





Abstract
N6-methyladenosine (m6A) modification on messenger RNAs (mRNAs) is deposited by evolutionarily conserved methyltransferases (writers). How individual m6A writers sculpt the overall landscape of the m6A methylome and the resulting biological impact in multicellular organisms remains unknown. Here, we systematically surveyed the quantitative m6A methylomes at single-nucleotide resolution and their corresponding transcriptomes in Arabidopsis (Arabidopsis thaliana) bearing respective impaired m6A writers. The m6A sites associated with the five Arabidopsis writers were located mostly within 3′ untranslated regions with peaks at around 100 bp downstream of stop codons. m6A predominantly promoted the usage of distal poly(A) sites but had little effect on RNA splicing. Notably, impaired m6A writers resulted in hypomethylation and downregulation of transcripts encoding ribosomal proteins, indicating a possible correlation between m6A and protein translation. Besides the common effects on mRNA metabolism and biological functions uniquely exerted by different Arabidopsis m6A writers compared with their counterparts in human cell lines, our analyses also revealed the functional specificity of individual Arabidopsis m6A writers in plant development and response to stresses. Our findings thus reveal insights into the biological roles of various Arabidopsis m6A writers and their cognate counterparts in other multicellular m6A methyltransferase complexes.
Introduction
Chemical modifications of RNA are emerging as important regulatory components of gene expression. Among all these modifications, N6-methyladenosine (m6A), formed by the methylation of the N6 position of internal adenosine residues, represents the most widespread internal modification in eukaryotic messenger RNAs (mRNAs). m6A influences nearly every aspect of mRNA metabolism, including pre-mRNA processing (Ke et al., 2015; Haussmann et al., 2016; Zhou et al., 2019), nuclear export (Fustin et al., 2013; Roundtree et al., 2017), translation (Wang et al., 2015; Zhou et al., 2015), and decay (Wang et al., 2014; Shen et al., 2016), thereby modulating a wide range of fundamental biological processes in eukaryotes.
Deposition of m6A to target mRNAs requires evolutionarily conserved methyltransferases, which are collectively termed m6A writers. In the model plant Arabidopsis (Arabidopsis thaliana), so far two types of m6A writers have been suggested to install m6A on mRNA. The first multicomponent writer complex possibly consists of the mRNA adenosine methylase (MTA) (Zhong et al., 2008), its homolog MTB (Růžička et al., 2017), FKBP12 INTERACTING PROTEIN 37KD (FIP37) (Shen et al., 2016), VIRILIZER (VIR), and HAKAI (Růžička et al., 2017). Like their mammalian counterparts, these components install m6A preferentially near the stop codon and 3′ untranslated regions (UTRs), which is predominantly responsible for m6A levels detectable in Arabidopsis. By contrast, FIONA1 (FIO1), the only Arabidopsis ortholog of the human methyltransferase METTL16, acts separately to deposit m6A modifications mainly in U6 small nuclear RNA and a subset of mRNAs contributing modestly to the overall m6A levels (Wang et al., 2022; Xu et al., 2022).
The effects of m6A on mRNA stability are the best-characterized roles of m6A in plants to date and ultimately influence various aspects of plant developmental processes and stress responses (Anderson et al., 2018; Wei et al., 2018; Parker et al., 2020; Arribas-Hernandez et al., 2021; Shao et al., 2021; Cheng et al., 2022). Notably, knocking out any of the components except for HAKAI in the Arabidopsis writer complex results in embryonic lethality (Tzafrir et al., 2004; Vespa et al., 2004; Zhong et al., 2008; Růžička et al., 2017), which implicates an essential role of m6A in plants, and also reveals a technical challenge of elucidating the function of various writer components in post-embryonic development. In multicellular organisms, including Arabidopsis, some studies have been performed to understand specific m6A methylomes and the relevant biological responses associated with the disruption of individual m6A writers in a writer complex (Bodi et al., 2012; Shen et al., 2016; Parker et al., 2020; Qin et al., 2022; Zhang et al., 2022). However, as these individual m6A methylomes were surveyed using different methodologies and experimental materials collected under different growth conditions, it is practically unreliable to compare these methylomes with distinct detection resolutions and biological backgrounds. Thus, by far, it remains largely unknown how individual m6A writers in a methyltransferase complex contribute to the overall landscape of m6A methylomes, the global transcriptome profiles, and the associated biological processes in a given developmental context.
Among the methodologies commonly used for mapping m6A methylomes, both the methylated RNA immunoprecipitation sequencing (MeRIP-seq/m6A-seq) and m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) combine immunoprecipitation using m6A-specific antibody with short-read cDNA-based sequencing (Dominissini et al., 2013; Linder et al., 2015). Although miCLIP can detect m6A at single-base resolution, it is unable to quantify differential modifications among different samples (Koh et al., 2019). In contrast, nanopore long-read direct RNA sequencing (DRS) offers a promising approach to quantitatively locate m6A methylation at single-nucleotide resolution and compare m6A methylomes across different conditions (Garalde et al., 2018; Parker et al., 2020; Pratanwanich et al., 2021). In nanopore DRS, RNA modifications cause changes in intensity levels of the electric current when a native, unamplified RNA strand traverses the protein nanopore, thus permitting precise localization of modified bases in a quantitative manner by computational methods (Liu et al., 2019; Lorenz et al., 2020).
Here we report a systematic atlas of quantitative m6A methylomes at single-nucleotide resolution and their corresponding transcriptomes mediated by the known individual components, including MTA, MTB, FIP37, VIR, and HAKAI, in the Arabidopsis m6A writer complex using nanopore DRS. Comparison of these methylomes and transcriptomes obtained from respective genetic materials grown under the same conditions permits us to uncover important characteristics of common or distinct m6A modification effects conferred by the different Arabidopsis m6A writers in the same developmental context, which provides insights into the biological roles of various Arabidopsis m6A writers and their cognate counterparts in other eukaryotic m6A methyltransferase complexes.
Results
m6A writers affect plant development
To systematically compare the effects of different m6A writers on plant development and m6A deposition, we created genetic materials with impaired expression or activity of MTB, VIR, and HAKAI in addition to the previously reported fip37-4 LEC1:FIP37 and AmiR-mta in which FIP37 and MTA were knocked down (Shen et al., 2016). First, we isolated two heterozygous T-DNA insertional mutants of mtb from ABRC, designated mtb-1/+ and mtb-2/+, which produced aborted mtb-1 and mtb-2 homozygous seeds (Supplemental Figure 1, A and B). We then partially complemented mtb-2 with the ABI3:MTB transgene, in which the coding sequence of MTB was driven by the embryo-specific promoter of ABA INSENSITIVE 3 (ABI3) (Parcy et al., 1994) to study the post-embryonic function of MTB. One mtb-2/+ ABI3:MTB line containing a single copy of the transgene based on the segregation ratio was selected for further characterization (Supplemental Figure 1B). Among its progenies, the homozygous mtb-2 ABI3:MTB seedlings were isolated for subsequent analyses according to their distinctive growth phenotype (Figure 1A). Second, as loss of function of VIR also led to embryonic lethality (Růžička et al., 2017), we generated its knockdown lines (AmiR-vir) by artificial microRNA (AmiR) interference (Supplemental Figure 1A) (Schwab et al., 2006). Third, we generated a mutant of HAKAI by CRISPR/Cas9-mediated gene editing using a single guide RNA targeting a region within the first exon of HAKAI. From the T3 population of the transformants, we selected a homozygous null mutant without the CRISPR/Cas9 transgene, hakai-3, which contains a 1-bp deletion of guanine (G) resulting in a premature stop codon immediately in the first exon (Supplemental Figure 1, A and C).
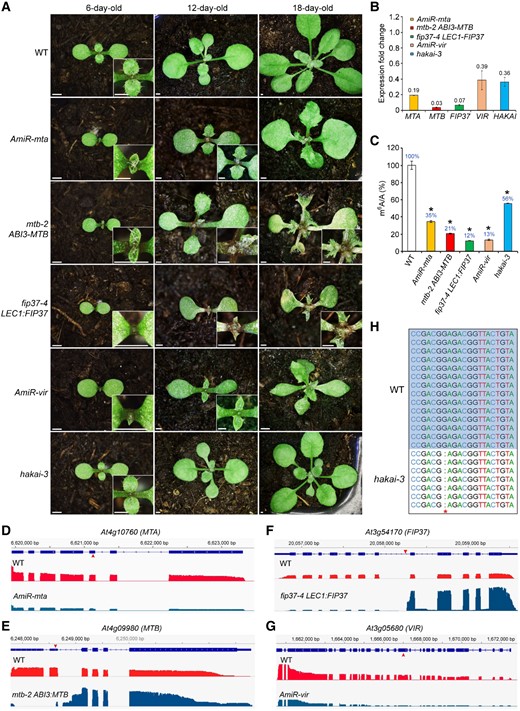
m6A writers affect plant development. A, Disruption of individual m6A writer genes except for HAKAI results in severe growth defects. The insets show enlarged views of the respective shoot apical meristem regions. Scale bars, 1 mm. B, RT–qPCR analysis of MTA, MTB, FIP37, VIR, and HAKAI expression, respectively, in 6-day-old AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, Amir-vir, and hakai-3 seedlings. The expression levels of each gene in 6-day-old wild-type seedlings were set as 1.0. Error bars, mean ± Sd; n = 3 biological replicates. C, m6A levels relative to those of adenosine (m6A/A) determined by LC-MS/MS analysis in mRNA isolated from 6-day-old seedlings in different genetic backgrounds. The m6A level in wild-type seedlings was set as 100%. Error bars, mean ± Sd; n = 2 biological replicates. Asterisks indicate significant differences in m6A levels between the indicated genetic materials and wild-type seedlings (two-tailed paired Student’s t-test, P < 0.01). D–G, Alignment of nanopore DRS reads for MTA (D), MTB (E), FIP37 (F), and VIR (G) showing either reduced transcript levels or truncated transcripts in respective genetic materials used in this study. Upward or downward red arrowheads indicate the AmiR targeting sites in MTA and VIR or the T-DNA insertion sites in mtb-2 and fip37-4, respectively. H, Nucleotide sequence alignment of HAKAI from wild-type and hakai-3 plants showing the 1-bp deletion of G in the first exon of HAKAI in hakai-3.
Next, we examined the post-embryonic phenotypes of AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, AmiR-vir, and hakai-3. All these plants, excluding hakai-3 exhibited a clear delay in leaf initiation and growth and produced leaves with irregular margins and abnormal outgrowth (Figure 1A). Strikingly, both mtb-2 ABI3:MTB and fip37-4 LEC1:FIP37 generated fasciated shoot apical meristems at the early seedling stage (Figure 1A). They subsequently developed necrotic lesions on the cotyledons and the first pair of leaves and eventually died without flowering (Figure 1A). AmiR-mta and AmiR-vir also produced large shoot apical meristems even with meristem outgrowth on lobed leaves visible at the late seedling development stage (Supplemental Figure 2A) but were eventually able to flower and set seeds. In contrast, like two previously reported hakai mutants (Růžička et al., 2017), hakai-3 did not show obvious growth defects during seedling development (Figure 1A and Supplemental Figure 2B).
In developing seedlings, MTB and FIP37 expression remained at very low levels in mtb-2 ABI3:MTB and fip37-4 LEC1:FIP37 compared to that in wild-type plants, respectively, while considerable reduction of MTA and VIR was observed in their respective AmiR lines (Figure 1B), confirming the impaired expression of m6A writer genes in these materials. Interestingly, we also observed reduced HAKAI expression in hakai-3 containing a point mutation (Figure 1B and Supplemental Figure 1C), implying possible nonsense-mediated decay of the mutated HAKAI transcripts harboring a premature stop codon or self-regulation of HAKAI. We then examined m6A levels in these genetic materials by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and revealed that m6A was reduced by 44%–88% compared to wild-type (Figure 1C). Levels of m6A reduction were associated with the phenotypic severity of these plants as exemplified by a comparison between fip37-4 LEC1:FIP37 displaying the lowest m6A levels and the most severe growth defects and hakai-3 bearing the lowest decrease of m6A levels without obvious growth defects (Figure 1, A and C). Taken together, these observations demonstrate that these m6A writers govern m6A deposition associated with plant development.
Nanopore DRS of m6A writer mutants
To quantitatively examine the occurrence and regulation of m6A at single-nucleotide resolution mediated by different writers, we performed nanopore DRS on three biological replicates of 6-day-old wild-type, AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, AmiR-vir, and hakai-3 seedlings. The total number of high-quality reads (quality score ≥ 7) for each sample ranged from 1.6 to 3.4 million, and at least 96.2% of these reads were successfully mapped to the TAIR10 reference transcriptome using Minimap2 (Li, 2018) (Supplemental Figure 3A). The majority of nanopore reads had a high-quality score at around 20 with an average read length of about 840–971 nt (Supplemental Figure 3B). The longest read length detected was 12,155 nucleotides corresponding to At2g17930 which spans 35 exons (Supplemental Figure 3C). The expression of 19,035 genes was identified with good reproducibility across replicates (Supplemental Figure 4). These observations indicate the high quality of our nanopore data set used for subsequent analyses. Moreover, nanopore reads confirmed reduced transcript levels of MTA and VIR, truncated transcripts of MTB and FIP37, and the 1-bp deletion of G in HAKAI in their respective genetic materials (Figure 1, D–H), further corroborating the reliability of our nanopore DRS reads.
Precise and quantitative detection of hypomethylated m6A sites
Information on m6A modification can be extracted from the nanopore reads through several computational algorithms (Huang et al., 2019; Lorenz et al., 2020; Parker et al., 2020; Pratanwanich et al., 2021), among which xPore not only identifies m6A sites at the single base resolution, but also quantifies the fraction of modified reads known as the modification rate, thus providing an unprecedented detail of epitranscriptome (Pratanwanich et al., 2021). We thus analyzed our nanopore reads using xPore to profile m6A sites at single-nucleotide resolution and to compute the differential modification rates (DMRs) representing the differences in modification rates between the respective writer genetic materials and wild-type seedlings. Based on stringent criteria (P < 0.001; one-directional signal shifts; detectable in all biological replicates), we identified 6,899, 6,022, 3,650, 6,594, and 3,015 high-confidence hypomethylated m6A sites in AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, AmiR-vir, and hakai-3, respectively (Figure 2A and Supplemental Figure 5, Supplemental Data Set 1). While most of the DMRs in hakai-3 were distributed between −0.50 and 0, the patterns in other genotypes were similar to the majority of DMRs being distributed between −1.00 and −0.25. The lowest number of hypomethylated sites and overall lower DMRs observed in hakai-3 were consistent with the mild reduction of m6A levels as measured by LC-MS/MS (Figure 1C). In total, 15,551 unique hypomethylated m6A sites were identified, corresponding to 4,315 protein-encoding genes (Supplemental Data Set 1). We randomly selected a few candidates and validated the sites using m6A–IP–qPCR (Supplemental Figure 6), indicating the authenticity of hypomethylated sites revealed in our study. Furthermore, a comparison of our data with the miCLIP data set (Parker et al., 2020) revealed that 44.3%–56.5% of our hypomethylated sites were within 5 nt of miCLIP peaks (Figure 2, B and C), while 57.5%–77.4% of our hypomethylated m6A sites were located within m6A peaks identified by m6A-seq data (Shen et al., 2016; Duan et al., 2017) (Supplemental Figure 7A). Non-overlapping sites revealed in our nanopore DRS versus the previous studies could be due to distinct approaches used for identifying m6A sites and/or plant materials grown at different conditions.
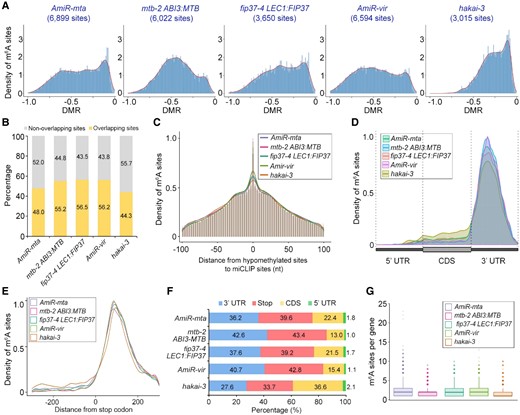
Identification of hypomethylated m6A sites in plants with impaired m6A writers. A, Density distribution of hypomethylated m6A sites and the corresponding DMRs identified from the comparison between the respective genotypes with impaired m6A writers and wild-type plants (P < 0.001). B, Overlap of hypomethylated sites found in this study with the published miCLIP data (Parker et al., 2020). C, Histogram illustrating the density distribution of hypomethylated m6A sites along the distance between our identified hypomethylated sites and miCLIP sites (Parker et al., 2020). D, Density distribution of hypomethylated m6A sites along transcripts divided into 5′ UTR, CDS and 3′ UTR. UTR, untranslated region; CDS, coding region. E, Density distribution of hypomethylated m6A sites at the stop codon region in respective genetic materials. F, Percentage of hypomethylated m6A sites along transcripts divided into 5′ UTR, stop (100 bp upstream or downstream of the stop codon), and other CDS and 3′ UTR regions in respective genetic materials. G, Box plot depicting the number of hypomethylated m6A sites per transcript in respective genetic materials. The median number of m6A sites for hakai-3 is 1, while the median number for other genotypes is 2. The third quartile and first quartile range of the data as well as the whiskers and outliers are also shown in the plot.
We next examined the distribution pattern of the hypomethylated sites along the transcripts and found that m6A was markedly enriched near the stop codon and 3′ UTR for all genetic materials examined (Figure 2D), which is in accordance with previous observations (Shen et al., 2016; Parker et al., 2020). A closer inspection revealed the enrichment of the hypomethylated sites at ∼100 bp downstream of the stop codon in all data sets (Figure 2E). Notably, the highest percentage of the hypomethylated sites were in the region 100 bp upstream or downstream of the stop codon in all genetic materials except for hakai-3, in which more than one-third of the hypomethylated sites were in the coding region (Figure 2F). Among hypomethylated transcripts found in this study, more than half of the genes contained only one hypomethylated m6A site in hakai-3, and this percentage was higher than those in other genetic backgrounds (Supplemental Figure 7B). In contrast, AmiR-mta had the highest percentage of genes (63%) containing more than one hypomethylated site (Figure 2G and Supplemental Figure 7B), reflecting the role of MTA as the major catalytically active subunit of the m6A methyltransferase complex.
Comparative m6A methylomes mediated by different m6A writers
To evaluate the sequence features of hypomethylated m6A sites associated with individual m6A writers, we performed motif search of a ± 20 bp region surrounding the hypomethylated site using MEME (Bailey and Elkan, 1994). The most frequently detected motif was RRACH (R = G/A, H = A/U/C) for all genotypes except for hakai where a more specific motif, ARACH, was identified (Figure 3A), implying that HAKAI is likely involved in m6A deposition on a smaller subset of target transcripts. Meanwhile, UGGAY (Y = U/C) and GGAUU were other significant motifs for a smaller number of consensus sequences (Supplemental Figure 7C).
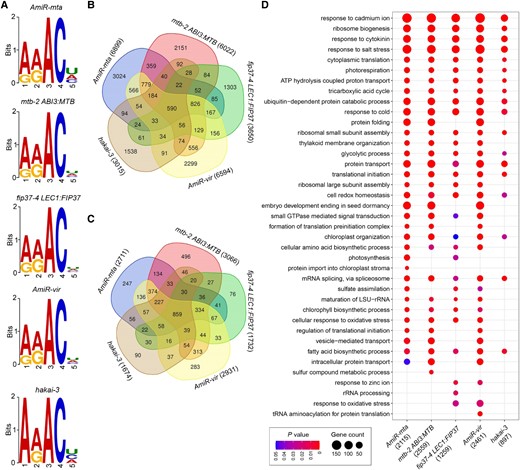
Functional enrichment analysis of hypomethylated transcripts. A, Sequence logos representing the top consensus motifs identified from hypomethylated m6A sites in the respective genotypes with impaired m6A writers. The top sequence logo RRACH (E value ≤ 2.5E−103; R = G/A, H = A/U/C) is identical for hypomethylated sites in plants with impaired MTA, MTB, FIP37 and VIR, while ARACH (E value < 2.5e−17) is the top sequence logo for the hypomethylated sites in hakai-3. The height of a nucleotide reflects its frequency. B and C, Venn diagrams depicting numbers of unique and overlapping hypomethylated sites (B) and transcripts bearing hypomethylated sites (C) among the five data sets. D, Functional enrichment analysis of transcripts containing hypomethylated m6A site(s) with DMR < −0.3. Top 30 GO terms for each genetic background were compared. The gene number for each genotype used in the enrichment analysis is indicated in the bracket.
Notably, we found that only 8.5%–19.6% (relative ratios of individual writers) of the hypomethylated sites were common to all five writers (Figure 3B and Supplemental Figure 8A), while on a transcript basis, the ratios of commonly modified transcripts were higher, ranging from 28.0% to 51.3% (Figure 3C). The ratios of common sites or transcripts were increased when we compared the profiles among a smaller number of writers. These observations indicate that individual m6A writers in the methyltransferase complex may modify different targets by acting in various combinations (Figure 3, B and C).
In contrast, each impaired m6A writer also contained some unique hypomethylated sites and transcripts, indicating their respective indispensable roles in the methyltransferase complex. For example, we found that 3,024 and 2,151 hypomethylated m6A sites were unique to AmiR-mta and mtb-2 ABI3:MTB, respectively (Figure 3B and Supplemental Figure 8, B and C). Since both MTA and MTB contain the core catalytic motif “DPPW” (Supplemental Figure 9A) within the MT-A70 methyltransferase domain that is essential for m6A methylation (Shih et al., 2019), MTA and MTB may function both cooperatively and independently to deposit m6A to their target transcripts. To this end, we examined their in vitro m6A methyltransferase activity and found that MTB exhibited an even stronger m6A methyltransferase activity than MTA (Supplemental Figure 9B). Gene ontology (GO) enrichment analysis of hypomethylated transcripts unique to impaired MTA and MTB also revealed some distinct GO terms between these two writers (Supplemental Figure 9C), indicating that they independently contribute to m6A modification associated with different aspects of plant growth and stress responses.
Functional enrichment analysis of hypomethylated transcripts
To understand the biological context of m6A modification, functional enrichment analysis was carried out on the hypomethylated genes associated with each impaired writer using clusterProfiler R package (Wu et al., 2021). Top GO terms that were common to all five writers included ribosome biogenesis, response to cytokinin, cytoplasmic translation, and responses to cadmium ion and salt stress (Figure 3D), demonstrating that these writers methylate target transcripts with similar functions in some key cellular processes and responses to stresses. As the overall reduction in the methylation rate was lower in hakai-3 than in the other genetic backgrounds (Figure 2A and Supplemental Data Set 1), the numbers of genes in these GO terms in hakai-3 were usually the lowest among all genotypes examined.
Notably, when we interrogated the list of hypomethylated genes for the enrichment of cellular compartments, those genes associated with chloroplast/plastid appeared as the most significantly enriched GO terms (Supplemental Figure 10A). Loss of methylation in these genes could contribute to the severe developmental defects observed in plants with impaired m6A writers except hakai-3 (Figure 1A). These genes included those encoding essential proteins that constitute a functional chloroplast. For example, CELL GROWTH DEFECT FACTOR 1 (AT5G23040; Supplemental Data Set 1) is required for chlorophyll synthesis (Reinbothe et al., 2015), which is a functional GO term appearing in all genotypes except hakai-3 (Figure 3D). As depletion of this gene results in embryo arrest at the globular stage (Kawai-Yamada et al., 2014), its hypomethylated status might be associated with developmental arrest displayed by the seedlings with impaired m6A writers except hakai-3. Another example is CRUMPLED LEAF (CRL, AT5G51020; Supplemental Data Set 1) which encodes a chloroplast protein and its mutation leads to severe developmental defects with the constitutive expression of stress-related genes (Hudik et al., 2014), which resembles the transcriptomes of plants with impaired m6A writers (Figure 4). Indeed, a comparison of their transcriptomes showed a moderate positive correlation [Spearman correlation coefficient (ρ) = 0.62–0.75] between crl and the plants with impaired m6A writers except for hakai-3 (Supplemental Figure 10B). While these observations provide circumstantial evidence linking chloroplast function with m6A modification, further experiments are required to test whether loss of m6A at chloroplast-related genes could be associated with chloroplast malfunction and the resulting severe developmental defects exhibited by the seedlings with impaired m6A writers except hakai-3.
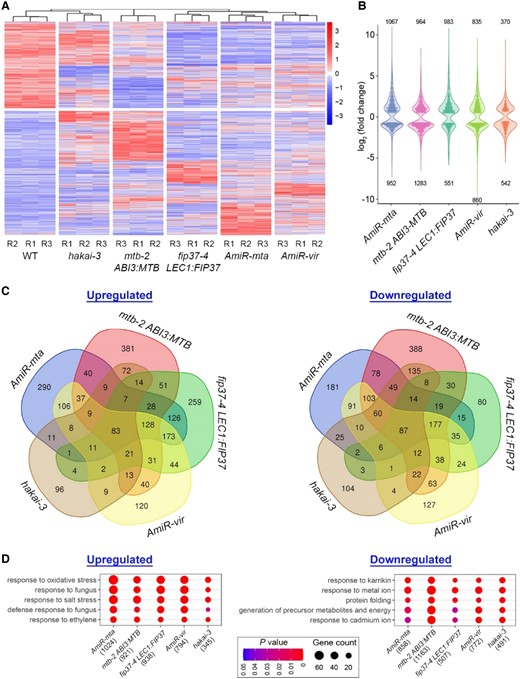
Effects of defective m6A deposition on the transcriptome. A, Heatmap showing the hierarchical clustering of differentially expressed genes (DEGs) between respective plants with impaired m6A writers and wild-type (WT) plants. B, Violin plot illustrating the fold change distribution of DEGs between the indicated genetic backgrounds and wild-type plants. The number of upregulated or downregulated genes for each data set is given above or below the plot, respectively. C, Venn diagram showing the numbers of unique and overlapping DEGs among different genetic backgrounds. DEGs were identified with log2 (fold change) > 0.5 or < −0.5 and P < 0.05. D, Top 5 GO terms that are common in the upregulated or downregulated gene list from different genetic backgrounds. The gene number for each genotype used in the enrichment analysis is indicated in the bracket.
Besides the common GO terms, we also found uniquely enriched GO terms for hypomethylated transcripts in plants with individual impaired writers (Supplemental Figure 11), indicating that individual m6A writers also specifically deposit m6A on different target transcripts involved in distinct biological processes in Arabidopsis.
Comparative transcriptomes affected by individual m6A writers
To understand the effects of individual m6A writers on the transcriptome, we performed differential gene expression analysis of the nanopore reads between impaired writers and wild-type plants using DESeq2 (Love et al., 2014). Hierarchical clustering of differentially expressed genes revealed that the transcriptomes of wild-type and hakai-3 plants were grouped together, while those of the other four genotypes contained two separate clusters with the mtb-2 ABI3:MTB transcriptome as an independent cluster (Figure 4A and Supplemental Data Set 2). Consistently, the mutation in HAKAI had the least disturbance on the transcriptome with the lowest number of differentially expressed genes among the genotypes with impaired m6A writers (Figure 4B). There were 211 and 264 commonly up- and downregulated genes, respectively, in all genotypes except hakai-3, while these numbers considerably dropped to only 83 and 87, respectively, if hakai-3 was included in the comparison (Figure 4C), substantiating that HAKAI influences a smaller group of genes than other writers.
GO analysis on differentially upregulated or downregulated transcripts revealed that responses to various abiotic and biotic stimuli, including oxidative stress, fungus, salt stress, karrikin and cadmium ion, were among the top five functional categories common to all genotypes (Figure 4D), implying that m6A greatly affects plant responses to external stimuli. Interestingly, in contrast to a smaller number of genes associated with two out of the five top GO terms in the downregulated list, all transcripts associated with the top five GO terms in the upregulated list were relevant to stress responses (Figure 4D), indicating that m6A modification is more prone to suppress the upregulation of stress-related transcripts, which likely occurs both directly and indirectly as some of these transcripts are hypomethylated (Supplemental Data Set 3).
Since response to salt stress was the top GO term associated with both the hypomethylated and differentially upregulated transcripts (Figures 3D and 4D), we tested the effect of defective m6A on plant response to salt stress. Seedlings with impaired m6A writers exhibited greater susceptibility to salt treatment than wild-type seedlings (Supplemental Figure 12), as shown in a previous study (Hu et al., 2021), implicating m6A in regulating salt stress responses.
We subsequently examined the top five enriched GO terms that were unique in each of the differentially expressed data sets (Supplemental Figure 13). For upregulated transcripts, genes associated with GO terms related to primary metabolism and photosynthesis were uniquely prevalent in AmiR-mta and fip37-4 LEC1:FIP37 transcriptomes, respectively. Growth-related GO terms were associated distinctly with upregulated transcripts in hakai-3, while some GO terms associated with stress and defense were only found in AmiR-vir. For downregulated transcripts, GO terms related to carbohydrate derivative biosynthesis, and several metabolic processes were prevalent in mtb-2 ABI3:MTB, while responses to hypoxia and oxygen were notable GO terms unique in hakai-3. These observations substantiate that individual m6A writers affect distinct aspects of plant growth and stress response despite some of their common functions.
m6A hypomethylation mainly decreases the stability of transcripts
To examine the potential association between m6A modification and mRNA abundance, we compared differentially expressed genes with hypomethylated genes. The transcripts containing hypomethylated m6A sites were predominantly downregulated in all plants with impaired m6A writers (Figure 5A), indicating that m6A modification mainly stabilizes mRNA in Arabidopsis. This is also consistent with a previous report showing that m6A primarily protects Arabidopsis mRNA from endonucleolytic cleavage (Anderson et al., 2018). Subsequent examination of the enriched GO terms for hypomethylated transcripts (DMR < −0.3) that were differentially regulated disclosed a greater overlap of functional categories among the different genotypes for hypomethylated transcripts that were downregulated than those upregulated (Supplemental Figure 14).
![m6a hypomethylation affects the stability of transcripts. A, Distribution of changes in expression of differentially expressed genes (log2 [fold change] > 0.5 or < −0.5; P < 0.05) with and without hypomethylated sites (DMR < −0.3) in various plants with impaired m6A writers. B, Paralogs of cytosolic RPs with hypomethylated m6A sites. A total of 207 out of 242 curated RP genes were identified to contain at least one hypomethylated m6A site in any of the impaired m6A writers. Heat map illustrates DMR and log2 (fold change) of the corresponding transcripts in various plants with impaired m6A writers compared to those of wild-type plants. C, RT–qPCR analyses of AT1G43170, AT3G52590 and TUB8 expression in 6-day-old seedlings in various genetic backgrounds treated by actinomycin D or DMSO (as mock treatment) for 0, 8, and 24 h. The relative expression of each gene at different time points was calculated by normalizing the gene expression in actinomycin D-treated samples against that in mock-treated samples. Asterisks indicate that the expression of AT1G43170 and AT3G52590 is significantly different between all plants with impaired m6A writers and wild-type plants after treatment for 8 and 24 h (two-tailed paired Student's t-test, P < 0.05). Error bars, mean ± Sd; n = 3 biological replicates.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/plphys/191/3/10.1093_plphys_kiad010/1/m_kiad010f5.jpeg?Expires=1747889458&Signature=23KKfVYnA6yXfMyE0RQiXAwyFGtwAgTkOnRf7Ak5jGOcByYdOzP0F97U4B76dn-BIeZ46gyLYMfi8ryMtqoimZDj4UgCPBzz04UQgxRrDBlt6nQXNrtsyX6UpY1kjc~kJ0TVzijE3LBnVdblpw47T739Pwsl5pZk-fCCXsvBHxGbevnmqbsT06bWhoazzwRzh29v2IVMzbp2vmKBl50krhgSvflUuUZhoJlKMEwDuqEuPauxFwCyxU~PnmR4WZHywD~hMehmi-ld5rtluymZEeoIy-M2yjUKoUkQ2a9kuZFgd3PhVOzENKllvYc6xh8TPAyPMDILOVpQG8plsSwFZQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
m6a hypomethylation affects the stability of transcripts. A, Distribution of changes in expression of differentially expressed genes (log2 [fold change] > 0.5 or < −0.5; P < 0.05) with and without hypomethylated sites (DMR < −0.3) in various plants with impaired m6A writers. B, Paralogs of cytosolic RPs with hypomethylated m6A sites. A total of 207 out of 242 curated RP genes were identified to contain at least one hypomethylated m6A site in any of the impaired m6A writers. Heat map illustrates DMR and log2 (fold change) of the corresponding transcripts in various plants with impaired m6A writers compared to those of wild-type plants. C, RT–qPCR analyses of AT1G43170, AT3G52590 and TUB8 expression in 6-day-old seedlings in various genetic backgrounds treated by actinomycin D or DMSO (as mock treatment) for 0, 8, and 24 h. The relative expression of each gene at different time points was calculated by normalizing the gene expression in actinomycin D-treated samples against that in mock-treated samples. Asterisks indicate that the expression of AT1G43170 and AT3G52590 is significantly different between all plants with impaired m6A writers and wild-type plants after treatment for 8 and 24 h (two-tailed paired Student's t-test, P < 0.05). Error bars, mean ± Sd; n = 3 biological replicates.
One stark example of the top functional category associated with hypomethylated transcripts that were downregulated includes those encoding the highly conserved ribosomal proteins (RPs) that make up ribosomal subunits involved in the cellular process of translation (Figure 5B). There are 79 RPs in Arabidopsis, with each encoded by several paralogs, thereby giving rise to ribosomes that are heterogeneous in their composition (Martinez-Seidel et al., 2020). Among the 242 genes encoding different RPs (Martinez-Seidel et al., 2020), transcripts corresponding to 207 genes (85.5%) that cover all RP families in Arabidopsis were hypomethylated in at least one type of the plants with impaired m6A writers (Supplemental Data Set 1). In particular, most of these hypomethylated RP transcripts (188 out of 207) were downregulated in at least one genotype (Supplemental Data Set 1), indicating that m6A methylation mainly stabilizes RP paralogs.
To verify the effect of m6A modification on mRNA decay of RP genes, we measured the levels of two randomly selected downregulated RP transcripts in 6-day-old seedlings with impaired m6A writers treated with the transcription inhibitor, actinomycin D. Both transcripts displayed obvious decreases in their cumulative RNA stability in these seedlings versus wild-type seedlings after actinomycin D treatment, whereas the stability of non-m6A targeted transcripts, such as β-tubulin (TUB8), remained almost unchanged (Figure 5C). This result confirms that m6A methylation decreases the decay of these two RP transcripts in seedlings. It will be interesting to further investigate whether the reduction in mRNA abundance for many RP paralogs mediated by impaired m6A methylation could influence their protein levels, thus affecting their participation in functional ribosomes and the subsequent translation processes.
m6A hypomethylation disrupts mRNA 3′-end processing
As m6A was evidently enriched near the stop codon and 3′ UTR (Figure 2D), we subsequently examined the impact of defective m6A deposition on two major steps of pre-mRNA processing, splicing and 3′-end polyadenylation. Only a small number of differential alternative splicing events, such as intron retention and exon skipping, were detected in plants with impaired m6A writers (Supplemental Figure 15A; Supplemental Data Set 4). In addition, the numbers of hypomethylated and non-hypomethylated transcripts that undergo different types of alternative splicing were mostly similar in the plants with defective m6A deposition (Supplemental Figure 15B). These observations suggest that m6A has a little direct effect on RNA splicing.
Polyadenylation of pre-mRNA is a key processing step in mRNA maturation and more than 70% of Arabidopsis genes have at least two poly(A) sites, leading to the expression of alternative polyadenylation (APA) isoforms (Wu et al., 2011). Since most APA sites were found within the 3′ UTR (Wu et al., 2011) where the majority of m6A sites are located (Figure 2D), we examined the effect of defective m6A deposition on APA and revealed clear defects in RNA 3′-end formation in 2159, 2168, 1414, 2545, and 595 genes, respectively, in the plants with different impaired m6A writers (Figure 6A and Supplemental Data Set 5). Strikingly, most of these transcripts (91.3%–94.6%) displayed a shift to the usage of proximal poly(A) sites (pPAS) (Figure 6A), which is also observed in the hypomorphic vir mutants (Parker et al., 2020). Nearly half of these genes (40.9%–51.6%; hypergeometric P value ≤ 1.49E−67) also contained hypomethylated m6A sites (Figure 6, B and C), implying that m6A could affect the usage of alternative poly(A) sites in plants. One example of hypomethylated genes with an increased pPAS usage in plants with impaired m6A writers except hakai-3 is AT1G77490 (Figure 6D), which encodes a thylakoid ascorbate peroxidase that plays a major role in regulating H2O2 level and thereby regulates H2O2-triggered retrograde signaling from chloroplasts to nucleus in response to stress (Maruta et al., 2012).
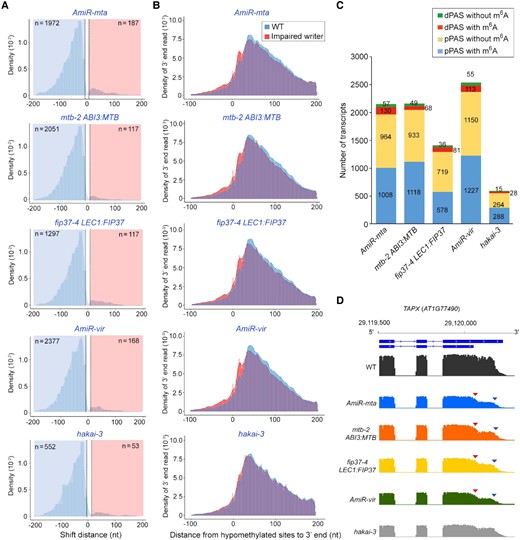
m6A affects poly(A) site selection. A, A shift to proximal 3′ end usage detected in all transcripts from various genotypes with defective m6A deposition. Histogram depicting the distance in nucleotides between the most reduced and most increased 3′ end positions for genes with significant changes in the 3′ end profile in various genotypes (Kolmogorov–Smirnov test, FDR < 0.05 and a threshold of 13 nt). B, A global shift toward proximal 3′ poly(A) site usage was observed in hypomethylated transcripts of the respective genotypes with impaired m6A deposition versus wild-type (WT) plants. The shift is less pronounced in the hakai-3 data set. The hypomethylated site is at position 0. C, Percentages of distal polyadenylation site (dPAS) and proximal polyadenylation site (pPAS) with or without m6A site. Alternate polyadenylation sites were detected by comparing the polyadenylation sites in various genotypes with those in WT plants. D, A representative gene, TPAX (AT1G77490), containing m6A in its 3′ UTR displays a shift from distal (dPAS; blue triangle) to proximal (pPAS; red triangle) 3′ poly(A) site usage in AmiR-MTA, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, and AmiR-VIR, but not obvious in hakai-3.
As m6A has been reported to protect the transcriptome integrity in plants (Pontier et al., 2019), we examined possible readthrough at poly(A) sites but found that very few transcripts were significantly affected in plants with impaired m6A writers (P value < 0.05; Supplemental Data Set 6). Overall, these results indicate a predominant effect of m6A modification on promoting the usage of distal poly(A) sites.
Comparison of m6A modification in plants and human cell lines
To gain a better understanding on the regulatory roles of m6A, we compared m6A features uncovered from this study with published data on the orthologs of plant writers in human cell lines, including METTL3 (ortholog of MTA), METTL14 (ortholog of MTB), Wilm's tumor 1-associating protein (WTAP) (ortholog of FIP37), VIRMA (ortholog of VIR), and HAKAI. As only one nanopore DRS data set on human embryonic kidney cell line with METTL3 knockout (HEK293T METTL3 KO versus HEK293T) was available (Pratanwanich et al., 2021), we also included published meRIP data derived from defective HeLa cell lines created either by CRISPR/CAS9 knockout of METTL3, METTL14 and VIRMA, or by siRNA knockdown of all five writers (Wang et al., 2014; Yue et al., 2018).
Although the m6A motif, RRACH, is conserved in animals and plants (Shao et al., 2021), the distribution of m6A enrichment along mRNA transcripts differed between Arabidopsis and human cell lines (Figure 2, E and 7A). m6A was highly enriched near stop codons with almost comparable distribution in surrounding coding regions and 3′ UTR in human HeLa cell lines (Wang et al., 2014; Yue et al., 2018) (Figure 7A), while m6A was mostly enriched in 3′ UTR with peaks downstream of stop codons in Arabidopsis (Figure 2E). A small number of m6A sites in coding regions of Arabidopsis mRNAs could be associated with weak effects of m6A modification on alternative splicing in Arabidopsis (Supplemental Figure 15A). In contrast, m6A effects on alternative splicing in animals are still under debate because of contradictory findings so far reported (Dominissini et al., 2012; Geula et al., 2015; Ke et al., 2017).
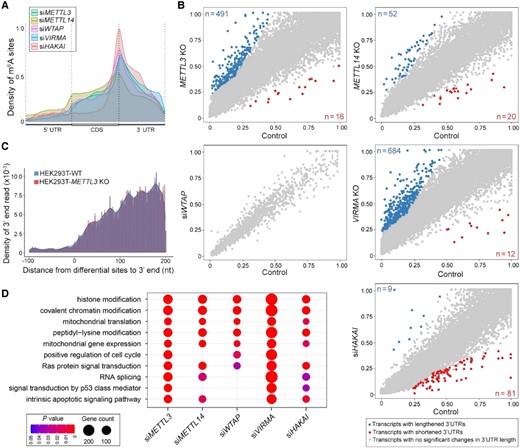
Features of m6A modification uncovered from human cell lines. A, Density distribution of hypomethylated m6A sites along transcripts divided into 5′ UTR, CDS and 3′ UTR. UTR, untranslated region; CDS, coding region. The analyses were performed on meRIP data of siRNA knockdown of the respective writers in human HeLa cell lines (8, 51). B, Effects of depletion of different m6A writers in human HeLa cell lines on the percentage of distal polyA site usage index (PDUI). meRIP data were generated using defective cell lines created either by CRISPR/CAS9 knockout of METTL3, METTL14, and VIRMA, or by siRNA knockdown of WTAP and HAKAI (Wang et al., 2014; Yue et al., 2018). DaPars was used for the de novo identification of APA (Xia et al., 2014) and the resulting scatter plots of PDUIs between defective and control cell lines are shown (FDR < 0.05 with at least two-fold increase or decrease). C, Knockout of METTL3 does not affect poly(A) site choice in the HEK293T cell line. The analysis was performed on nanopore DRS data (Pratanwanich et al., 2021). D, Functional enrichment analysis of human transcripts containing hypomethylated m6A sites using the meRIP data shown in A. Top 30 GO terms for each data set were compared. A complete list of the resulting GO terms is given in Supplemental Figure 16.
Besides the known m6A effect on accelerating mRNA decay in HeLa cells (Wang et al., 2014; Yue et al., 2018), we further assessed the influence of defective m6A deposition on the APA of mRNAs. Although there was a pronounced 3′ UTR lengthening of transcripts associated with hypomethylated peaks resulting from knockout of METTL3 or VIRMA (Yue et al., 2018) (Figure 7B), the numbers of affected transcripts were much lower than those found in their counterparts in Arabidopsis (Figure 6A). In addition, such an effect was either undetectable in siWTAP (Wang et al., 2014) or less obvious in the knockout of METTL14 and depletion of HAKAI (Yue et al., 2018) (Figure 7B). Surprisingly, analysis of APA in HEK293T cell line defective in METTL3 using nanopore DRS did not reveal m6A effect on poly(A) site choice (Pratanwanich et al., 2021) (Figure 7C). While these paradoxical findings indicate that different human cell lines and sequencing approaches may affect the detection of the effects of various m6A writers on APA, the overall results so far demonstrate a less prevalent effect of m6A on APA in human cell lines than in plants.
We also performed functional enrichment analysis on the hypomethylated genes associated with each impaired writer in human HeLa cell lines (Wang et al., 2014; Yue et al., 2018). Top GO terms common to all five writers included those associated with histone modification, chromatin modification, mitochondrial protein and gene expression, and protein modification (Figure 7D and Supplemental Figure 16). These categories are different from the top enriched functional GO terms in Arabidopsis (Figure 3D) implying different biological roles of m6A modification in plants and human cell lines.
Discussion
The m6A writer complex comprising multiple components catalyzes m6A deposition to target mRNAs in eukaryotes. Although some studies have been performed to understand the roles of individual writer components in determining m6A deposition on their targets, variations in biological materials and sequencing approaches have so far prevented a systematic survey on the common or distinct functions of various writer components in sculpting the landscape of m6A methylome in a given biological context. In this study, we have carried out nanopore DRS to compare the methylomes and transcriptomes generated from respective Arabidopsis genetic materials where the five writer components are impaired. As these plants were grown and harvested under the same conditions, the quantitative m6A methylomes at single-nucleotide resolution and their corresponding transcriptomes permit an unambiguous elucidation of different and complementary roles of individual writer components in determining m6A methylomes and the associated biological effects in Arabidopsis (Figure 8).
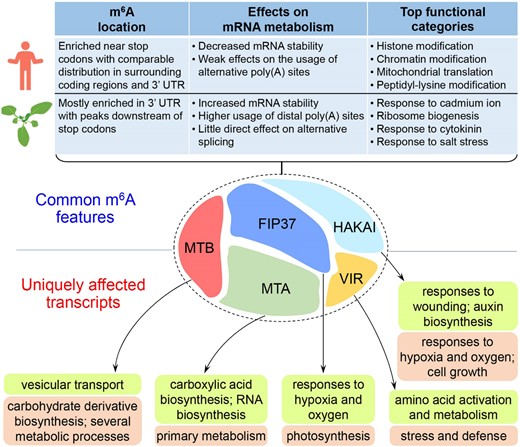
A schematic diagram summarizing the common and unique effects of m6A modification conferred by the different Arabidopsis m6A writers in the same developmental context. The upper panel shows the comparison of the common features of m6A modification, including m6A location, its effects on mRNA metabolism, and top functional categories of hypomethylated transcripts, exhibited by the five Arabidopsis m6A writers and their orthologs in human cell lines. The lower panel displays the top biological processes for the transcripts uniquely associated with individual Arabidopsis m6A writers. Biological processes pertaining to hypomethylated and differentially expressed transcripts for each m6A writer are listed in lime and light orange boxes, respectively.
Among the known components of the m6A writer complex in Arabidopsis, HAKAI has the lowest effects on m6A modification and plant development. First, unlike other writers, knocking out HAKAI does not cause obvious phenotypes during seed development and post-embryonic development (Tzafrir et al., 2004; Vespa et al., 2004; Zhong et al., 2008; Růžička et al., 2017) (Figure 1A and Supplemental Figure 2B). Second, among the plants bearing the impaired writers, disruption of HAKAI in hakai-3 results in the lowest reduction of m6A levels (Figure 1C) associated with the lowest number of hypomethylated sites (Figure 2A). Third, the transcriptome of wild-type plants more closely resembles that of hakai-3 than other impaired writers (Figure 4A), demonstrating that HAKAI influences a smaller number of transcripts than other writers. These pieces of evidence suggest that compared to the other four writers, HAKAI mediates m6A deposition to the least number of transcripts, conferring a minimal effect on the landscape of m6A methylome and transcriptome as well as the resulting plant phenotypes.
Despite various strengths exhibited by the five writers on m6A deposition, their effects on mRNA exhibit some common features uniquely in Arabidopsis compared with those in human cell lines (Figure 8). Most of the m6A sites associated with the five Arabidopsis writers are located within 3′ UTR with peaks at 100 bp downstream of stop codons, which contrasts with a comparable distribution of m6A sites in coding regions and 3′ UTR surrounding the peaks near stop codons in human cell lines. The differential m6A localization pattern is coupled with distinct effects on mRNA metabolism between Arabidopsis and human cell lines. m6A deposited by the Arabidopsis writers mainly stabilizes mRNA and promotes the usage of distal poly(A) sites, whereas m6A accelerates mRNA decay but has a less prominent effect on determining APA sites in human cell lines, and if any, m6A promotes the usage of pPAS (Wang et al., 2014; Yue et al., 2018; Pratanwanich et al., 2021). In addition, Arabidopsis writers deposit m6A on the transcripts with similar functions in some key biological processes, such as ribosome biogenesis and response to stresses. These functions are different from the top enriched ones associated with m6A writers in human cell lines. Together, these common features displayed by Arabidopsis m6A writers imply that different components of the m6A methyltransferase complex may have evolved to act in concert to install m6A on some key transcripts, which affects RNA metabolism and the resulting biological functions distinctively in multicellular plants.
Besides the common features, our analyses of nanopore DRS have also revealed the functional specificity of individual Arabidopsis m6A writers. The plants bearing individual impaired m6A writers all contain a group of unique hypomethylated and differentially expressed transcripts that are absent in other genetic backgrounds. These transcripts are enriched for different biological processes (Figure 8), indicating that the individual m6A writers specifically affect certain aspects of plant development and response to stresses by directly catalyzing m6A modification of the relevant transcripts and/or indirectly influencing gene expression as a result of m6A modification.
In addition to enhancing mRNA stability, it is possible that m6A modification influences other RNA metabolic processes, such as nuclear transport and translation, especially for many transcripts that are hypomethylated without any changes in their steady-state levels (Figure 3, D and 5A). This possibility is supported by two observations. First, there is a predominant effect of m6A modification on promoting the usage of distal poly(A) sites in Arabidopsis. This alters 3′ UTR length that could influence post-transcriptional gene regulation, including nuclear export and protein translation (Tian and Manley, 2017; Turner et al., 2018). Second, a high proportion of hypomethylated transcripts that are differentially expressed in the plants with impaired writers are those encoding RPs (Figure 5B), which are essential subunits in various functional ribosomes for translation. This observation implies that alteration in protein translation could be associated with m6A effects on many transcripts because of defective ribosomes. This potential role of m6A in ribosomal code is likely specific to plants as there is a lack of mRNAs encoding RPs among the m6A-containing mRNAs in yeast and mammalian cell cultures (Schwartz et al., 2014).
In conclusion, our study has provided unprecedented insights into the common and unique features of the methylomes and the corresponding transcriptomes associated with the five m6A writers in the Arabidopsis m6A methyltransferase complex. In addition to some common effects on mRNA metabolism uniquely exerted by these Arabidopsis writers versus their human counterparts, our findings also reveal the functional specificity of individual Arabidopsis writers in different biological processes. Further elucidation of the mechanisms underlying the target specificity of m6A deposition for various m6A writers in multicellular organisms, including Arabidopsis, requires in-depth investigation of the function and dynamics of RNA modification-related proteins that interact with an individual or grouped m6A writers in given biological contexts.
Materials and methods
Plant materials and growth conditions
Arabidopsis (Arabidopsis thaliana) plants were grown on soil or Murashige and Skoog (MS) plates under a 16 h light/8 h dark-light cycle at 23°C ± 2°C. Plants used were in the Columbia (Col) background. Seeds of mtb-1 (CS16197) and mtb-2 (CS850592) were obtained from the Arabidopsis Biological Resource Center (http://www.arabidopsis.org/).
Vector construction and plant transformation
To construct ABI3:MTB, the coding sequence of MTB was inserted downstream of the 5.0 kb ABI3 promoter in the modified pENTR vector (Shen et al., 2016). To construct AmiR-vir, the AmiR was designed by WMD3 Web MicroRNA Designer (http://wmd3.weigelworld.org/). Based on the AmiR sequence, a set of four primers were generated and used for PCR amplification according to the published protocol (Schwab et al., 2006). The resulting PCR fragment was digested with EcoRI and BamHI and cloned into a modified pENTR vector containing the 2 × 35S promoter. To construct CAS9-HAKAI, the CAS9-sgRNA cassette was amplified and cloned into the modified pENTR vector. The resulting vector was digested with BbsI and ligated with the synthesized sgRNA oligos. Agrobacterium tumefaciens-mediated transformation of Arabidopsis was performed with the floral dipping method. To generate mtb-2/+ ABI3:MTB, the ABI3:MTB construct was transformed into mtb-2/+ plants and selected by kanamycin on MS plates. T1 transformants harboring both ABI3:MTB and mtb-2/+ were maintained as independent lines, from which one representative line that contained only a single ABI3:MTB transgene based on the segregation ratio was selected for further investigation. AmiR-vir and CAS9-HAKAI were transformed into wild-type Col plants and selected by Basta on soil.
Expression analysis
Total RNA was isolated with the RNeasy Plus Mini Kit (Qiagen) and reversed transcribed with the M-MLV Reverse Transcriptase (Promega). Reverse transcription-quantitative PCR (RT–qPCR) was performed with three biological replicates on the 7900HT Fast Real-Time PCR systems (Applied Biosystems) using PowerUp SYBR Green Master Mix (Applied Biosystems). The differences between the cycle threshold (Ct) of target genes and the Ct of TUBULIN 2 (TUB2) (ΔCt = Cttarget gene—CtTUB2) served as an internal control for determining the normalized expression of target genes. Primers used for gene expression analysis are listed in Supplemental Data Set 7.
Measurement of m6A level via LC-MS/MS
Total RNA of 6-day-old seedlings of wild-type, AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, AmiR-vir, and hakai-3 was extracted using Trizol reagent (Invitrogen). mRNA was subsequently isolated using Dynabeads Oligo(dT)25 (Invitrogen)and was digested to single ribonucleosides following a published protocol (Thüring et al., 2017). Multiple reaction monitoring quantification was performed on a NanoLC 425 (Eksigent) coupled to a QTrap 6500 mass spectrometer (SCIEX) to detect A and m6A with mass transitions at 268.0–136.0 and 282.0–150.1, respectively. Solvent A was 2% (v/v) acetonitrile with 0.1% (v/v) formic acid, while solvent B consisted of 98% (v/v) acetonitrile with 0.1% (v/v) formic acid. The samples were resolved on a Kinetex F5 analytical column (2.6 μm, 0.3 × 100 mm, Phenomenex) using an elution gradient of 0%–30% solvent B over 5 min at a flow rate of 8 µl/min.
Nanopore DRS
Total RNA was extracted from three biological replicates of 6-day-old seedlings of wild-type, AmiR-mta, mtb-2 ABI3:MTB, fip37-4 LEC1:FIP37, AmiR-vir, and hakai-3 using Trizol reagent (Invitrogen). mRNA was then isolated using the Dynabeads mRNA purification kit (Invitrogen). The quality and integrity of mRNA were assessed with an Agilent Bioanalyzer system. Each library was prepared with 750 ng of mRNA using the nanopore direct RNA sequencing kit (SQK-RNA002, Oxford Nanopore Technologies). The prepared libraries were loaded onto FLO-MIN106 flow cells and sequenced with the GridION sequencer. The raw fast5 data were basecalled by Guppy 4.2.3 with the high accuracy mode to generate FASTQ files.
Identification of m6A sites
The FASTQ reads were mapped to the reference transcriptome of TAIR10 by Minimap2 2.20 (Li, 2018). Alignment was subsequently converted to a BAM file by Samtools (Li et al., 2009) and Nanopolish Eventalign v0.13 (Loman et al., 2015) was subsequently used for signal segmentation. Both aligned reads and events with the reference annotation (Ensemble plant release 50) were processed with xPore (Pratanwanich et al., 2021) to determine differential m6A modification sites.
Differential expression analysis with nanopore reads
Read counts for wild-type plants and plants with impaired m6A writers were performed with Subread v2.0.3 and featureCounts v2.0.2 in the long-read mode (Liao et al., 2014, 2019). Differentially expressed genes were then identified using DESeq2 v1.32 (Love et al., 2014). Heatmaps for transcriptomics data were created via the pheatmap package using log-transformed expression value (http://rpackages.ianhowson.com/cran/pheatmap/). Hierarchical clustering of genes and samples in heatmaps was done using the complete-linkage clustering method and Euclidian distances.
Identification of alternative 3′ end positions
The poly (A) and mRNA regions were segmented using the poly (A) script of Nanopolish v0.13 (Loman et al., 2015) that identifies authentic 3′end for each read. To identify the differential usage of each position between wild-type plants and plants with impaired m6A writers, we performed the Kolmogorov–Smirnov test followed by multiple testing correction with the Benjamini–Hochberg method. The differential 3′ end usage results were filtered for FDR < 0.05 and normalized to the total number of reads. The difference between normalized numbers of plants with impaired m6A writers and wild-type plants for each site was used to obtain the minimum and maximum change in 3′UTR length, which represents the most reduced and increased 3′ end usage sites, respectively. The difference between the most reduced and increased 3′ end usage sites was then obtained to estimate the direction and distance of each change.
Alternative splicing analysis
Full-length alternative isoform analysis of RNA (FLAIR v1.5) (Tang et al., 2020) was performed on the nanopore reads to detect the differential alterative splicing events between wild-type plants and plants with impaired m6A writers.
m6A–IP–qPCR
m6A–IP–qPCR was performed using 6-day-old seedlings in different genetic backgrounds as previously described (Dominissini et al., 2013; Shen et al., 2016). m6A-containing candidate genes and an internal control (TUB2) mRNA were detected by qPCR on the 7900HT Fast Real-Time PCR systems (Applied Biosystems) with PowerUp SYBR Green Master Mix (Applied Biosystems). The primers used for m6A–IP–qPCR analysis are listed in Supplemental Data Set S7.
Analysis of human data
MeRIP data (GSE102493 and GSE46705) (Wang et al., 2014; Yue et al., 2018) were downloaded from GEO (Supplemental Data Set 8, A–E). The raw SRA files are converted to FASTQ files using SRA Toolkit. The data was processed and quality-controlled by nf-core/rnaseq pipeline (https://github.com/nf-core/rnaseq). The reads were aligned to human hg19 genome (version GRCh37.87) by HISAT2 (Kim et al., 2019). The differential methylation peaks between wild-type and the respective cell lines with defective writers were detected using exomePeak (Meng et al., 2013) and the peak distribution was generated by Guitar (Cui et al., 2016). Hypomethylated peaks with log2(fold change) < 0 and FDR < 0.05 were selected for further analysis. DaPars was used for the de novo identification of APA (Xia et al., 2014) with FDR < 0.05 and at least two-fold increase or decrease of PDUIs between wild-type and defective cell lines (Supplemental Data Set 8, F–J). For human nanopore DRS data (PRJEB40872) (Pratanwanich et al., 2021), the FASTQ files were downloaded from ENA. Nanopore reads were aligned to the hg19 genome (version GRCh37.87) using minimap2 (Li, 2018). The APA sites were identified as described for our Arabidopsis nanopore data, while the m6A peaks were retrieved from published data sets (Pratanwanich et al., 2021).
Accession numbers
Sequence data from this article can be found in the Arabidopsis Information Resource (https://www.arabidopsis.org) under the following accession numbers: MTA (AT4G10760), MTB (AT4G09980), FIP37 (AT3G54170), VIR (AT3G05680), and HAKAI (AT5G01160).
Data availability
All data from this study are available in the main text and Supplemental Information. The nanopore direct RNA sequencing data described in this study have been deposited in NCBI Sequence Read Archive (SRA) database with the accession numbers, PRJNA815899 (plants with impaired m6A writers) and PRJNA749003 (wild-type plants).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Generation of various genetic materials where the expression or activity of MTB, VIR, and HAKAI is impaired.
Supplemental Figure S2. Phenotypes of 1-month-old AmiR-mta, AmiR-vir, and hakai-3.
Supplemental Figure S3. Analysis of nanopore DRS reads.
Supplemental Figure S4. Heatmap showing the correlation of gene expression across three biological replicates of all six samples in different genetic backgrounds.
Supplemental Figure S5. Distribution of differential methylation rate (DMR) and P value for hypomethylated sites identified from the comparison between the respective genotypes with impaired m6A writers and wild-type plants.
Supplemental Figure S6. Verification of m6A sites identified through nanopore DRS analysis for five randomly selected candidates.
Supplemental Figure S7. Characteristics of hypomethylated sites identified in plants with impaired m6A writers.
Supplemental Figure S8. Examples of hypomethylated sites with corresponding P values in genes identified in plants with impaired m6A writers.
Supplemental Figure S9. MTA and MTB possess m6A methyltransferase activity in vitro.
Supplemental Figure S10. Enrichment analysis of hypomethylated genes for cellular components.
Supplemental Figure S11. Functional enrichment analysis of hypomethylated transcripts that are unique in individual genetic backgrounds.
Supplemental Figure S12. Susceptibility of plants with impaired m6A writers to salt stress.
Supplemental Figure S13. Top 5 GO terms that are distinct in the upregulated or downregulated gene list (log2 [fold change] > 0.5 or < −0.5; P < 0.05) from different genetic backgrounds.
Supplemental Figure S14. Functional enrichment analysis of differentially expressed transcripts (log2 [fold change] > 0.5 or < −0.5; P < 0.05) containing hypomethylated m6A site(s) (DMR < −0.3).
Supplemental Figure S15. m6A has a minimal effect on alternative splicing of transcripts.
Supplemental Figure S16. Functional enrichment analysis of human transcripts containing hypomethylated m6A site(s).
Supplemental Data Set S1. List of hypomethylated sites identified from genetic materials with impaired m6A writers.
Supplemental Data Set S2. Differentially expressed genes in genetic materials with impaired m6A writers compared to wild-type plants [log2(Fold change) ≤ −0.5 or log2(Fold change) ≥ 0.5; P < 0.05].
Supplemental Data Set S3. Stress-related transcripts that are hypomethylated and upregulated in genetic materials with impaired m6A writers compared to wild-type plants.
Supplemental Data Set S4. List of differential alternative splicing events identified from genetic materials with impaired m6A writers (P < 0.05).
Supplemental Data Set S5. List of genes with significant alteration in 3′ UTR length identified from genetic materials with impaired m6A writers (FDR < 0.05).
Supplemental Data Set S6. List of differential readthrough transcripts identified from genetic materials with impaired m6A writers (Fisher's exact test: P < 0.05).
Supplemental Data Set S7. List of primers used in this study.
Supplemental Data Set S8. Human genes with differential methylation peaks [A–E; log2(fold change) < 0 and FDR < 0.05] or distal polyA site usage index (PDUI; F–J; FDR < 0.05 and at least two-fold increase or decrease of PDUIs).
Acknowledgments
The authors thank the Arabidopsis Biological Resource Centre for providing seeds and Genome Institute of Singapore, A*STAR for the nanopore direct RNA sequencing service. The authors thank the Protein and Proteomics Centre (PPC) in the Department of Biological Sciences, National University of Singapore for the LC-MS/MS service.
Funding
This work was supported by the National Research Foundation Competitive Research Programme (NRF-CRP22-2019-0001), Singapore Food Story R&D Programme (SFS_RND_SUFP_001_04), and the intramural research support from Temasek Life Sciences Laboratory and National University of Singapore.
References
Author notes
C.E.W. and S.Z. contributed equally.
Senior author.
L.S. and H.Y. conceived and designed this study. C.E.W., Y.Z., Z.W.T., A.Y., and L.S. performed the experiments. S.Z. and T.X. performed the bioinformatics analysis of the nanopore DRS data. C.E.W., S.Z., T.X., L.S., and H.Y. analyzed the data. C.E.W., L.S., and H.Y. wrote the paper. All authors read and approved the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/general-instructions) is: Hao Yu ([email protected]).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}