history from the ancestral genome to the modern karyotype through the minimum number of rearrangements (including inversion, deletion, fusion, division, and translocation; Murat et al. 2012, 2014). Based on this approach, ancestral karyotypes of rosid crops, including the Caricaceae, Brassicaceae, Malvaceae, Fabaceae, Rosaceae, Salicaceae, and Vitaceae families, were inferred, respectively (Murat et al. 2015). More recently, a telomere-centric model was proposed to explain chromosome rearrangement and fusion initially based on the analysis of Poaceae and Arabidopsis (Wang et al. 2015a, 2015b, 2015c). Supposing that a chromosome's DNA crossed-over near its 2 telomeres, the resolution of the crossing-over may remove the telomeres to produce a transitional free-end chromosome, which may then be inserted into another chromosome. Alternatively, crossing-over may occur between 2 different chromosomes near their telomeres, and the resolution of the crossing-over may result in removing the involved telomeres and merging 2 chromosomes into a larger one. During the processes, the B chromosome may be formed by removed telomeres, and the loss of the B chromosome results in chromosome number reduction. The telomere-centric model was applied to infer ancestral karyotypes in Poaceae, Brassicaceae, Malvaceae, Apiaceae, Fabaceae, and Cucurbitales (Wang et al. 2015a, 2015b, 2015c; Zhuang et al. 2019; Song et al. 2021; Wang et al. 2022). Besides, the model facilitates the inference of evolutionary trajectories from very ancient proto-chromosome to extant chromosomes.
Here, using the telomere-centric model, we inferred that the Lamiales common ancestor has 11 proto-chromosomes and reconstructed the evolutionary trajectories from the major core eudicot common ancestor to the Lamiales common ancestor and then from the Lamiales common ancestor to the extant chromosomes in Lamiales plants under consideration. Actually, the inference of the Lamiales proto-chromosomes was based on the comparative analysis B. alternifolia and C. americana chromosomes. Notably, further analysis with the other Lamiales plants, J. mimosifolia and J. sambac, provides supportive evidence to the above inference in that most of the inferred proto-chromosomes have perfect extant derivatives, ignoring small-scale DNA rearrangement and loss, in these plants. The other inferred proto-chromosomes also have very good gene collinearity in extant chromosomes, even to the length of each considered proto-chromosome. Grossly, these above facts support the credibility of the present inference of Lamiales proto-chromosomes and their evolutionary trajectories using the telomere-centric model.
Materials and methods
Plant genome data materials
Genome data of Lamiales species and grape were downloaded from relevant public databases (Supplementary Table S1). Python scripts were used to process data format to facilitate subsequent research.
Phylogenetic analysis
We used the gene functional analysis tool OrthoFinder to construct the species phylogenetic tree (Emms and Kelly 2019). Amino acid sequences of Lamiales species and outgroup V. vinifera were used to do the analysis. OrthoFinder is run to find homologous gene groups, and diamond (Buchfink et al. 2015) is used as the comparison method. FastTree is used to build phylogenetic trees. MEGA-X (Kumar et al. 2018) was used to display and edit the alignment, tree production, and beautification. Default parameters were adopted with the adopted software items, if not specified in the context.
Multiple sequence alignment and inference of gene collinearity
The first step is to perform multiple sequence alignment. According to the sequence alignment tool BLAST (Altschul et al. 1990), select the -blastp module to perform homology alignment within and between genomes of the selected species’ genomic protein sequences. The E-value threshold of the output result is set to 1e−5 to accommodate the duplicated genes produced by paleopolyploidization ∼10 Mya, and the output file format (-outfmt) is set to 6 during the specific operation.
Based on the previously obtained homologous sequence alignment result file (Blast file), combined with the genome annotation file (Gff file) and chromosome length file (Lens file), we used the - d module implemented in WGDI (Sun et al. 2022) to draw homologous gene dot plots. The dots of different colors (red, blue, and gray) in the dot plots represent the level of similarity of their gene pairs. According to the dot plots of homologous genes, homologous collinearity was inferred within each genome and between genomes. Next, we used the -icl module to perform collinearity analysis and obtained the collinearity regions, described by scores, statistical significance, collinear gene numbers, etc.
Calculation of synonymous nucleotide substitutions
According to the collinear genes obtained previously, the Ks values were calculated by combining the cds and pep files. Here, the -Ks module in WGDI was used. This module used Muscle software (Edgar 2021) to perform protein binding based on the protein sequence, used pal2pal.pl to convert the protein binding into codon binding based on the CDS sequence, and finally calculated Ks using yn00 from PAML (Yang 2007).
Calculation of 4DTV value
Run MCScanX to obtain collinear gene pairs, and calculate 4DTV values by combining cds and pep files (Wang et al. 2012). Here, we used the ParaAT program and related scripts (calculate_4DTV_correction.pl and axt2one-line.py) to calculate 4DTV values (Zhang et al. 2012). Then, we used RStudio for visualization and created corresponding graphs.
Construction of ancestral karyotypes and chromosome evolution trajectories
The homologous gene dot plot can intuitively display the gene arrangement positions on each chromosome of a species and can be used to check the ortholog and paralog relationships within a single species genome and between different species genomes, exploring the process of chromosome fusion, breakage, and rearrangement, providing convenience for inferring the formation of ancestral chromosomes and reconstructing modern chromosome evolution history. According to the previously proposed telomere-centric chromosome reconstruction model, its several manifestations in homologous gene dot plots are shown in Supplementary Fig. S14. Supplementary Fig. S14A shows that with species B as a reference, the chromosome B1 of species B can find a complete corresponding chromosome A1 in species A, indicating that they originated from the same ancestor chromosome; Supplementary Fig. S14B shows the EEJ model, where 1 chromosome A1 of species A is formed through EEJ by 2 chromosomes B1 and B2 of species B; Supplementary Fig. S14C shows the NCF model, in which the formation of chromosome A1 of species A is due to the nesting of chromosome B2 of species B into B1, leading to NCF; Supplementary Fig. S14D shows the RTA model, in which the 2 chromosomes A1 and A2 of species A are due to RTA between the 2 chromosomes B1 and B2 of species B.
Author contributions
X.W. and J.W. designed and conceptualized the work. J.W. and X.W. prepared the draft manuscript. J.W., B.S., M.Y., F.H., H.Q., H.Z., Y.J., Y.L., and Z.W. prepared the figure. X.W. and J.W. revised the manuscript critically for important intellectual content. All authors edited and reviewed the final version.
Supplementary data
The following materials are available in the online version of this article.
Supplementary Fig. S1. Homologous gene dot plot in the genome of J. mimosifolia.
Supplementary Fig. S2. Homologous genes dot plot in the genome of B. alternifolia.
Supplementary Fig. S3. Homologous genes dot plot in the genome of C. americana.
Supplementary Fig. S4. Homologous genes dot plot in the genome of T. grandis.
Supplementary Fig. S5. Homologous genes dot plot between the genomes of J. mimosifolia and C. americana.
Supplementary Fig. S6. Homologous genes dot plot between the genomes of J. mimosifolia and T. grandis.
Supplementary Fig. S7. Homologous genes dot plot between the genomes of J. mimosifolia and A. marina.
Supplementary Fig. S8. Homologous genes dot plot between the genomes of J. mimosifolia and B. alternifolia.
Supplementary Fig. S9. Explanation and differentiation of orthology, paralogy, and outparalogy.
Supplementary Fig. S10. Local homologous gene dot plots.
Supplementary Fig. S11. Homologous gene dot plot in the genome of A. marina.
Supplementary Fig. S12. Homologous gene dot plot between the genomes of J. mimosifolia and S. cusia.
Supplementary Fig. S13. Distribution of 4DTV values among collinearity genes in or between compared genomes.
Supplementary Fig. S14. Supposed gene collinearity shown in homologous gene dot plots and corresponding chromosome rearrangements.
Supplementary Table S1. Species genome data information and download sources.
Supplementary Table S2. Statistics of colinear gene pairs and blocks of colinear genes.
Supplementary Table S3. Kernel function analysis of Ks distribution related to duplication events within each genome and between selected genomes (before evolutionary rate correction).
Supplementary Table S4. Best-matched results for Blast (using Jmi2 as an example, corresponding to chromosome Bal3).
Supplementary Table S5. Best-matched results for Blast (using Jmi2 as an example, corresponding to chromosome Bal18).
Funding
This project was financially supported by the National Natural Science Foundation of China (32070669) and financial support from the Bureau for Human and Social Resources Security of Tangshan Municipal to X.W.
Data availability
If you need our data set, you can contact the corresponding author.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
References
Author notes
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) is Xiyin Wang ([email protected]).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}