





Abstract
Manipulation of gene expression is central to understanding gene function, engineering cell behavior, and altering biological traits according to production demands. Nuclease-dead Cas9 (dCas9), a variant of active Cas9, offers a versatile platform for the precise control of genome function without DNA cleavage. Notably, however, an effective and universal dCas9-based transcriptional repression system remains unavailable in plants. The noncanonical histone acetyltransferase TENDRIL-LESS (CsTEN) is responsible for chromatin loosening and histone modification in cucumber (Cucumis sativus). In this study, we engineered a gene regulation tool by fusing TEN and its truncated proteins with dCas9. The full-length dCas9-TEN protein substantially repressed gene expression, with the N-terminal domain identified as the core repression domain. We subsequently validated the specificity and efficacy of this system through both transient infection and genetic transformation in cucumber and Arabidopsis (Arabidopsis thaliana). The electrophoretic mobility shift assay (EMSA) revealed the ability of the N-terminal domain of TEN to bind to chromatin, which may promote target binding of the dCas9 complex and enhance the transcriptional repression effect. Our tool enriches the arsenal of genetic regulation tools available for precision breeding in crops.
Introduction
Gene expression regulation encompasses a multitude of intricate mechanisms governing vital biological processes such as signal transduction, chromatin remodeling, and RNA transport and degradation. The advent of clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated nuclease 9 (Cas9)-mediated gene editing in eukaryotic cells (Cong et al. 2013) has sparked a profound transformation across various domains of life sciences, which has been extensively applied in the genetic engineering field (Sanchez-Rivera and Jacks 2015; Zhang et al. 2019; Dubois and Roudier 2021; Liu et al. 2022).
The emergence of gene regulation tools derived from the CRISPR/Cas9 system foreshadowed a new era of biological understanding (Sternberg and Doudna 2015). Among these derivatives, the nuclease-dead Cas9 (dCas9), containing a D10A mutation in the RuvC1 domain and an H840A mutation in the HNH domain, retains its DNA binding capacity while preventing DNA cleavage (Gilbert et al. 2013). The inactivated form, cooperating with different transcriptional activators or repressors, is capable of modulating gene expression with high precision, referred to as CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), respectively. Moreover, dCas9-based regulation offers the unique advantages of influencing native gene expression without introducing exogenous copies, enabling manipulation of chromatin through dCas9-effector fusions, allowing facile genome-wide screens, and minimizing off-target risks associated with traditional RNA interference methods.
In recent years, scientists have developed various dCas9-derived transcriptional activation tools, such as dCas9-VP64-EDLL, dCas9-TV, SAM, SunTag, and CRISPR-ACT3.0 (Konermann et al. 2015; Li et al. 2017; Lowder et al. 2018; Papikian et al. 2019; Pan et al. 2021). Nevertheless, the comparable system for transcriptional repression is still limited, especially in the plant realm (Hiratsu et al. 2003; Piatek et al. 2015; Vazquez-Vilar et al. 2016; Li et al. 2020). This discrepancy underscores the pressing need for the expeditious development of an effective and versatile repression tool, mirroring the strides witnessed in activation tools. Recent years have seen the development of enhanced dCas9 repressors by deploying various transcriptional repression domains in mammalian systems (Yeo et al. 2018; Alerasool et al. 2020; Veggiani et al. 2022). However, over the past decade, only 2 Arabidopsis (Arabidopsis thaliana) endogenous transcriptional repression domains, namely BRD and SRDX motifs, have been proven effective for the construction of dCas9 repression tools in plants, including dCas9-BRD (Vazquez-Vilar et al. 2016), dCas9-SRDX (Piatek et al. 2015; Vazquez-Vilar et al. 2016), and dCas9-3SRDX (Lowder et al. 2015). The characterization and deployment of other transcriptional repression domains would provide a new possibility for developing novel dCas9-derived transcriptional repression tools in plants.
In our preceding investigations, we unveiled TENDRIL-LESS (CsTEN), a histone acetyltransferase (HAT) protein that orchestrates chromatin loosening and gene activation in cucumber (Cucumis sativus) (Wang et al. 2015). This intriguing finding led us to explore the fusion of TEN with dCas9, aiming to create a gene activation tool. To our astonishment, the resultant fusion protein exhibited gene repression capabilities, as evidenced by its effect on the expression of the target gene, CsACO1, and the corresponding tendril-free phenotype in transgenic cucumber (Yang et al. 2020). This unexpected outcome highlighted the repression capability and versatility of dCas9-TEN, even in transgenic plants. Further structural analysis of the TEN protein guided us to identify the N-terminal domain as the critical effector responsible for pronounced inhibitory effects on the proximal promoter and gene body region.
The TEN protein was reported to contain HAT activity, facilitating chromatin accessibility. The acetylation modification in histone core can directly alter histone-DNA interaction (Tessarz and Kouzarides 2014; Yang et al. 2020). In our study, we explored the specific binding ability of the N-terminal domain of TEN (the N protein) to the assembled DNA region. Additionally, we conducted a comprehensive investigation into its specificity and adaptability across genes with diverse expression levels in the cucumber and Arabidopsis protoplasts. Our research involves the development of precision gene regulation tools, offering a perspective on advanced gene regulation techniques and precision breeding in crops.
Results
A dCas9-derived transcriptional repression system
It was previously demonstrated that the cucumber (C. sativus) tendril identity gene TEN encodes a noncanonical HAT, targeting gene body enhancers, loosening chromatin, and further activating gene expression (Wang et al. 2015; Yang et al. 2020). Here, we tentatively fused TEN to dCas9 (dCas9-TEN) to improve target gene expression. In addition, we fused dCas9 to the P300 activation domain, a classical mammalian acetyltransferase (Hilton et al. 2015), and used dCas9-HA as a control (Fig. 1A). We selected CsACO1 as a test case, as perturbations of its expression levels would affect tendril development and result in easily identifiable phenotypes. We targeted 2 sites in CsACO1 (Fig. 1B), namely T1 (promoter) and T4 (gene body), with each sgRNA cloned in the pBSE402 vector. We individually transformed each vector into cucumber cotyledons using Agrobacterium tumefaciens-mediated transformation based on an “optimal infiltration intensity” strategy (Xin et al. 2022). Surprisingly, the results showed the opposite: dCas9-TEN repressed CsACO1 expression by 92% compared with the control when targeting the T4 site (Fig. 1C). Consistently, stable cucumber transgenic plants showed a tendril-free phenotype that was more pronounced than the aco1 mutant we previously observed (Yang et al. 2020). However, dCas9-P300 transgenic plants had no discernible change in CsACO1 expression and morphology (Fig. 1, C and D), which may be attributed to the limited efficacy of a single mammalian effector in plant cells, as documented in an earlier study (Lee et al. 2019). Notably, relative CsACO1 expression was normal when dCas9-TEN was directed to the T1 site, as was tendril development (Fig. 1, C and D), underscoring the importance of screening suitable target sites while also indicating that the overexpression of dCas9-TEN per se has no effect on cucumber tendril development.
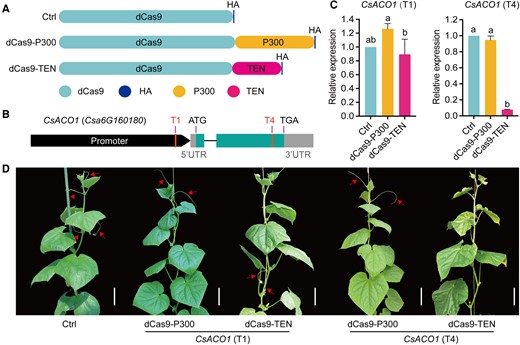
The dCas9-TEN fusion protein inhibits transcription by targeting the proximal promoter and gene body of the target gene. A) Diagrams of dCas9 and the dCas9 fusion proteins dCas9-P300 and dCas9-TEN. P300, activation domain. HA, hemagglutinin epitope tag. B) Diagram of the CsACO1 locus, showing the location of the corresponding sgRNAs (T1 and T4). C) dCas9-TEN represses relative CsACO1 expression when targeting the T4 site. D) Representative photographs showing the tendril-free phenotype of dCas9-TEN transgenic plants targeting the T4 site, while other transgenic plants produce normal tendrils. Images were digitally extracted for comparison. Scale bars, 10 cm; Arrows point to normally growing tendrils. Values are shown as means ± Sd (n = 3). Different lowercase letters indicate significant differences among means at P < 0.05 (1-way ANOVA with post hoc Tukey test).
Identification of functional domain of TEN protein
To define the core effector domain of the TEN protein, we fused dCas9 to different truncated domains (N, TCP, and C domains) in the pBSE402 vector (Fig. 2A). These vectors were individually transformed into cucumber through A. tumefaciens-mediated infection. We observed robust transcriptional repression from dCas9-TEN, dCas9-N, and dCas9-N+C, decreasing CsACO1 expression levels by 93%, 87%, and 96%, respectively, compared with the control. In contrast, other dCas9 fusion proteins lacking the N domain showed weak repression (Fig. 2B). The tendril phenotype of the same transgenic plants was consistent with the extent of transcriptional repression, indicating that the N domain plays an essential role in transcriptional repression (Fig. 2C).
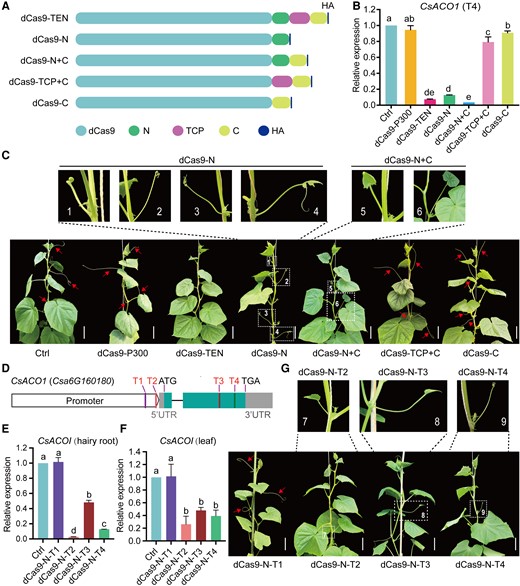
The N domain is the main functional inhibitory domain. A) Diagrams of dCas9 fused to full-length TEN or different truncated proteins of TEN. N, N-terminal domain; TCP, TCP domain; C, C-terminal domain. B) Relative CsACO1 expression shows that the N domain is the main transcriptional inhibitory domain of TEN. C) Representative photographs showing the phenotype of different transgenic plants harboring the indicated vector transformed in cucumber. All dCas9 variants fused to the N domain (dCas9-TEN, dCas9-N, and dCas9-N+C) and targeting the T4 site resulted in a tendril-less or modified tendril phenotype. Images were digitally extracted for comparison. Scale bars, 10 cm; Arrows point to normal tendrils, and the enlarged images and numbers (1 to 6) in the upper panel originated from the white dashed frames in the bottom panel. D) Diagram of the CsACO1 locus, showing the location of the corresponding sgRNAs (T1, T2, T3, and T4). E) and F) Relative CsACO1 expression when dCas9-N is targeted to 1 of the 4 sites shown in panel D in the cucumber hairy root system (E) and stable cucumber transformations (F). G) Representative photographs showing the phenotype of transgenic plants with dCas9-N targeted to each of the 4 sites in CsACO1. Images were digitally extracted for comparison. Scale bars, 10 cm; red arrows point to normally developed tendrils, and the enlarged images and numbers (7 to 9) in the upper panel originated from the white dashed frames in the bottom panel. Values are shown as means ± Sd (n = 3). Different lowercase letters indicate significant differences among means at P < 0.05 (1-way ANOVA with post hoc Tukey test).
Afterward, we tested 2 additional target sites at different positions along CsACO1, with T1 and T2 located in the promoter and T3 and T4 in the gene body (Fig. 2D). Expressing the vectors via Agrobacterium rhizogenes-mediated hairy root transformation (Boisson-Dernier et al. 2001), we observed that dCas9-N strongly downregulates CsACO1 expression when targeted to the T2, T3, and T4 sites, achieving repression levels of 48% to 97% (Fig. 2E). Stable transformation of the same constructs produced similar results, with dCas9-N inducing transcriptional repression of CsACO1 of 52% to 74% when targeted to the T2, T3, and T4 sites (Fig. 2F), resulting in the abnormal tendril phenotype. In contrast, CsACO1 expression and the phenotype of transgenic plants expressing dCas9-N-T1 remained normal (Fig. 2, F and G). Therefore, we hypothesize that the transcription initiation or elongation was substantially hindered by the binding of dCas9-N at the T2, T3, and T4 sites but not at the T1 site, which could be far from the transcription start site (TSS).
The versatility of dCas9-N in cucumber
A versatile repression system should effectively repress various genes at different basal expression levels and be compatible with multiple transformation methods. To assess the versatility of our dCas9-based repressor tool, we selected 4 target genes (Fig. 3A), 3 with high (Csa6G450370), medium (Csa7G398090), and low (Csa6G010030) expression levels, and the 4th candidate, Csa2G238880, specifically highly expressed in roots based on previous RNA-seq data (Li et al. 2011). For each gene, we tested 3 target sites (T1 to T3) via hairy root transformation (Fig. 3, B to E) and transient cotyledon infection (Supplementary Fig. S1, A to D). We observed that dCas9-N serves as a robust repressor for most targets compared with control plants, and transformation harboring dCas9 for all 4 tested genes. Notably, dCas9-N efficiently downregulated target gene expression using a single sgRNA by more than 50% at all tested target sites, while the efficiencies of transcriptional repression mediated by dCas9 were more variable (Fig. 3, B to E; Supplementary Fig. S1, A to D). This suggests that dCas9-N may be less sensitive to target genes and target site selection.
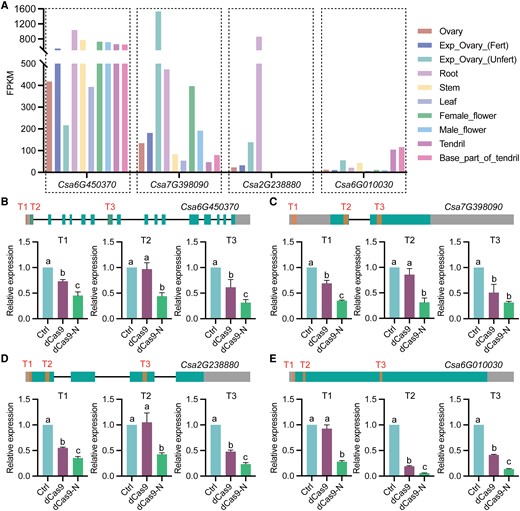
Verification of the repression effects of dCas9-N on genes with different levels of background expression in cucumber. A) Expression patterns of the 4 selected cucumber genes, Csa6G450370, Csa7G398090, Csa2G238880, and Csa6G010030 (Li et al. 2011). B) to E) dCas9-N inhibits the expression of Csa6G450370 (B), Csa7G398090 (C), Csa2G238880 (D), and Csa6G010030 (E) when targeted to different sites (T1, T2, and T3) through the hairy root system. Ctrl, WT plants as control. Data are shown as means ± Sd (n = 3). Different lowercase letters indicate significant differences among means at P < 0.05 (1-way ANOVA with post hoc Tukey test).
The universality and effectiveness of dCas9-N
in Arabidopsis
To expand the applicability of dCas9-N to other species, we employed Arabidopsis due to its ease of transformation, allowing for multiple experimental combinations for validation and optimization. At first, we tested whether placing the N domain of TEN upstream of dCas9 to construct N-dCas9 would also induce effective targeted transcriptional repression (Fig. 4A). Four previously reported target sites in AtFLS2 were chosen (Fig. 4B) (Li et al. 2017). We found that N-dCas9 mediated effective transcriptional repression of the target gene in Arabidopsis with efficiencies comparable with those of dCas9-N as well as dCas9-3SRDX, a commonly used dCas9 repressor in plants (Supplementary Fig. S1, E to H). Our data revealed that N-dCas9 acts as a mild repressor in transient transformation in protoplasts. We further attempted to increase the repression strength by multimerizing the N domain, generating the constructs N-dCas9, 2N-dCas9, and 4N-dCas9 (Supplementary Fig. S2A), to target each of the 4 sites in AtFLS2. However, additional copies of the N domains did not statistically strengthen the repression (Supplementary Fig. S2B). The immunoblot analysis revealed that adding extra N domains may compromise the stability of the fusion protein (Supplementary Fig. S2C), in agreement with previous reports (Gilbert et al. 2013; Li et al. 2017).
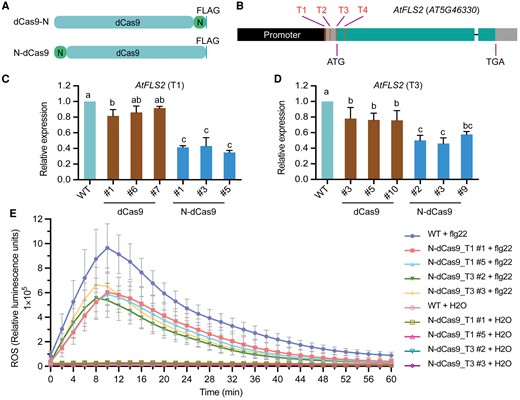
Universality and effectiveness of N-dCas9 in Arabidopsis via transient and stable transformation. A) Diagrams of dCas9-N and N-dCas9. B) Diagram of the AtFLS2 locus showing the position of the sgRNAs (T1 to T4). C) and D) Relative AtFLS2 expression when N-dCas9 is targeted to the T1 (C) or T3 (D) site by stable transformation. Different transgenic lines are shown. Data are shown as means ± Sd (n = 3). Different lowercase letters indicate significant differences among means at P < 0.05 (1-way ANOVA with post hoc Tukey test). E) ROS production in response to the pathogen elicitor flagellin 22 (flg22). Data are shown as means ± Sd (n = 8 experiments).
Based on the results of the transient assay, we generated stable Arabidopsis transgenic lines in which dCas9 and N-dCas9 were individually targeted to the T1 and T3 sites of AtFLS2, yielding results comparable with the transient assay (Fig. 4, C and D). Arabidopsis FLS2 encodes an immune receptor that perceives flg22, the highly conserved epitope in the N-terminal region of the bacterial elicitor flagellin (Gómez-Gómez and Boller 2000). We verified that the transgenic plants with N-dCas9-mediated AtFLS2 repression all exhibit compromised reactive oxygen species (ROS) production in responses to flg22 (Fig. 4E). Collectively, these results demonstrated that our dCas9 tool based on the N domain can efficiently induce targeted transcriptional repression in Arabidopsis.
The specificity of N-dCas9/dCas9-N tools in Arabidopsis and cucumber
A critical concern for the CRISPR/Cas9 technology is its specificity. To assess the specificity of our transcriptional repression tool, we identified the genes with potential off-target sites of the sgRNAs used to target the 6 genes (Supplementary Table S3) and examined their transcript levels by RT-qPCR (Supplementary Figs. S3 to S5). The relative expression of potential off-target genes indicates that the dCas9-N/N-dCas9 tool exhibits commendable specificity with minimal impact on off-target sites. Furthermore, we evaluated the genome-wide transcriptional repression specificity using RNA-seq. To this end, we targeted N-dCas9 to T1 and T3 sites of AtFLS2 in Arabidopsis protoplasts and targeted dCas9-N to T1 sites of Csa2G238880 in cucumber hairy roots. RT-qPCR and RNA-seq data validated effective downregulation of target gene expression (Fig. 5, A to F). For transcriptome profiles, the result shows a strong linear relationship in the gene expression pattern across samples expressing or not expressing N-dCas9/dCas9-N (Fig. 5, G to I). Pearson's correlation coefficients are 0.966 for the AtFLS2 T1 site and 0.965 for the AtFLS2 T3 site in Arabidopsis (Fig. 5, G and H). The global transcriptome profiles of the cucumber hairy roots after A. rhizogenes-mediated transformation showed a similar conclusion (Pearson's correlation coefficient 0.963; Fig. 5I). The RNA-seq results suggest that gene expression at the whole genome level is not broadly influenced by N-dCas9/dCas9-N in Arabidopsis and cucumber. However, a small subset of data points deviates slightly from the diagonal line, which is probably due to the secondary effect of target gene downregulation and the complex intrinsic regulatory network of gene expression since no predictable sgRNA off-target binding sites could be identified within the proximal promoter and the gene body of these genes (Fig. 5, G to I; Supplementary Table S3). More detailed specificity analyses with rational experimental design and control are needed to comprehensively assess the specificity of our transcriptional repression tool in the future.
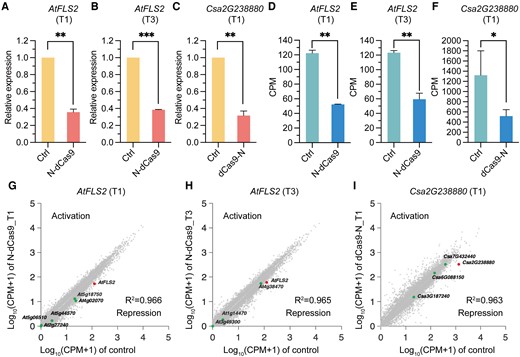
The specificity of the N-dCas9/dCas9-N tools in Arabidopsis and cucumber. A) to C) RT-qPCR validation of the relative expression levels of AtFLS2 when N-dCas9 is targeted to T1 (A) and T3 (B) sites in Arabidopsis protoplasts and the expression level of Csa2G238880 when dCas9-N is targeted to T1 site (C) in cucumber hairy roots. Data are shown as means ± Sd (n = 2). Differences between 2 groups were tested using the Student's t test (*P < 0.05, **P < 0.01, ***P < 0.001). D) to F) Expression level of AtFLS2 when N-dCas9 is targeted to T1 (D) and T3 (E) sites in Arabidopsis protoplasts and expression level of Csa2G238880 when dCas9-N is targeted to T1 site (F) in cucumber calculated using RNA-seq CPM values. Data are shown as means ± Sd (n = 2). Differences between 2 groups were tested using the Student's t test (*P < 0.05, **P < 0.01, ***P < 0.001). G) and H) Pearson's correlation analysis with RNA-seq data reveals the specificity when N-dCas9 is targeted to the T1 (D) and T3 (E) sites of AtFLS2 in Arabidopsis protoplasts. I) Pearson's correlation analysis with RNA-seq data reveals the specificity when dCas9-N is targeted to the T1 site of Csa2G238880 in cucumber hairy roots. On-target and potential off-target genes are marked in each plot respectively, in G to I.
N protein promotes the transcriptional repression probably by binding to the chromatin
To investigate the potential mechanism of N protein, we designed an experiment to explore how chromatin is influenced by the repressor N protein as a part of the intragenic enhancer-binding protein. For nucleosome assembly, we first purified N protein (Fig. 6A) and expressed and purified each unit of histone octamer, H2A, H2B, H3, and H4 (Fig. 6B). All parts of histones were assembled in refolding buffer with an equimolar amount and purified through a gel filtration column. The octamer elution was marked with a yellow square and dispensed into 7 tubes (C1 to C7) (Supplementary Fig. S6). Each column of eluted histone was collected and examined with SDS-PAGE (Fig. 6C). Next, we assembled the nucleosome by mixing histone octamer and a free DNA probe, which was examined by agarose gel electrophoresis. A lagged band represents the completed assembly of the nucleosome (Fig. 6D). Electrophoretic mobility shift assay (EMSA) was performed with purified N protein, demonstrating its specific binding ability to nucleosome-binding DNA (Fig. 6, A and E). It suggests that the N protein promotes dCas9-mediated transcriptional repression, most likely by binding to the DNA fragment within the chromatin.
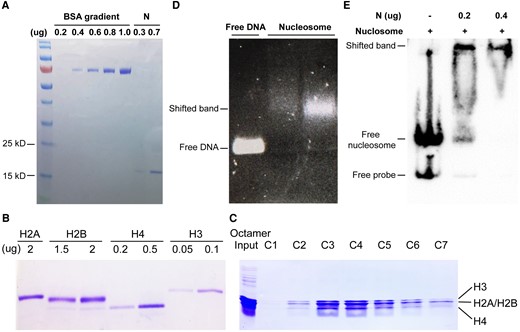
N promotes transcription repression, probably by binding to the chromatin. A) The BSA standard marker quantified the concentration of purified N protein in SDS-PAGE. B) SDS-PAGE shows the expression of the purified histone protein from BL21 (DE3) pLysS cells, staining by Coomassie brilliant blue (CBB). C) The corresponding octamer elution from the gel filtration column in Supplementary Fig. S3A was examined with SDS-PAGE. D) DNA gel electrophoresis shows the shift band of the assembled nucleosome compared with free DNA. E) EMSA shows the binding ability of N protein to assembled nucleosomal DNA.
In conclusion, we developed an effective and versatile gene expression suppression tool (dCas9-N/N-dCas9) and demonstrated its robustness and effectiveness by both transient infecting and stable genetic transformation in cucumber and Arabidopsis. N protein enhanced the inhibition of gene expression, probably by binding to the DNA assembled in the chromatin, without causing obvious disruption to the expression of other genes. dCas9-N could be useful for precisely regulating target gene expression and facilitating crop breeding.
Discussion
CRISPR/dCas9 technology has ushered in a transformative era in the realm of functional genomics. Renowned for its simplicity, efficacy, and cost-effectiveness, this tool enables the precise activation or repression of gene expression (Xu and Qi 2019). In this study, we introduced a potential gene regulatory tool, N-dCas9/dCas9-N, a dCas9-derived fusion protein capable of reducing gene expression by obstructing RNA polymerase binding or elongation when precisely targeted to the proximal promoter and gene body region. Impressively, N-dCas9 also exhibits a capacity for endogenous gene repression in Arabidopsis, with efficiencies comparable with those of the previously reported dCas9-3SRDX repressor in plants. A noteworthy aspect of this inquiry is the unexpected enhancement of the CRISPRi effect upon fusion of dCas9 with the N domain of the TEN protein, which is recognized for its HAT function. The underlying mechanism remains enigmatic and merits further exploration.
Further investigation unveiled the combination of dCas9 with different truncated domains of the TEN protein, exhibiting different levels of inhibition on the expression of the CsACO1 gene. Notably, transgenic cucumbers harboring the N domain exhibited a modified tendril or tendril-free phenotype, a manifestation potentially influenced by environmental factors that also warrants subsequent investigation.
Within this context, our finding underscores the selectivity of transcriptional repression mediated by dCas9-N or N-dCas9 across diverse genes. Nevertheless, not all target loci are susceptible to dCas9-N or N-dCas9 repression, as manifested by previously reported regulatory tools (Gilbert et al. 2013; Lowder et al. 2015; Piatek et al. 2015; Vazquez-Vilar et al. 2016). The efficacy of our dCas9 tool may be influenced by the genomic context of the target site, including the existence of endogenous transcriptional regulators and the local epigenomic status. Therefore, choosing functional target sites would be critical to effective transcriptional repression. While no very comprehensive study summarizing rules for CRISPRi target selection in plants has yet been reported, we empirically consider some parameters that would facilitate this process. Firstly, all possible target sites on the target gene should be assessed using bioinformatic packages or online platforms (Xie et al. 2017) to evaluate off-target probabilities and identify specific candidates. In terms of genomic location, we primarily focus on targets proximal to the TSS, particularly those located within the window of DNA from −50 to +300 bp relative to the TSS, a region reported to confer better CRISPRi activity in human cells (Gilbert et al. 2014). Targeting other regions within the gene body distal to the TSS is also effective in our study (Figs. 2 and 3), which was not observed for other dCas9 repressors in plants, but we will try to avoid designing targets in the 3′ UTR region and beyond. Additionally, targets with nucleotide homopolymers (AAAA, GGGG, UUUU) or extreme GC contents (lower than 30% or higher than 70%) should be discarded, as these characteristics were believed to have negative effects on repression activity (Gilbert et al. 2014). Although these design guidelines may be useful for the implementation of our dCas9 repression tool, considering the complexity of gene regulation, we strongly recommend conducting a preliminary experiment on 2–4 target sites within the target gene before practical application to ensure its efficacy. Alternatively, researchers can overcome inefficiencies in repression associated with poor target selection by simply targeting the same gene with an array of different sgRNAs (Lowder et al. 2015; Piatek et al. 2015; Vazquez-Vilar et al. 2016; Yeo et al. 2018).
In our experiments with Arabidopsis, a comprehensive comparison of inhibitory effects between N-dCas9 and dCas9-N revealed comparable repression efficacies but distinct protein expression levels. Additionally, fusion of multiple copies of the N domain with the dCas9 protein did not augment inhibitory effects. Western blot analysis revealed that it may be caused by the degradation of fused proteins with repeated domains and lead to instability of the entire protein system, which is also observed in similar instances (Gilbert et al. 2013; Li et al. 2017).
We verified the effectiveness and versatility of dCas9-N when targeted to a single locus through transient and stable transformation in cucumber and Arabidopsis and confirmed its specificity by RNA-seq. However, the potential for multiplex targeting of a single gene or multigene corepression by dCas9-N remains unexplored within whole plants in this study. Consequently, optimizing the performance of corepression via our repression tool may emerge as a future research direction, promising to provide comprehensive strategies to enhance the transcriptional repression efficacy of dCas9-N in plants. Building on previous studies, TEN is an intragenic enhancer-binding transcription factor, facilitating chromatin accessibility. Additionally, the modification to histone could alter the nucleosome structure and dynamics (Tessarz and Kouzarides 2014). Therefore, we can envisage the mechanism where the binding of the assembled DNA on chromatin could probably affect the DNA and histone interaction, thus hindering the initiation or elongation of the transcription process. The interaction of the N domain with other protein factors might be distinct between different target sites, potentially leading to varying inhibitory effects on transcriptional elongation and transcriptional repression efficiencies.
In this study, our investigation also yielded the creation of tendril-free cucumber germplasm through precise targeting of the tendril-regulated gene CsACO1 with dCas9-TEN fusion proteins. Traditional considerations of cucumber tendrils as energy-intensive structures led to their manual removal in contemporary agricultural practice, a laborious and time-consuming endeavor. This study also holds potential as a germplasm resource, envisaging a simplified breeding approach for cucumber and serving as a reference for breeding other crops.
Materials and methods
Plant materials and growth conditions
Wild-type (WT) Arabidopsis (A. thaliana) Col-0 plants were used in this study, which were grown in a plant growth room under photoperiods of 12 h light (75 μmol m−2 s−1) at 23 °C and 12 h dark at 21 °C with 65% constant humidity. The cucumber (C. sativus) inbred line CU2 (South China type) was used in this study, which was planted in a greenhouse under 16 h light/8 h dark at a temperature from 18 °C to 24 °C.
sgRNA design and plasmid construction
For cucumber plasmid construction, the sgRNA was designed in the CRISPR-GE web (http://skl.scau.edu.cn) and inserted into the pBSE402 vector as previously described (Xing et al. 2014). The short DNA fragment was assembled into pBSE402 using the restriction enzymes BsaI and T4 Ligase (New England Biolabs). The constructed recombinant vector was subjected to DNA sequencing for sequence verification and was transformed into A. tumefaciens strain EHA105. All sgRNA sequences are shown in Supplementary Table S1.
For transient expression in Arabidopsis protoplasts, the coding sequences of dCas9 and the N-terminal domain of TEN (N) were assembled in different orientations into the NcoI/PstI-digested HBT vector (Li et al. 2013) to obtain HBT-dCas9-N and HBT-N-dCas9 plasmids by recombination-based cloning using the ClonExpress MultiS One-Step Cloning Kit (Vazyme). The 3 × SRDX coding sequence was fused to the 3′ end of dCas9 by PCR as part of a mega-primer to generate HBT-dCas9-3SRDX. Two and 4 copies of the N domain coding sequence were fused to the 5′ end of dCas9 to construct HBT-2N-dCas9 and HBT-4N-dCas9, respectively, by the Golden Gate cloning method (Engler et al. 2008). The sgRNA expression plasmids were constructed by inserting annealed oligos of guide sequences into the BsaI-digested pUC119-sgRNA plasmid (Xiong et al. 2022), which already includes an AtU6-26 promoter and the sgRNA scaffold.
To construct binary plasmids for Arabidopsis stable transformation, firstly, the expression cassette of dCas9 or N-dCas9 was amplified from the corresponding HBT plasmid and inserted into the pFGC-RCS vector (Li et al. 2013) through the StuI and SbfI sites. Subsequently, the sgRNA cassette cut out from the pUC119-sgRNA plasmid was further inserted into the AscI site of the pFGC vector.
RT-qPCR analysis
Total RNA was isolated using an RNA extraction kit (Qiagen, Cat. #74104). First-strand cDNA was synthesized from 1 μg total RNA using the M-MLV Reverse Transcriptase (Promega, Cat. #M1705). Primer specificity was checked by sequencing and BLAST analysis. RT-qPCR assays were performed on an ABI 7900 (Applied Biosystems) using SYBR Premix (Roche, Cat. No. 04707516001), according to the manufacturer's instructions. Three independent biological replicates were performed. Relative gene expression was performed using the comparative 2−ΔΔCt method. CsACTIN2 or AtACTIN2 was used as the internal reference gene. The primers are listed in Supplementary Table S2.
A. rhizogenes–mediated hairy root transgenic system in cucumber
A. rhizogenes-mediated hairy root transformation of cucumber was carried out as previously described (Boisson-Dernier et al. 2001). Cucumber CU2 seeds were soaked in distilled water at 50 °C for 30 min. The seed coats were then removed with forceps, and the naked seeds were surface sterilized with 75% (v/v) ethanol for 15 seconds, followed by 0.4% (v/v) sodium hypochlorite solution for 15 minutes, and then rinsed 4 times with sterile water. These sterilized seeds were germinated and grown on MS medium for 1 week. Germinated cotyledons were sectioned into multiple square fragments, ∼1 × 1 cm in size, and soaked with diluted A. rhizogenes strain Ar. Qual (OD600 = 0.05) for 20 min. After the liquid culture was removed, explants were placed on MS solid medium, covered with sterilized filter paper, and allowed to co-cultivate for 2 days in the dark at 23 °C. Next, the explants were transferred to MS solid medium containing 200 mg/L timentin for 2 weeks in the light. After 7 days of incubation, the hairy roots with green fluorescent protein (GFP) were collected for subsequent experiments.
Protoplast isolation and transfection in Arabidopsis
Four-week-old Arabidopsis were used for protoplast isolation. Briefly, Arabidopsis leaves were cut into 0.5 mm strips and digested in 10 mL enzyme solution containing 1.5% cellulase R10 (Yakult Honsha, w/v), 0.4% macerozyme R10 (Yakult Honsha, w/v), 0.4 M mannitol, 20 mM MES (pH 5.7), 20 mM KCl, 10 mM CaCl2, and 0.1% BSA for 3 h. After mixing with 10 mL of W5 solution containing 154 mM NaCl, 125 mM CaCl2, 5 mM KCl, and 2 mM MES (pH 5.7), the digestion mixture was filtered through a 75 μm Falcon cell strainer. Cells were collected by centrifugation for 2 min at 100 × g at room temperature, resuspended in 10 mL of W5 solution, and rested on ice for 30 min. After centrifugation for 1 minute, the cell pellet was resuspended in the MMg solution containing 0.4 mM mannitol, 15 mM MgCl2, and 4 mM MES (pH 5.7) to a concentration of 2 × 105 cells per mL. DNA transfection was carried out in a 5 mL round-bottom microcentrifuge tube, where 400 μL protoplasts were gently mixed with 20 μL plasmid (2 μg/μL) expressing dCas9-derived repressor, 20 μL plasmid (2 μg/μL) expressing sgRNA, and 440 μL PEG solution containing 40% PEG4000 (v/v), 0.2 M mannitol, and 0.1 M CaCl2. For comparison of the expression of dCas9 repressors, an additional 1 μL GFP-expressing plasmid (2 μg/μL) was transfected. The mixture was incubated at room temperature for 5 minutes before the transfection was quenched by adding 1.6 mL of W5 solution. Transfected cells were collected by centrifugation for 3 min at 100 × g at room temperature, resuspended in 200 μL W5 solution, and transferred to 2 mL WI solution containing 0.5 M mannitol, 20 mM KCl, and 4 mM MES (pH 5.7) for 16 h incubation at room temperature in the dark for gene expression detection. Finally, protoplasts were harvested by centrifugation for 3 min at 200 × g for subsequent RT-qPCR and Western blot assays.
Stable genetic transformation
For cucumber, small guide RNA (sgRNA) design and plasmid construction were performed as previously introduced above. The transformation of cucumber was carried out as previously described (Xin et al. 2022). Explants with GFP fluorescent shoots were selected and transferred to jars for rooting. Rooted seedlings were planted in the soil to promote growth and development.
For Arabidopsis, the recombinant pFGC binary plasmids were introduced into A. tumefaciens GV3101 cells through electroporation, which were in turn used for floral dip-mediated Arabidopsis transformation (Clough and Bent 1998). Transgenic T1 plants were selected on the soil soaked with 1:2,000 diluted Basta herbicides (Amresco) containing glufosinate ammonium.
Immunoblotting
Crude protein extracts were extracted from transfected protoplasts using extraction buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 4 M urea, 10% glycerol [v/v], 5 mM DTT, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1 × protease inhibitor cocktail [Roche]). Proteins were resolved in an 8% SDS-PAGE gel and transferred to a PVDF membrane. dCas9 proteins and GFP (as transfection control) were immunoblotted with anti-FLAG horseradish peroxidase (HRP)-conjugated antibodies (Sigma-Aldrich) at 1:10,000 dilution.
ROS burst assay
Immunity-associated ROS bursts were detected using a luminol-HRP-based chemiluminescence assay. Briefly, leaf discs with a diameter of 4 mm were cut out with a punch from the leaves of 6-week-old WT or T2 transgenic Arabidopsis plants expressing N-dCas9 and sgRNA targeting AtFLS2. Two pieces of leaf discs were taken from each plant, one for immune elicitor treatment and the other for H2O control. Each leaf disc was floated on 200 μL of sterile water overnight in individual wells of a 96-well plate. On the next day, after removal of the liquid, a 100-μL HRP-luminol reaction mixture containing 20 μg/mL HRP (Sigma-Aldrich), 40 μg/mL luminol (Sigma-Aldrich), and 100 nM flg22 elicitor was added to each well. The ROS burst was immediately measured in a time-course manner using a Varioskan LUX microplate reader (Thermo Scientific). The luminescence of each well was recorded one by one with an integration time of 1 s for each reading. As a result, each well was measured every 2 minutes for 30 times in total. The kinetics of ROS production were determined by plotting the average luminescence units from all the leaf discs under the same condition over the reading period. Every time point is the mean value of 8 leaf discs.
RNA-seq analysis
Arabidopsis protoplasts expressing N-dCas9 and sgRNA-AtFLS2 or empty vector and cucumber hairy roots transformed with dCas9-N and sgRNA-Csa2G238880 were collected for RNA extraction with the RNAiso Plus reagent (TaKaRa). A total of 3 μg of RNA per sample was used for the RNA-seq library construction. RNA-seq was carried out on the Illumina HiSeq X platform with 150-bp paired-end reads at BioMarker Technologies (Beijing, China). For gene expression analyses, the gene count was calculated by using counts per million (CPM), and the genes with P > 0.05 were filtered out. The average CPM value of 2 biological replicates was used to calculate the gene expression level. Pearson's correlation coefficient was calculated using Microsoft Excel.
Recombinant protein expression and purification (Escherichia coli His-N121)
The recombinant protein His-N121 was cloned into vector pET22b with 6 × His-tag and then transformed into Escherichia coli BL21 (DE3). After induction with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 5 h at 26 °C, cells were harvested and then affinity purified (25 mM Tris-HCl, pH 7.5, 500 mM NaCl) with an Ni2+-chelating Sepharose Fast Flow column (Amersham Biosciences) in the lysis buffer. Purified supernatant was analyzed by SDS-PAGE.
Histone expression and purification
Recombinant histones (H2A, H2B, H3, and H4) were generated in BL21 (DE3) pLysS cells. The colonies containing expressed vectors were cultured in LB medium at 37 °C until the optical density reached 0.6 at 600 nm. Induction was carried out by adding 0.5 mm of IPTG for 2 hours. Cells were harvested through centrifugation at 5,000 × g at 4 °C for 15 min and suspended in a lysis buffer (50 mM Tris, 100 mM NaCl, 1 mM EDTA, 1 mM 2-mercaptoethanol, pH 7.5). Sonication was used to lyse the cells, followed by another centrifugation at 30,000 × g at 4 °C for 15 min. Sediments were washed by resuspending in 100 mL of wash buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM 2-mercaptoethanol, 1% v/v, Triton X-100) and then centrifuged for 10 min at 4 °C at 23,000 × g. This process was repeated once with wash buffer and twice with lysis buffer. The resulting pellet was dissolved in 30 mL of unfolding buffer (consisting of 7 mL of guanidium hydrochloride, 20 mL of Tris at pH 7.5, and 10 mL of DTT) for 1 h at room temperature. Following centrifugation for 15 minutes at 4 °C at 12,000 rpm, the supernatant was used for SDS-PAGE analysis.
Histone octamer and nucleosome assembly
For octamer reconstitution, equimolar amounts of individual histones in unfolding buffer were mixed and then dialyzed into refolding buffer (2 M NaCl, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 5 mM 2-mercaptoethanol) at 4 °C for 12 h. Purification was conducted with a Superdex S200 gel filtration column (Cytiva) and diluted by slowly pumping in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). The collected octamers were checked for purity by SDS-PAGE and then stored at −80 °C.
For nucleosome assembly, a molar ratio of 1:1.05 of histone octamers and 180 bp (5S rDNA) DNA templates was mixed in 2 M NaCl buffer and then dialyzed for 16 h at 4 °C in refolding buffer. A peristaltic pump was used to continuously dilute by slowly pumping in dialysis buffer to lower the concentration of NaCl from 2 to 0.6 M. The assembled nucleosomes were collected after final dialysis in Hepes-EDTA buffer (10 mM Hepes, pH 8.0, 0.1 mM EDTA) for 4 h and visualized on 2% (w/v) agarose gels.
EMSA
The recombinant His-N121 protein was purified from BL21 (DE3) E. coli. DNA probes were amplified and labeled with the biotin 3′ end DNA labeling kit (Thermo Scientific). The binding buffer contains 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10 mM KCl, 1 mM MgCl2, 10 μM ZnCl2, 0.5 mg/mL BSA, and 0.02 mg/mL poly(deoxyinosinic-deoxycytidylic) sodium salt. EMSA was performed following the instructions for the LightShift Chemiluminescent EMSA kit (Thermo, Cat. No. 20148).
Data analysis
All RT-qPCR analyses were conducted with 3 biological replicates and 3 technical replicates. The 2−ΔΔCt method was used to calculate relative gene expression, and all data were shown as means ± Sd; the significant differences among groups were tested using 1-way ANOVA with a post hoc Tukey test (P < 0.05). Differences between 2 groups were tested using the Student's t test (*P < 0.05, **P < 0.01; ns, no significance). Statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software Inc., San Diego, CA).
Accession numbers
Gene accession numbers are provided in the supplemental tables.
Acknowledgments
We thank Prof. Huiming Chen for providing cucumber seeds. We also thank Dr. Weijie Jiang for providing a greenhouse for the development and phenotypic investigation of transgenic cucumbers.
Author contributions
X.Y. and J.F.L. contributed to the experimental design and supervised the study. B.W., Z.L., T.X., and K.Z. contributed to genetic transformation and gene expression detection. B.W., Z.L., and K.Z. were involved in the cucumber and Arabidopsis phenotype investigations. B.W., Z.L., K.Z., and J.G. were involved in the construction of plasmids. Z.Z. contributed to the purification of histone and the assembly of octamer. X.Y., B.W., X.L., and Z.L. analyzed the data. B.W. and X.L. were involved in writing the manuscript.
Supplementary data
The following materials are available in the online version of this article:.
Supplementary Figure S1. Effects of transient transformation of dCas9-N on the relative expression of various genes in cucumber and Arabidopsis.
Supplementary Figure S2. Comparison of dCas9 repressor variants with different copy numbers of the N domain.
Supplementary Figure S3. RT-qPCR analysis of putative off-target genes when dCas9-N was targeted to CsACO1 and Csa6G450370 by transient transformation of cucumber cotyledons.
Supplementary Figure S4. RT-qPCR analysis of putative off-target genes when dCas9-N was targeted to Csa7G398090, Csa2G238880, and Csa6G010030 by transient transformation of cucumber cotyledons.
Supplementary Figure S5. RT-qPCR analysis of putative off-target genes when N-dCas9 was targeted to AtFLS2 by transient transformation of Arabidopsis protoplasts.
Supplementary Figure S6. Assembly and purification of histone octamers.
Supplementary Table S1. sgRNA sequences used in this study.
Supplementary Table S2. RT-qPCR primers used in this study.
Supplementary Table S3. Putative off-target sites of all target genes in this study.
Funding
This work is supported by the National Natural Science Foundation of China (32172606 and 31922076 to X.Y., 32202498 to X.L., and 32300339 to Z.L.), the National Key Research and Development Program of China (2019YFA0906204), the Beijing Joint Research Program for Germplasm Innovation and New Variety Breeding (G20220628003-03), the Chinese Academy of Agricultural Sciences Innovation Project (CAAS-ZDRW202103), the Science and Technology Program of Beijing (Z231100003723005), and the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2023-IVF).
Data availability
The sequencing data associated with this study are stored and available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under PRJNA1077600 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1077600).
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
References
Author notes
Bowen Wang, Xiaolin Liu and Zhenxiang Li contributed equally.
The author responsible for the distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) is Xueyong Yang ([email protected]).
Conflict of interest statement. The Author declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}