





Abstract
The macronutrient phosphorus is essential for plant growth and development. Plants have evolved multiple strategies to increase the efficiency of phosphate (Pi) acquisition to protect themselves from Pi starvation. However, the crosstalk between Pi homeostasis and plant development remains to be explored. Here, we report that overexpressing microRNA399 (miR399) in maize (Zea mays) is associated with premature senescence after pollination. Knockout of ZmPHO2 (Phosphate 2), a miR399 target, resulted in a similar premature senescence phenotype. Strikingly, we discovered that INDETERMINATE1 (ID1), a floral transition regulator, inhibits the transcription of ZmMIR399 genes by directly binding to their promoters, alleviating the repression of ZmPHO2 by miR399 and ultimately contributing to the maintenance of Pi homeostasis in maize. Unlike ZmMIR399 genes, whose expression is induced by Pi deficiency, ID1 expression was independent of the external inorganic orthophosphate status, indicating that ID1 is an autonomous regulator of Pi homeostasis. Furthermore, we show that ZmPHO2 was under selection during maize domestication and cultivation, resulting in a more sensitive response to Pi starvation in temperate maize than in tropical maize. Our study reveals a direct functional link between Pi-deprivation sensing by the miR399-ZmPHO2 regulatory module and plant developmental regulation by ID1.
Background: Phosphorus is an essential macronutrient for plant growth and development. To cope with phosphorus limitation, plants have evolved sophisticated strategies to coordinate phosphorus acquisition, scavenging, and recycling. Maize is an important crop and cultivated widely for both staple food and industrial usage. Although phosphorus is a major constituent of the fertilizers required to sustain high yields in maize, global phosphorus resources are quickly diminishing and may be exhausted in the near future.
Question: We sought to understand the underlying molecular mechanisms behind phosphate acquisition and utilization in maize to develop varieties with high-phosphorus use efficiency.
Findings: We observed that overexpressing microRNA399 (miR399) or knocking out of its target gene ZmPHO2 results in an apparent leaf senescence phenotype after pollination. We further discovered that the transcription factor INDETERMINATE1 (ID1) functions as an autonomous upstream regulator of phosphorus homeostasis by suppressing the transcription of ZmMIR399 genes, thus decreasing the accumulation of mature miR399 and alleviating the cleavage of ZmPHO2 transcripts, ultimately contributing to the maintenance of phosphorus homeostasis in maize. Our work establishes a regulatory connection between phosphorus nutrient homeostasis and vegetative–reproductive development in maize. More importantly, we show that ZmPHO2 underwent strong selection during maize domestication and cultivation. This study provides genetic resources for improving maize phosphorus use efficiency and breeding low phosphorus-tolerant maize varieties by editing the ID1-miR399-ZmPHO2 module.
Next steps: We will attempt to generate ID1-modified plants to fine-tune flowering time and phosphorus uptake in maize. The specific functions of each MIR399 family member may also be interrogated by generating genome-edited mutants. As we discovered that Bx genes involved in benzoxazinoid biosynthesis are also affected by miR399, we hope to further explore the mechanism of miR399 functioning as a negative regulator of plant immunity.
Introduction
Phosphorus is present in every living cell and its functions cannot be substituted by other nutrients (Marschner 1995). As 1 of 17 essential nutrients, phosphorus is required for almost all developmental processes of plants and is considered to be 1 of 3 primary macronutrients, along with nitrogen and potassium (Bindraban et al. 2020). Nevertheless, most cultivated land worldwide has poor phosphorus availability, and levels of inorganic orthophosphate (Pi), which plants take up through their roots, are suboptimal in soils for vegetative growth and crop productivity (Raghothama 1999; López-Arredondo et al. 2014). Along with other fertilizers, Pi must be routinely applied to farmland to support healthy crop growth. However, Pi fertilizers used for agriculture are mostly produced from mined rock Pi, which is nonrenewable and predicted to decline in availability before the end of this century (Vance et al. 2003; Baker et al. 2015). Thus, modifying crop plants to make them amenable to soils with a restricted supply of Pi is vital if high crop yields, which are necessary to support the food demands of a continuously increasing world population, are to be maintained.
Pi-efficient plants would reduce the requirement for Pi fertilizers, thereby ameliorating their overuse while concurrently enhancing yield. The investigation and manipulation of genes involved in Pi acquisition, remobilization, and metabolism are potential strategies for the development of Pi-efficient crops, which might facilitate agricultural sustainability. To date, a series of Pi starvation-related genes and regulators have been characterized in the model plant Arabidopsis (Arabidopsis thaliana), including genes encoding transcription factors such as PHOSPHATE STARVATION RESPONSE1 (PHR1) (Rubio et al. 2001; Maeda et al. 2018), PHR1-LIKE1 (Bustos et al. 2010), WRKY75 and ZAT6 (Devaiah and Raghothama 2007; Devaiah et al. 2007a; Devaiah et al. 2007b), the long noncoding RNA INDUCED BY PHOSPHATE STARVATION1 (IPS1) (Franco-Zorrilla et al. 2007), microRNA399 (miR399) (Bari et al. 2006; Chiou et al. 2006; Pant et al. 2008), and members of the PHOSPHATE (PHO) gene family (Aung et al. 2006; Stefanovic et al. 2007). In particular, PHO2 (Phosphate 2), which encodes a ubiquitin-conjugating E2 enzyme (UBC), is known to be a miR399 target and is involved in Pi homeostasis (Bari et al. 2006). Under Pi-sufficient conditions, PHO2 targets Pi transporters for poly-ubiquitination and degradation, thereby maintaining optimal Pi uptake (Liu et al. 2012; Li et al. 2017; Sega et al. 2020). By contrast, Pi deficiency stress induces miR399 accumulation, which downregulates the abundance of PHO2 transcripts via cleavage (Bari et al. 2006), in turn allowing the accumulation of Pi transporters to enhance Pi acquisition and translocation (Aung et al. 2006). The miR399-PHO2 module also regulates Pi homeostasis in rice (Oryza sativa), maize (Zea mays), wheat (Triticum aestivum), and other species (Chiou et al. 2006; Hu et al. 2011; Xu et al. 2013; Du et al. 2018). Long noncoding RNAs PILNCR1 (Pi-deficiency-induced long noncoding RNA1) in maize, or IPS1 in Arabidopsis, serve as miR399 sponges to reduce miR399-guided cleavage of PHO2 RNA and help plants adapt to Pi deficiency (Franco-Zorrilla et al. 2007; Du et al. 2018). Previous studies have demonstrated that the induction of MIR399 under Pi-limited conditions is repressed in an Arabidopsis mutant, phr1 (Bari et al. 2006), showing that PHR1 functions as an upstream regulator of miR399 in the Pi-deprivation response. Similarly, overexpression of ZmPHR1 in maize or OsPHR1 and OsPHR2 in rice resulted in increased Pi uptake ability and elevated Pi content under Pi-limited conditions (Zhou et al. 2008; Wang et al. 2013b). Although PHR1, miR399, and PHO2 define a conserved regulatory module that maintains Pi homeostasis in various plant species (Bari et al. 2006), other transcription factors, such as OsPTF1 (Pi starvation-induced transcription factor 1) in rice and ZmPTF1 in maize, have also been functionally characterized with respect to low-Pi stress responses (Yi et al. 2005; Li et al. 2011). Even so, characterizing more genes involved in Pi homeostasis and elucidating their regulatory networks could help improve phosphorus use efficiency to facilitate plant acclimation to changing Pi environments.
Maize is not only an important crop for food and feed but also a source of primary compounds for industrial innovation (Andorf et al. 2019). It has been widely cultivated in tropical and temperate soils worldwide, following domestication from its wild progenitor, teosinte parviglumis (Z. mays ssp. parviglumis), which was originally grown in the lowland, subtropical environment of the Balsas River basin (Guerrero, México) (Matsuoka et al. 2002). In the early stage of domestication, adaptation of parviglumis from the lowland environments to low temperature and soils with low phosphorus content in highland environments drove the genetic divergence between parviglumis and mexicana (Z. mays ssp. Mexicana, another teosinte subspecies) (Aguirre-Liguori et al. 2019; Barnes et al. 2022), suggesting that the soil phosphorus status is a key driver of local adaptation in maize. Under current cultivation conditions, especially in acidic and alkaline soils, large amounts of Pi fertilizers are applied to maize fields, allowing maize plants to become well-established and mature in a suitable timeframe, thus improving overall yield (Khan et al. 2020; Qaswar et al. 2020). However, despite these advantages, the long-term use of large amounts of fertilizers might weaken the nutrient (such as Pi) uptake systems of plants during cultivation (Liu et al. 2021). Thus, research on Pi regulators is needed, with the ultimate aim of optimizing the acquisition and use of Pi by plants.
Here, we report that overexpressing miR399 in maize resulted in typical Pi toxicity phenotypes, as manifested by premature senescence in mature leaves after pollination. We also generated Zmpho2 mutants by clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated nuclease 9 (Cas9)-mediated editing and determined that knocking out the ZmPHO2 gene, a miR399 target, consistently produced a premature leaf senescence phenotype similar to that of the transgenic miR399-overexpression lines. Transcriptome profiling and targeted metabolite measurements in miR399-overexpression and miR399-knockdown transgenic lines revealed that, in addition to affecting the Pi response and leaf senescence, miR399 might mediate pathogen and insect defense in maize by regulating the expression of Bx genes in the benzoxazinoid biosynthesis pathway. We further demonstrated that an upstream regulator of the miR399-ZmPHO2 module, INDETERMINATE1 (ID1), can inhibit the transcription of ZmMIR399 genes and hence the accumulation of mature miR399, to alleviate the repression of ZmPHO2 by miR399, and ultimately maintain Pi homeostasis in maize. ID1 expression was unperturbed by low-Pi stress, suggesting that ID1 acts as an autonomous Pi regulator in maize. We conclude that mutation of ID1 triggers the miR399-ZmPHO2 pathway, a conserved Pi response module in plants, to promote Pi acquisition, providing the necessary Pi supply to support the vegetative gigantism of the id1 mutant. We also showed that ZmPHO2 was under selection during maize domestication and cultivation, resulting in a more sensitive response to Pi starvation in temperate maize than tropical maize. We speculate that, during maize domestication or subsequent cultivation, Pi uptake ability might have degenerated as maize spread into high-Pi environments, resulting from prolonged and excessive Pi application in pursuit of improvements in yield. Our study establishes a regulatory connection between nutrient homeostasis and vegetative development in maize.
Results
Transgenic maize plants overexpressing miR399 display an apparent leaf senescence phenotype after pollination
The miR399 family in maize contains a total of 10 members (i.e. miR399a-j) generated by ten ZmMIR399 genes and can be divided into 6 subgroups based on their respective mature miRNA sequences (Zhang et al. 2009) (Supplemental Fig. S1). To investigate the biological roles of the miR399 family in maize, we subcloned 2 types of mature 21-nt miR399 sequences (miR399a/c/h and miR399e/i/j) in the same vector using a 2-hit artificial miRNA design (Supplemental Fig. S2A). Because the mature miR399 species have very similar sequences (Supplemental Fig. S1A) and certain mismatches between a miRNA and its target sequence are allowed (Bologna and Voinnet 2014), this overexpression construct probably represents the overexpression of the entire miR399 family. We chose 3 independent transgenic lines (named miR399-OE#1, miR399-OE#2, and miR399-OE#3) for experiments after confirming the accumulation of mature miR399 by RNA gel blot analysis (Fig. 1A). The total phosphorus content was significantly higher in shoots of miR399-overexpressing transgene-positive plants (miR399-OEP#1-#3) compared with their isogenic siblings without the transgene, i.e. miR399 transgene-negative plants (miR399-OEN#1-#3) derived from the same transformation event (Fig. 1B; Supplemental Fig. S3, A and B), suggesting that accumulation of miR399 induces enhanced Pi acquisition. By contrast, we observed no significant difference for phosphorus content in roots of transgene-positive and transgene-negative plants, resulting in a higher shoot-to-root Pi content ratio in miR399-overexpressing transgene-positive plants (Fig. 1B; Supplemental Fig. S3, A and B). Consistent with a previous report (Du et al. 2018), leaf necrosis at the margins of old leaves appeared at the seedling stage of miR399-overexpressing transgene-positive plants grown in hydroponic solution with a sufficient Pi supply (Supplemental Fig. S4); this phenotype was previously reported to be a typical Pi-toxicity phenotype (Du et al. 2018). To investigate whether miR399 accumulation affects maize developmental and agronomic traits, we grew 3 independent miR399-overexpression lines in the field (Liaoning, 41.8° N, 123.4° E) along with their corresponding isogenic control siblings. All 3 miR399 overexpression transgene-positive lines showed markedly earlier leaf senescence than their corresponding transgene-negative siblings, especially after pollination (Fig. 1C; Supplemental Fig. S3, C and D), but did not exhibit any significant differences on agronomic traits (Supplemental Fig. S5). Concomitantly, the miR399-overexpression transgenic lines showed significantly lower chlorophyll content across the entire mature leaf, from base to tip (Fig. 1D; Supplemental Fig. S3, E and F). Collectively, these results indicate that miR399 functions as a positive regulator of phosphorus uptake and that overexpression of miR399 induces an apparent premature senescence syndrome after pollination in maize.
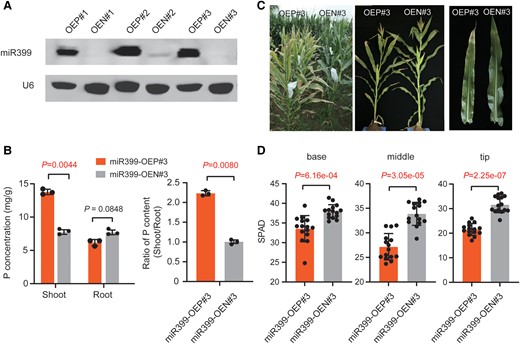
Phenotype of field-grown miR399-overexpressing transgenic plants. A) RNA gel blot showing that mature miR399 accumulates in 3 independent transgenic lines (named OEP#1–OEP#3). A total of 10 µg total RNA from seedlings of each line was loaded per lane, and the membrane was hybridized with a biotin-labeled miR399 probe. U6 served as a loading control. OEP and OEN represent transgene-positive and transgene-negative plants, respectively, derived from the same transformation event. B) Total phosphorus (P) contents in shoots and roots of miR399-OEP and miR399-OEN lines. A representative transgenic strain, miR399-OE#3, was chosen and 3 biological replicates for each genotype were prepared for total P concentration measurement. Values are means ± SD; the P-values were determined by Student's t-test (Supplemental Data Set S6). C) Premature senescence phenotypes of miR399-OEP#3 and miR399-OEN#3 plants grown in the field. A field scene and representative plants at 15 d after pollination are shown. D) Quantification of total chlorophyll content (shown as SPAD values) in different parts (base, middle, and tip) of mature ear leaves from miR399-OEP#3 and miR399-OEN#3 after pollination. A total of 15 plants were measured for chlorophyll content for each genotype. Values are means ± SD; the P-values were determined by Student's t-test (Supplemental Data Set S6). SPAD represents total chlorophyll content, which was measured with a Konica Minolta SPAD 502 Plus instrument.
Short tandem target mimicry (STTM) technology (Tang et al. 2012; Tang and Tang 2013), which was developed as an improvement upon the original target mimicry approach, is an effective tool to block miRNA activity or cause miRNA degradation (Peng et al. 2018). The STTM construct usually contains 2 imperfect miRNA target sites with a CTA trinucleotide bulge in the center linked by a 31-nt to 96-nt short spacer (Yan et al. 2012). To generate miR399-knockdown transgenic plants (miR399-STTM), we designed a STTM construct containing 2 noncleavable miR399-binding sites targeting 2 different mature miR399 species corresponding to miR399a/c/h and miR399e/i/j (Supplemental Fig. S2B). The abundance of miR399 decreased in 2 independent miR399-STTM transgene-positive lines (miR399-STTMP#1 and miR399-STTMP#2) compared with their isogenic transgene-free siblings (miR399-STTMN#1 and miR399-STTMN#2), confirming the successful knockdown of endogenous miR399 (Supplemental Fig. S6A). However, phosphorus accumulation in shoots was not significantly different between miR399-STTM transgene-positive and transgene-negative lines (Supplemental Fig. S6B). The lack of a change in phosphorus content might be due to the naturally low levels of miR399 at the seedling stage under normal growth conditions, which would minimize any differences in miR399 levels between miR399-STTMP and miR399-STTMN lines. We thus grew homozygous miR399-STTMP transgenic lines in the field (Liaoning, 41.8° N, 123.4° E) for phenotypic observation along with their corresponding isogenic control siblings. We observed no visible developmental or morphological differences between the transgene-positive and their corresponding transgene-negative lines (Supplemental Fig. S6C), suggesting that a minor reduction in miR399 levels does not affect maize development under normal conditions. Growing miR399-STTM plants under Pi-sufficiency/-deficiency soil regimes needs to be performed in the future for further phenotypic characterization.
Knocking out ZmPHO2, a miR399 target, results in premature senescence
PHO2, which encodes a UBC enzyme, has been experimentally confirmed to be a target of miR399 in various species, including Arabidopsis, rice, barley (Hordeum vulgare), soybean (Glycine max), and maize (Fujii et al. 2005; Bari et al. 2006; Chiou et al. 2006; Pant et al. 2008; Hu et al. 2011; Hackenberg et al. 2013; Xu et al. 2013; Du et al. 2018). In maize, miR399b directs the cleavage of ZmPHO2 mRNA, which contains 6 tandem miR399-binding sites in its 5′ untranslated region (5′ UTR) (Fig. 2A), based on transient expression, and 5′ rapid amplification of cDNA ends (5′ RACE) assays (Du et al. 2018). To validate this relationship for other miR399s, we performed transient coexpression assays in maize protoplasts with different miR399 precursors as effectors, together with a firefly luciferase (LUC) reporter construct harboring the 5′ UTR of ZmPHO2 upstream of LUC and downstream of a minimal cauliflower mosaic virus (CaMV) 35S promoter (Supplemental Fig. S7A). Besides the observation that miR399b significantly repressed the activity of LUC derived from the reporter construct, we confirmed that miR399a/c/h, miR399e/i/j, and miR399f significantly inhibit LUC activity (Supplemental Fig. S7B), suggesting that these miR399 species can act through the miR399-binding sites in the 5′ UTR of ZmPHO2. We examined ZmPHO2 expression by RT-qPCR in miR399-OE and miR399-STTM transgenic lines and observed the expected expression changes, with significant downregulation and upregulation in miR399-OE and miR399-STTM transgenic lines, respectively (Fig. 2B), demonstrating that ZmPHO2 is an authentic target of miR399. However, so far, we lack genetic evidence for the function of ZmPHO2 in the regulation of Pi homeostasis and plant development in maize. To examine the function of ZmPHO2, we knocked out ZmPHO2 in the maize inbred line ZZC01 using CRISPR/Cas9-mediated mutagenesis (Doudna and Charpentier 2014; Belhaj et al. 2015; Ma et al. 2015). We noticed that the annotations of ZmPHO2 show different gene structures among different versions of the maize genome (GRMZM2G381709 in AGPv3, Zm00001d038972 in AGPv4, and Zm0001eb295490 in AGPv5; Supplemental Fig. S8). We thus used public RNA-seq data and transcription start site data to reevaluate the gene structure of ZmPHO2 (Supplemental Fig. S8). We determined that the gene structures annotated by Du et al. (2018) and AGPv5 highly agree, with the same translation start site and similar exon–intron junctions, and thus adopted this annotation for generating knockout mutants (Supplemental Fig. S8). We designed 2 single-guide RNAs (sgRNAs) to target the coding region immediately after the translation start site within the first exon of ZmPHO2 (Fig. 2A). Genotyping and sequence analyses identified 3 homozygous mutants harboring fragment insertions and/or deletions around the sgRNA target sites (Fig. 2C). The 3 homozygous, potentially null mutants consistently showed a typical Pi toxicity phenotype and exhibited premature senescence (Fig. 2D). The leaf senescence phenotypes were more severe than those of the miR399-OE transgenic lines such that very few seeds could be harvested.
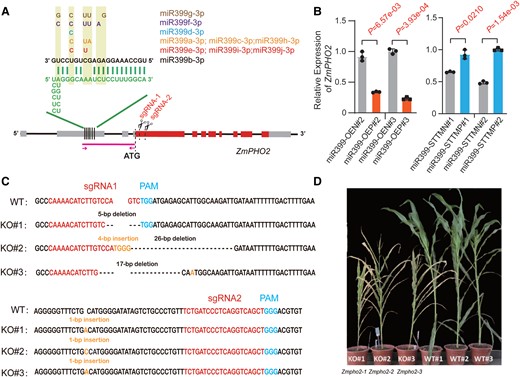
CRISPR/Cas9-mediated mutagenesis of ZmPHO2 causes premature senescence in maize. A) Diagram of the ZmPHO2 locus. Gray box, untranslated regions; red box, coding regions. Six predicted miR399 target sites (4 of them were experimentally validated by Du et al. (2018)) are located in the 5′ UTR. The sequences of the 6 sites are shown in green and those of the various miR399 members are shown in multiple colors. sgRNA-1 and sgRNA-2 represent 2 single-guide RNAs for CRISPR/Cas9-mediated editing. The magenta line segment with arrows indicates the 5′ UTR fragment used in dual-luciferase assays in Supplemental Fig. S7 and Fig. 7D. B) Relative ZmPHO2 expression as determined by RT-qPCR in miR399-OE and miR399-STTM transgene-positive lines and the corresponding negative controls. The P-values were calculated by Student’s t-test (Supplemental Data Set S6). ZmPHO2 expression was normalized to that of ZmUBI. C) Sequences of 3 homozygous knockout lines with insertions/deletions that truncate the ZmPHO2 open reading frame (named KO#1, KO#2, and KO#3). The wild-type (WT) sequence is shown at the top. sgRNA and protospacer-adjacent motif (PAM) sequences are in red and blue, respectively. The deletions are indicated by dashes, while the inserted nucleotides are shown in orange. D) Gross morphology of 3 Zmpho2 knockout lines and 3 wild-type lines.
miR399 might mediate pathogen and insect defense in maize, in addition to affecting Pi response and leaf senescence
To characterize the gene network(s) through which miR399 might influence maize development and physiology, we carried out a comparative transcriptome analysis of mature leaves (V5 stage) between homozygous miR399-OEP#3 and miR399-STTMP#2 and their corresponding transgene-negative siblings (miR399-OEN#3 and miR399-STTMN#2) with 3 replicates, which showed high reproducibility (Supplemental Fig. S9). By comparison to their corresponding transgene-negative siblings, we identified 2,559 and 1,480 differentially expressed genes (DEGs) in miR399-OE#3 and miR399-STTM#2, respectively (fold change ≥ 1.5 & FDR ≤ 0.05; Fig. 3A; Supplemental Data Sets S1 and S2). Annotating these DEGs highlighted some genes involved in Pi homeostasis, such as the Pi transporters ZmPT2 (Phosphate transporter 2) (Vasconcelos et al. 2022) and ZmPHO1;2b (Phosphate transporter PHO1-2-like) (Ma et al. 2021), as well as the Pi-salvaging/recycling genes ZmPAP21 (Purple acid phosphatase 21) (González-Muñoz et al. 2015) and ZmGPX-PDE9/11/14 (Glycerophosphodiester phosphodiesterase 9/11/14) (Wang et al. 2021) (Supplemental Data Sets S1 and S2), and some leaf senescence-associated genes, including SAG12 (Senescence-associated gene 12) and transcription factor genes belonging to the NAC and WRKY families (Breeze et al. 2011; Stigter and Plaxton 2015) (Supplemental Data Sets S1 and S2). In addition, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses showed that these DEGs are significantly enriched in GO terms or pathways associated with secondary metabolism, including oxylipin metabolism, lignin biosynthesis, and linoleic acid metabolism, to name a few (Supplemental Fig. S10; Supplemental Data Sets S3 and S4). Of these DEGs, we defined 971 genes as downregulated DEGs (down-DEGs) in miR399-OE plants, with significantly lower expression levels in miR399-OEP#3 compared with miR399-OEN#3 (Fig. 3A). Similarly, we identified 729 upregulated DEGs (up-DEGs) in miR399-STTM plants, with significantly higher expression levels in miR399-STTMP#2 than in miR399-STTMN#2 (Fig. 3B). Overlap analysis between the down-DEGs in miR399-OE and up-DEGs in miR399-STTM identified a small subset of high-confidence genes (102 genes; Fig. 3C), whose expression was negatively correlated with miR399 levels. Detailed annotation of this small subset of genes revealed a number of Pi-related genes, including ZmPHO2 (Fig. 3D; Supplemental Data Set S2). Pathway enrichment analysis of these miR399-associated genes in reference to the GO and KEGG databases showed that the term “benzoxazinoid biosynthesis” is the most strongly enriched (Fig. 3E).
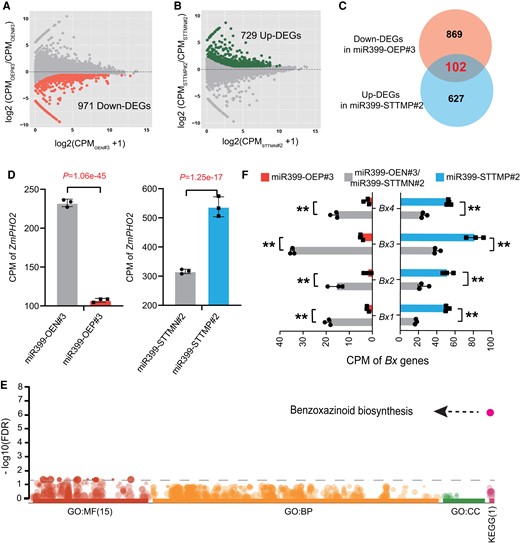
RNA-seq analysis reveals that miR399 might mediate pathogen and insect resistance in maize, in addition to affecting Pi responses. A) Differentially expressed genes (DEGs) in the miR399-OE transgene-positive line (miR399-OEP#3) compared with its transgene-negative line (miR399-OEN#3). A total of 971 downregulated genes (down DEGs) were detected in miR399-OEP#3. B) DEGs in the miR399-STTM transgene-positive line (miR399-STTMP#2) compared with its transgene-negative line (miR399-STTMN#2). A total of 729 upregulated genes (up DEGs) were detected in miR399-STTMN#2. C) Overlap of the downregulated DEGs in miR399-OEP#3 and upregulated DEGs in miR399-STTMP#2. D)ZmPHO2 expression in miR399-OE and miR399-STTM transgene-positive lines (miR399-OEP#3 and miR399-STTMP#2) and their corresponding transgene-negative lines (miR399-OEN#3 and miR399-STTMN#2). P-values were calculated by DESeq2 after multiple correction of the FDR. E) Gene ontology (GO) and KEGG pathway enrichment analyses of the 102 overlapping DEGs. Benzoxazinoid biosynthesis was the most highly enriched pathway. BP, biological process; MF, molecular function; CC, cellular component groups of GO. The total number of enriched categories in the GO or KEGG group is indicated in parentheses. F) Expression of several key Bx genes (Bx1 to Bx4) in the benzoxazinoid biosynthesis pathway in the miR399-OE and miR399-STTM transgene-positive lines and their corresponding transgene-negative lines. **P ≤ 0.01.
Benzoxazinoids are a class of indole-derived protective and allelopathic secondary metabolites that are involved in antifungal and insect defense responses in plants (Frey et al. 2009). 2,4-Dihydroxy-1,4-benzoxazin-3-one (DIBOA) and its 7-methoxy analog 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIMBOA) are the predominant benzoxazinoids in plants (Frey et al. 1997; Frey et al. 2009), and these compounds have been found in many plants including maize, wheat, and rye (Secale cereale) (Frey et al. 2009). The benzoxazinoid biosynthetic pathway is well-characterized in maize (Supplemental Fig. S10A). Metabolic profiling previously revealed that levels of both DIBOA-glucoside (DIBOA-Glc) and DIMBOA-glucoside (DIMBOA-Glc), the storage forms of DIBOA and DIMBOA, were markedly lower in maize lines tolerant to Pi limitation, which are able to take up Pi more efficiently under low Pi conditions (Luo et al. 2019). In our present study, we determined that Bx genes (Bx1-Bx9) involved in benzoxazinoid biosynthesis are significantly downregulated in miR399-OE transgene-positive lines, as shown by the RNA-seq data and RT-qPCR validation (Fig. 3F; Supplemental Figs. S11, B and S12), which should result in a reduced accumulation of benzoxazinoid compounds. To test this idea, we performed a targeted high-performance liquid chromatography (HPLC) assay with DIBOA and DIMBOA as standards. We measured significantly lower levels of both DIBOA and DIMBOA in miR399-OE plants, thus validating our hypothesis (Supplemental Fig. S13A). By contrast, the expression levels of most Bx genes tended to be upregulated and the metabolites (DIBOA and DIMBOA) accumulated to higher levels in miR399-STTM plants (Fig. 3F; Supplemental Figs. S12 and S13B). Collectively, these results support the conclusion that miR399 might influence pathogen and insect defense in maize, in addition to its effects on the Pi response and leaf senescence. However, how the expression of Bx genes is modulated in miR399-OE or miR399-STTM transgenic lines is still an open question. A similar scenario was noted in rice, where Pi accumulation induced by overexpressing OsMIR399f or high levels of Pi fertilization resulted in enhanced susceptibility to infection by the blast fungus Magnaporthe oryzae (Campos-Soriano et al. 2020), demonstrating that miR399 functions as a negative regulator of plant immunity, possibly through an indirect mechanism.
Mutation of ID1 promotes miR399 expression and facilitates Pi accumulation in maize
ID1 is a monocotyledon-specific zinc-finger transcription factor that plays an essential role in regulating the transition to flowering in maize. Loss-of-function id1 mutants experience a prolonged period of vegetative growth causing an extreme delay in flowering, ultimately resulting in vegetative gigantism (Singleton 1946; Colasanti et al. 1998; Colasanti and Sundaresan 2000). Although the id1 mutation severely alters the ability of maize to undergo the transition to reproductive growth, homozygous id1 plants are virtually identical in growth rate and morphology to wild-type maize plants until the point of floral transition (Singleton 1946; Colasanti et al. 1998; Coneva et al. 2007). Transcriptome profiling revealed that miR399 accumulated to abnormally high levels in both mature and immature leaves of the id1-m1 mutant at the floral transition stage (Minow et al. 2018). By reanalyzing the small RNA-seq data from this earlier study (Minow et al. 2018), we confirmed that members of the miR399 family (including miR399a/c/h, miR399e/i/j, miR399b, and miR399f) are significantly more abundant in mature leaves of the id1-m1 mutant (Fig. 4A; Supplemental Fig. S14). In immature leaves, miR399e/i/j also accumulated to higher levels in the id1-m1 mutant than in the wild type (Supplemental Fig. S14), while other members were slightly more abundant or unchanged. Using the corresponding RNA-seq data (Minow et al. 2018), we established that ZmPHO2 transcript levels are significantly downregulated in the id1-m1 mutant (Supplemental Fig. S15). Considering the nutritional requirements for vigorous vegetative growth and extremely delayed flowering, we speculated that the upregulation of miR399 in the id1 mutant might help provide more Pi to sustain the extra vegetative growth of this mutant. This hypothesis prompted us to examine the relationship between ID1 and the miR399-ZmPHO2 regulatory module.
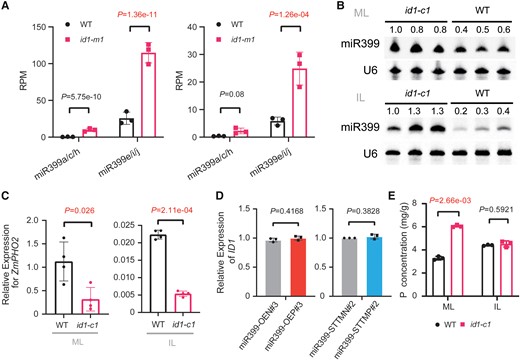
Mutation of ID1 promotes miR399 accumulation and facilitates Pi accumulation in maize. A) Abundance of miR399a/c/h and miR399e/i/j in the id1-m1 mutant and wild type (WT) based on small RNA-seq data (Minow et al. 2018). The left and right panels represent results of small RNA-seq in mature leaves and immature leaves, respectively. RPM, reads per million. Values are means ± SD. P-values were calculated by DESeq2 after multiple correction of the FDR. B) RNA gel blot detection of miR399 in mature leaves (ML) and immature leaves (IL) of the id1-c1 mutant and the wild type (WT). U6 served as an internal control. The numbers indicate the abundance of miR399 relative to the first lane. C)ZmPHO2 expression in mature leaves (ML) and immature leaves (IL) of WT and the id1-c1 mutant (n = 3), as determined by RT-qPCR. ZmPHO2 expression was normalized to that of ZmUBI. D)ID1 expression levels in miR399-OE (OE#3) transgenic lines and miR399-STTM (STTM#2) transgenic lines (n = 3). ID1 expression was normalized to that of ZmUBI. E) Total phosphorus (P) content in mature leaves (ML) and immature leaves (IL) of the id1-c1 mutant and wild type (WT) at the floral transition stage. The values in panels C) to E) are means ± SD. Student's t-test (Supplemental Data Set S6) was employed to calculate the P-values.
To independently confirm the small RNA-seq results, we created a new id1 mutant by CRISPR/Cas9-mediated gene editing and obtained an edited plant with a large deletion in the ID1 coding region (Supplemental Fig. S16A). Genotyping and sequencing of the targeted genomic region in T2 populations derived from 1 T1 heterozygous mutant allowed the identification of 1 homozygous mutant without the Cas9 transgene. The homozygous mutant, which was named id1-c1, showed lower expression of ID1 (Supplemental Fig. S16B) and vigorous vegetative growth and extremely delayed flowering in the field reminiscent of the original id1-m1 mutant (Supplemental Fig. S16, C and D). These molecular and phenotypic characteristics indicated that we had successfully obtained an id1 mutant by gene editing. RNA gel blot analysis confirmed that miR399 levels are significantly increased in mature and immature leaves at the floral transition stage of the id1-c1 mutant (Fig. 4B). In addition, consistent with the reported RNA-seq data (Supplemental Fig. S15) (Minow et al. 2018), we confirmed that ZmPHO2 expression is significantly downregulated in the id1-c1 mutant relative to the wild type (i.e. the corresponding transgene-free and editing-free sibling) by RT-qPCR (Fig. 4C).
We examined ID1 expression in miR399-OE and miR399-STTM transgenic lines by RNA-seq and RT-qPCR but did not detect a significant difference compared with the wild type (Fig. 4D; Supplemental Fig. S17), which supports the notion that ID1 is genetically upstream of the miR399-ZmPHO2 module. We also measured the total phosphorus content of the id1-c1 mutant at the floral transition stage and detected 2-fold higher phosphorus levels in mature leaves compared with the wild type (Fig. 4E). In other tissues, including immature leaves, roots, leaf sheaths, and stems, we observed no significant changes in phosphorus content (Supplemental Fig. S18). Altogether, these observations show that loss of ID1 function facilitates Pi accumulation in mature leaves, perhaps via the miR399-ZmPHO2 regulatory module. Increased Pi accumulation might provide an adequate nutritional reserve for the vigorous vegetative growth of the id1 mutant.
ID1 directly binds to the promoters of ZmMIR399c and ZmMIR399j to repress their transcription
We determined the levels of the primary transcripts (pri-miR399s) derived from the ZmMIR399a, ZmMIR399c, ZmMIR399h, ZmMIR399e, ZmMIR399i, and ZmMIR399j loci, which are responsible for the production of miR399a/c/h and miR399e/i/j. We determined that, in both mature and immature leaves of the id1-c1 mutant, ZmMIR399c and ZmMIR399j levels are markedly upregulated, while the expression of other ZmMIR399 genes was slightly upregulated or unchanged (Fig. 5A), indicating that the enhanced accumulation of miR399a/c/h and miR399e/i/j in the id1-c1 mutant was mainly due to ZmMIR399c and ZmMIR399j expression, respectively. We examined whether ID1 might regulate the transcription of ZmMIR399c and ZmMIR399j via a dual-LUC reporter assay, using the coding sequence of ID1 driven by the 35S promoter (p35S:ID1) as the effector and LUC driven by either the ZmMIR399c or the ZmMIR399j promoter (proMIR399c:LUC and proMIR399j:LUC), as the reporter (Fig. 5B). We observed that the LUC activity derived from the proMIR399c:LUC and proMIR399j:LUC reporter constructs is significantly repressed in maize protoplasts when the p35S:ID1 effector was cotransfected (Fig. 5B).
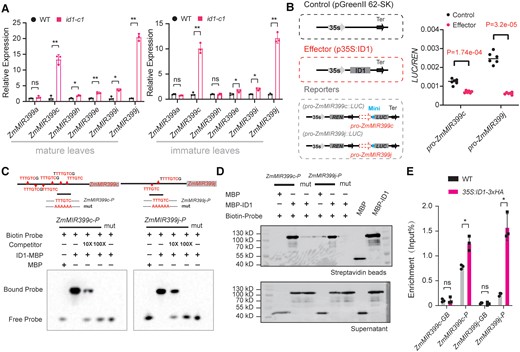
ID1 functions as an upstream regulator by binding directly to the promoter regions of ZmMIR399c and ZmMIR399j. A) Expression levels of 6 primary miR399 precursors (ZmMIR399a, c, h, e, i, j) in mature leaves and immature leaves of the id1-c1 mutant and wild type (WT) by RT-qPCR. Expression levels were normalized to that of ZmUBI. Values are means ± SD; the P-values were determined by Student's t-test (Supplemental Data Set S6). B) Schematic diagram showing the effector and reporter constructs used in the transient transcription activity assays. REN, Renilla luciferase; LUC, firefly luciferase; Mini: minimal 35S promoter. Values means ± SD based on the technical replicates (n = 6) of 1 representative out of 3 independent experiments; Student's t-test (Supplemental Data Set S6) was used to calculate the P-values. C) EMSA showing the binding of ID1 to the ZmMIR399c and ZmMIR399j promoters. Top, distribution of predicted ID1-binding motifs in the ZmMIR399c and ZmMIR399j promoter. The black lines indicate the biotin-labeled probes (ZmMIR399c-P and ZmMIR399j-P) used in EMSA and in vitro DNA pull-down experiments. Bottom, EMSAs showing binding of ID1 protein to the predicted motifs in the ZmMIR399c and ZmMIR399j promoters. “mut” represents biotin probes with the binding motif mutated (Supplemental Data Set S5). D) DNA pull-down analysis showing the binding of ID1 to ZmMIR399c and ZmMIR399j promoters at the predicted motifs. Biotin-labeled probes, ZmMIR399c-P and ZmMIR399j-P, were used to pull down recombinant MBP (control) and MBP-ID1, which were then detected by immunoblotting using an anti-MBP antibody in both the beads and the supernatant. E) Chromatin immunoprecipitation (ChIP) showing the association of ID1 with the ZmMIR399c and ZmMIR399j promoters in vivo. Chromatin from the leaves of 35S:ID1-3×HA transgenic or wild-type (WT) plants was immunoprecipitated with anti-HA Trap. The enrichment (IP%) of the 2 promoter fragments (ZmMIR399c-P and ZmMIR399j-P shown in C) was determined by qPCR. Asterisks indicate significant difference between the 35S:ID1-3×HA transgenic line and wild type (WT) (*P ≤ 0.05; Student's t-test, Supplemental Data Set S6). The gene body regions of ZmMIR399c and ZmMIR399j (ZmMIR399c-GB and ZmMIR399j-GB) were used as negative controls.
The transcription factor ID1 was previously shown to selectively bind to an 11-bp consensus element by in vitro DNA selection and amplification-binding assays and DNA-binding experiments (Kozaki et al. 2004). Transient expression of a construct encoding a fusion between ID1 and the green fluorescent protein (ID1-GFP) in Nicotiana benthamiana leaves showed that ID1 mainly localizes in the nucleoplasm (Supplemental Fig. S19), in line with its potential role as a transcription factor. Sequence analysis of the 2-kb regions upstream of the annotated 5′ ends of ZmMIR399c and ZmMIR399j revealed 6 and 2 copies, respectively, of a typical ID1-binding motif (Fig. 5C; Supplemental Fig. S20). To assess whether ID1 can bind directly to these promoter regions, we purified a recombinant maltose-binding protein-tagged version of ID1 (MBP-ID1) in Escherichia coli (Supplemental Fig. S21) and performed electrophoretic mobility shift assays (EMSA) and in vitro DNA pull-down experiments by incubating biotin-labeled probes with recombinant MBP-ID1. We determined that ID1 binds to DNA fragments containing the motifs, while the corresponding nonbiotin-labeled competitors neutralized this binding activity (Fig. 5, C and D). Mutation of the motif completely abolished ID1 binding (Fig. 5, C and D). We also utilized a 35S:ID1-3×HA transgenic line (Supplemental Fig. S22) to conduct a chromatin immunoprecipitation (ChIP) experiment in leaf tissue. Immunoprecipitation with an anti-HA antibody detected a specific enrichment of ID1-3×HA at the promoter regions of ZmMIR399c and ZmMIR399j that encompass the probe sequences (Fig. 5, C and E). Taken together, these results demonstrate that ID1 directly binds to the promoter regions of ZmMIR399c and ZmMIR399j and represses their transcription.
We further investigated the response of ID1 expression to low-Pi stress by growing the maize inbred line B73 in hydroponic solution containing normal Pi (NP; 250 μM KH2PO4) or low Pi (LP; 5 μM KH2PO4). We imposed low-Pi treatment for 3 d or 5 d prior to the reproductive transition stage when ID1 is specifically expressed. Mature and immature leaves were collected and used for total RNA isolation and expression analysis at the floral transition stage. We first confirmed that the abundance of Pi-responsive miR399 and the long noncoding RNA PILNCR1 is elevated under low-Pi conditions (Supplemental Fig. S23), indicating the efficacy of treatment in the hydroponic system in imposing quantitative Pi stress on the plants. However, we did not detect expression changes for ID1 under low-Pi conditions (Supplemental Fig. S24), indicating that ID1 is not Pi responsive, but rather an autonomous regulator in maize.
Based on all the above observations, we propose that loss of ID1 function triggers the miR399-ZmPHO2 pathway, which enhances Pi acquisition and transport, and finally leads to higher Pi levels that are able to support the vegetative gigantism of the id1 mutant (Fig. 6). This situation contrasts with that of wild-type plants, where ID1 lowers miR399 levels by repressing the transcription of ZmMIR399c and ZmMIR399j, which in turn maintains Pi homeostasis (Fig. 6), underscoring ID1 as an autonomous Pi regulator upstream of the miR399-ZmPHO2 signaling pathway.
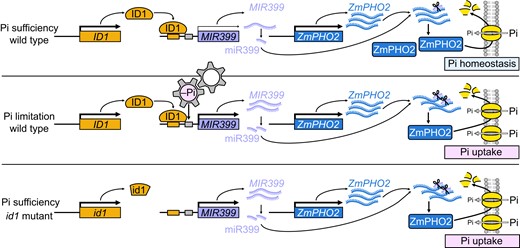
Proposed model for the function of ID1 as a Pi homeostasis regulator. In the absence of Pi stress (upper panel), ID1 represses the expression of ZmMIR399 genes to maintain in vivo Pi homeostasis and normal metabolism. Under Pi limitation (middle panel), the expression of ID1 is not responsive. However, ZmMIR399 expression can be directly upregulated by the response of Pi limitation, which finally promotes Pi uptake through downregulating ZmPHO2. When ID1 is mutated (lower panel), derepression of ZmMIR399 promotes the accumulation of mature miR399, with elevated miR399 levels knocking down the expression of ZmPHO2, resulting in enhanced Pi acquisition and transport. The size of the black arrows indicates expression levels.
ZmPHO2 was under selection during maize domestication
Crop domestication has not only been shaped by artificial selection but also by natural selective pressures (Martín-Robles et al. 2019). Natural selection under modern agricultural conditions, which differ from wild habitats in the availability of resources, might have led to changes in crops for both their external morphology and internal metabolism. Cultivated maize originated in the tropical environment of southwestern Mexico and has since spread widely to temperate zones having topsoil properties that differ from those of tropical areas. In tropical and subtropical regions, such as the Cerrado region in Brazil, acidic soils with low Pi availability are prevalent (Goedert 1983; Aguirre-Liguori et al. 2019). Under these conditions, without human intervention, the ancestral maize may have needed a more efficient Pi uptake system to ensure its survival. In humid temperate regions, Pi is more concentrated at the surface, a considerable portion being in the form of organic compounds (Lynch and Brown 2001). Under these conditions, cultivated maize can have better access to Pi. Furthermore, the use of fertilizer, which tends to be overapplied by farmers to guarantee adequate levels of crop yield and quality, makes Pi easily available. Thus, both natural and artificial selection pressures might have promoted changes in the Pi uptake/transport system during domestication or postdomestication dispersal of maize. We speculated that ZmPHO2, as a major Pi regulator, might have been targeted by selection as maize became domesticated and spread to temperate zones.
To test this hypothesis, we first analyzed the nucleotide diversity around ten ZmMIR399 genes and ZmPHO2 using the third-generation Z. mays haplotype map (HapMap 3), which includes ultrahigh-density single nucleotide polymorphism (SNP) data for various maize and teosinte accessions (Bukowski et al. 2018). We observed that the regions around ZmMIR399c and ZmMIR399j show a slight decrease in nucleotide diversity in maize and landraces compared with the teosinte group (Supplemental Fig. S25). Notably, the 5′ UTR of ZmPHO2, in which the miR399-binding sites are located, exhibited a clear drop in nucleotide diversity in maize compared with landraces or teosinte, with the maize group retaining only 29% of the genetic diversity of the teosinte group (Fig. 7A). We confirmed this observation by resequencing a ∼1.6-kb 5′ UTR fragment in 26 diverse maize inbred lines and 14 teosinte accessions (Z. mays ssp. parviglumis) (Supplemental Table S1). On average, we determined that maize (π = 0.00266) retained 33.7% of the nucleotide diversity of teosinte (π = 0.00788; Fig. 7B), with 2 specific regions (∼1.2 kb and ∼400 bp upstream of the ATG) showing very low nucleotide diversity in maize relative to teosinte (Supplemental Fig. S26). Coalescent simulations that incorporate the domestication bottleneck (Eyre-Walker et al. 1998; Tian et al. 2009) detected a significant deviation from expectation under a neutral domestication bottleneck for this resequenced region (P < 0.5; Fig. 7B), indicating that the 5′ UTR of ZmPHO2 was affected by selection. Although the miR399-binding sites do not show any sequence differences among the resequenced maize and teosinte accessions, we detected SNPs around the miR399-binding sites, which might affect the accessibility of miR399 and thus alter ZmPHO2 transcript levels. To test this hypothesis, we first predicted RNA secondary structures of a 5′ UTR fragment containing the miR399-binding sites (including 100 bp upstream of the first binding site and 100 bp downstream of the 6th binding site) with representative maize and teosinte sequences. Three conserved SNPs between maize and teosinte were present in this region and were predicted to affect the RNA secondary structures (Fig. 7C). Thus, it is possible that the SNPs affect the ability of miR399 to access its binding sites. To test whether the SNPs affect the activity of miR399, we performed transient coexpression assays in maize protoplasts using the minimal 35S promoter driving a 1.6-kb partial 5′ UTR fragment of ZmPHO2 (indicated by the magenta line segment in Fig. 2A) from maize and teosinte cloned upstream of LUC as reporters; we selected the miR399c precursor as a representative that we cloned as effector (see details in Materials and Methods). We observed that overexpression of the miR399c precursor effectively reduced LUC activity in both the maize and teosinte 5′ UTR reporters (Fig. 7D), which is consistent with the negative regulatory role of miR399 on ZmPHO2. Interestingly, miR399c showed higher inhibition of LUC activity derived from the maize 5′ UTR fragment than that of teosinte (Fig. 7D), suggesting that the 3 SNPs around the miR399-binding sites do affect the accessibility of miR399 to its binding sites in the 5′ UTR. The stronger repression in the maize 5′ UTR context may result in lower expression of ZmPHO2 in maize compared with teosinte; however, other regulatory elements may also affect ZmPHO2 expression.
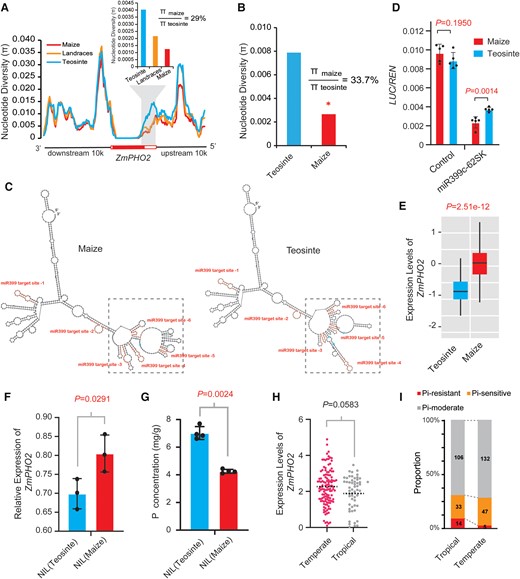
Evidence of selection at ZmPHO2. A) The 5′ regulatory region of ZmPHO2 exhibits low nucleotide diversity (π) in maize relative to teosinte, as analyzed with the maize HapMap v3 SNP data. The nucleotide diversity of maize, teosinte, and landraces is shown with red, blue, and orange lines or bars, respectively. White and red rectangles in the gene structure of ZmPHO2 under the line plot indicate the UTR and coding regions, respectively. B) Resequencing data for 26 maize inbreds and 14 teosinte accessions indicating that maize retained an average of the 33.7% nucleotide diversity seen in teosinte in the resequenced 5′ UTR. *P < 0.05 from the coalescent simulations. C) RNA secondary structures predicted by the RNAfold WebServer. The region from 100 bp upstream of the first miR399-binding site to 100 bp downstream of the 6th miR399-binding site of representative maize and teosinte lines was used for structure prediction. Red, miR399-binding sites; blue, conserved SNPs between maize and teosinte. D) Transient coexpression assays in maize protoplasts using miR399c as the effector and ZmPHO2 5′ UTRmaize:LUC and ZmPHO2 5′ UTRteosinte:LUC as reporters. Values are means ± SD based on the technical replicates (n = 5); two-tailed Student's t-test was used to calculate the P-values (Supplemental Data Set S6). E)ZmPHO2 expression levels in maize and teosinte, based on data from Wang et al. (2018). F)ZmPHO2 expression levels in ZmPHO2-NILmaize and ZmPHO2-NILteosinte as examined by RT-qPCR, normalized to that of ZmUBI. Values are means ± SD. Student's t-test (Supplemental Data Set S6) was employed to calculate the P-value. G) Total phosphorus (P) content in ZmPHO2-NILmaize and ZmPHO2-NILteosinte. Values are means ± SD. Student's t-test (Supplemental Data Set S6) was employed to calculate the P-value. H)ZmPHO2 expression levels in the root tissues of tropical and temperate inbred maize lines, based on data from (Kremling et al. 2018). P-values were calculated by a Kolmogorov–Smirnov test (Supplemental Data Set S6). I) Proportion of Pi-tolerant, Pi-sensitive, and Pi-moderate lines in tropical and temperate maize groups. The data are from (Luo et al. 2019).
To holistically examine the potential cis-regulatory effects on ZmPHO2 expression caused by maize and teosinte sequence divergence, we searched for cis-eQTL (expression quantitative trait locus) at ZmPHO2 using a maize–teosinte recombinant inbred line (RIL) population (Wang et al. 2018). We detected a strong cis-eQTL for ZmPHO2, with the maize allele showing a significantly higher expression level than the teosinte allele (Fig. 7E). To validate the effects of this cis-eQTL on ZmPHO2 expression, we developed a pair of near-isogenic lines (NILs) across the ZmPHO2 region from the same RIL population: the expression of ZmPHO2 was significantly higher in ZmPHO2-NILmaize harboring the maize allele of ZmPHO2 than in ZmPHO2-NILteosinte carrying the teosinte allele in an otherwise homogeneous maize (W22 inbred line) background (Fig. 7F). Thus, the cis-eQTL likely contains positive elements outside of the ∼1.6-kb 5′ UTR fragment used for the transient coexpression assay that enhance ZmPHO2 expression in maize such that the negative effects of the SNPs near the miR399-binding sites are overridden. However, the limited resolution of the cis-eQTL prevented us from pinpointing the position of such positive elements. In agreement with a role for ZmPHO2 as a negative regulator of Pi uptake, ZmPHO2-NILmaize lines exhibited a significantly lower total Pi level than ZmPHO2-NILteosinte lines (Fig. 7G). These results indicate that ZmPHO2 may have been a target of selection during maize domestication, leading to weaker Pi uptake in cultivated maize. We further examined the difference in expression of ZmPHO2 between temperate and tropical maize inbred lines using published RNA-seq data of a maize association panel (Kremling et al. 2018). This analysis revealed that the temperate lines tend to have higher ZmPHO2 expression levels than tropical lines in root tissues (Fig. 7H), suggesting that adapting maize from tropical regions to temperate zones was associated with an expression divergence of ZmPHO2. In addition, a recent study evaluated low-Pi tolerance in another maize association panel, which also included different sources of maize inbred lines (Fang and Luo 2019). Using the classification criteria of that study, we noticed a greater percentage of low-Pi-sensitive lines and a lower percentage of low-Pi-tolerant lines in the temperate maize subgroup than in the tropical maize subgroup (Fig. 7I), indicating that the temperate maize inbreds are more sensitive to low-Pi stress. Combined with the higher expression of ZmPHO2 in temperate maize relative to tropical maize, these observations suggest that Pi uptake ability might have degenerated as maize spread into temperate zones with high Pi cultivation environments from original tropical habitats. Taken together, the above results suggest that ZmPHO2 expression was affected by domestication or subsequent cultivation and may therefore have mediated the change of Pi uptake system during maize cultivation.
Discussion
Maize yield is largely limited by Pi poor availability in most cropping soils worldwide. Therefore, application of phosphorus fertilizers to cultivated crop plants is essential to ensure high-yielding agriculture. Given that global phosphorus resources are finite and rapidly diminishing, identifying Pi homeostasis regulators and dissecting the regulatory mechanism of Pi homeostasis are fundamental for developing Pi-efficient cultivars. In this study, we report that ID1, previously known as a floral transition regulator, can directly bind to the promoters of ZmMIR399 genes to inhibit their transcription, thus reducing the accumulation of mature miR399 and alleviating the repression of ZmPHO2 imposed by miR399. We conclude that ID1-miR399-ZmPHO2 contributes to the maintenance of Pi homeostasis in maize as a regulatory module. Interestingly, ID1 expression was independent of external Pi limitation, suggesting that ID1 is an autonomous regulator of Pi homeostasis. Our study revealed a direct functional link between Pi-limitation sensing by the miR399-ZmPHO2 regulatory module and plant developmental regulation by ID1, although further understanding of whether ID1 is integrated with other Pi regulators is still needed. Importantly, we also established that ZmPHO2 was under selection during maize domestication and cultivation, resulting in more sensitivity to Pi limitation in temperate maize than in tropical maize. We speculate that the degeneration of nutrient uptake ability might be a common phenomenon as maize spread to temperate zones with adequate soil nutrients.
ID1, as an upstream repressor, might regulate the miR399-ZmPHO2 module independently of known regulators
Several regulators involved in maintaining Pi homeostasis have been identified in plants (Rubio et al. 2001; Bari et al. 2006; Chen et al. 2007; Devaiah et al. 2007b; Zhou et al. 2008; Devaiah et al. 2009; Wang et al. 2013b). AtPHR1, a MYB-type transcription factor, was the first regulator demonstrated to mediate the Pi-starvation response; it plays a central role in transcriptional regulation of a large subset of Pi limitation-responsive genes, including ZmMIR399 (Bari et al. 2006). In the promoter regions of ZmMIR399 genes, AtPHR1 binds to the predicted PHR1-specific binding sequence (P1BS), resulting in positive regulation of miR399 accumulation. In a similar manner, PHR1 orthologs have been reported to act as transcription factors upstream of the miR399-PHO2 regulatory module in several plant species, including maize, rice, and wheat (Zhou et al. 2008; Wang et al. 2013a; Wang et al. 2013b). In maize, overexpression of ZmPHR1 significantly increases Pi content in shoots under low-Pi conditions (Wang et al. 2013b). By investigating the change in expression of ZmPHR1 and its closest ortholog, ZmMYB78, we determined that both genes show similar transcript levels in immature leaves and mature leaves, and between the id1-c1 mutant and wild-type plants (Supplemental Fig. S27), suggesting that ZmPHR1 and ZmMYB78 are not genetically downstream of ID1. PILNCR, a Pi deficiency-induced long noncoding RNA previously identified in maize, regulates the function of miR399 by competitively binding and sequestering the miRNA (Du et al. 2018). We also assessed the expression of PILNCR by RT-qPCR and measured similar expression levels in both the id1-c1 mutant and wild type (Supplemental Fig. S27). Combined with the experimental evidence showing the binding of ID1 to ZmMIR399 promoters, these expression results support the notion that ID1 is a direct upstream regulator of ZmMIR399 transcription. It is possible that ID1 regulates the miR399-ZmPHO2 module independently of ZmPHR1.
ID1 controls Pi homeostasis, likely through systemic movement of miR399, in maize and possibly other crops
Plants take up mineral nutrients from the rhizosphere via roots and the nutrients are subsequently distributed to aboveground shoots. The demand by shoots for nutrients and their supply from roots need to be properly coordinated to maintain the normal growth and development of plants. Long-distance signaling pathways can be triggered by internal or external factors as a component of adaptive responses when the nutrient status in shoots fluctuates (Forde 2002; Atkins and Smith 2007, 2007). Thus, when a change in internal nutrient status in shoots is sensed, this information is transduced by long-distance signals to roots, which ensures the integration of nutritional demands in shoots with physiological changes in roots. Many nutrient uptake systems involved in nutrient assimilation and mobilization in roots are regulated by demand from the shoot via shoot-derived signals (Marschner 1995).
In Arabidopsis, miR399 generated in shoots serves as a long-distance signal that represses PHO2 expression in roots under Pi limitation conditions, resulting in the activation of Pi uptake and translocation (Lin et al. 2008; Pant et al. 2008). In maize, ZmPHO2 transcripts in roots can be knocked down by miR399, whose precursor MIR399 is expressed in the vascular systems, ultimately leading to Pi overaccumulation in shoots (Du et al. 2018). In the present study, we also found that more Pi accumulated in shoots of miR399-overexpressing transgenic maize than in the wild type (Fig. 1B), in line with observations in Arabidopsis (Lin et al. 2008). However, in our miR399-OE transgenic maize, it is not clear whether miR399 is transported to roots to enhance Pi uptake there, because of the constitutive accumulation of mature miR399. In Arabidopsis, N. benthamiana, and soybean, micrografting experiments have been employed to determine the shoot-to-root movement of mature miRNAs (Pant et al. 2008; Kasai et al. 2010; Huen et al. 2018; Li et al. 2021). However, it is still very challenging to perform micrografting in maize, although a recent breakthrough was reported showing that the embryonic hypocotyl allows grafting in most monocotyledonous orders (Reeves et al. 2022). Accordingly, it might soon be possible to investigate the shoot-to-root movement of miR399 in maize as well.
The spatial patterns of ID1 expression led us to speculate that its role in regulating Pi homeostasis involves mobile miR399. ID1 is expressed exclusively in developing leaves (Colasanti et al. 1998), where it is expected to repress the expression of ZmMIR399 genes. In id1 mutants, the repression of ZmMIR399 transcription imposed by ID1 is released and miR399 is expected to be produced more in developing leaves. It is then likely that miR399 travels from leaves to roots, where its target gene ZmPHO2 is downregulated and Pi uptake is enhanced to support the vigorous vegetative growth and extremely delayed flowering of the id1 mutant. Interestingly, a mutant in the rice ortholog of maize ID1, early heading date2 (ehd2), also shows extremely delayed flowering with a giant plant architecture (Matsubara et al. 2008; Wu et al. 2008), which needs nutrients to support this extra vegetative growth. Given the conservation of the ID1, miR399, and PHO2 genes (Bari et al. 2006; Chiou et al. 2006; Matsubara et al. 2008; Wu et al. 2008), this regulatory system might be conserved between maize and rice. However, no ID1 orthologs have been identified in Arabidopsis (Colasanti et al. 2006). Thus, ID1 might be a monocotyledon-specific factor that orchestrates plant development and Pi acquisition to maintain growth homeostasis.
The nutrient uptake system may have been altered during maize cultivation to the current soil environments
Maize is well known to require large amounts of nitrogen, phosphorus, and potassium for optimal performance, which puts a high demand on the nutrient uptake system. After diverging from Balsas teosinte 9,000 yr ago through domestication and artificial selection, cultivated maize has been widely adopted as a crop plant in the fertile, highly productive plains of the temperate regions, where it is often grown with the application of fertilizer (Sluyter and Dominguez 2006; Hastorf 2009; Piperno et al. 2009). Compared with modern maize, teosinte grows in the wild in a variable and challenging environment in subtropical southwestern Mexico, where plants are only exposed to occasional and inadequate fertilization from recessional flooding and mineralization (Iltis et al. 1979; Piperno et al. 2007; Hastorf 2009; Zhu et al. 2010). Thus, in addition to the dramatic morphological changes in plant size and architecture, domestication probably also caused alterations in maize metabolism and in its responses to the soil environments, such as nitrogen, potassium, and phosphorus surplus. It is also well known that most breeders involved in maize improvement preferentially focused on yield and disease resistance, which was essential to meet the demand for food due to continued growth of the human population. To increase crop yields, farmers have typically applied large quantities of synthetic fertilizer to the soil, which might induce the degeneration of plant nutrient uptake systems over long periods. In this study, we confirmed that the 5′ UTR of ZmPHO2, encoding a central Pi homeostasis regulator, was under selection during maize domestication or cultivation (Fig. 7, A and B; Supplemental Fig. S26). In the context of a partial 5′ UTR (∼1.6 kb), the maize allele showed stronger repression by miR399 (Fig. 7D). However, the maize allele of ZmPHO2 tended to have a higher expression level than the teosinte allele, according to the eQTL data from a maize–teosinte RIL population (Fig. 7E) (Wang et al. 2018) and our investigation with a pair of maize–teosinte NILs across the ZmPHO2 region (Fig. 7F). Therefore, the expression difference of ZmPHO2 between maize and teosinte alleles did not result from differences in miR399-mediated RNA cleavage. In our transient coexpression assay (Fig. 7D), only a fragment of the ZmPHO2 5′ UTR was used, while additional regulatory elements could have resided in the promoter or elsewhere in the longer 5′ UTR that spans 4 exons intervened by 3 introns (Fig. 2A). Although eQTL mapping with a linkage population identified a cis-eQTL for ZmPHO2, the limited resolution prevented us from pinpointing the exact location of the causative elements. Thus, it is possible that the detected cis-eQTL was located in either the promoter or the 5′ UTR outside of the cloned fragment used for transient coexpression assays. Identifying the causative elements will be necessary to further explain the expression difference of ZmPHO2 between maize and teosinte alleles. Even so, the higher expression of ZmPHO2 is consistent with the prevalence of low Pi-sensitive lines in the temperate maize relative to the tropical maize subgroup (Fig. 7, H and I). Thus, the Pi uptake system might have degenerated in modern cultivars, resulting in more low Pi-sensitive maize cultivars, during maize cultivation.
Similar situations might have occurred in maize cultivation in local soils with long-term application of N fertilizer. Indeed, a previous report indicated that the maize progenitor, teosinte, uses nitrogen more efficiently than modern cultivated maize (Gaudin et al. 2011). In rice, high nitrogen supply was shown to have resulted in a prevalence of low nitrogen-use-efficiency (NUE) cultivars (Liu et al. 2021). The allelic variation in TEOSINTE BRANCHED 1, CYCLOIDEA AND PROLIFERATING CELL FACTOR 19 (OsTCP19) contributes to the geographical adaptation to local soil nitrogen. The OsTCP19-H allele, which confers higher nitrogen uptake ability, was frequently observed in wild rice populations in natural habitats that usually have low nitrogen content (Liu et al. 2021). However, it has largely been lost in modern cultivars during rice domestication or cultivation in nitrogen-rich regions (Liu et al. 2021), suggesting that OsTCP19-H was subjected to relaxed selection pressure due to the nitrogen-rich soil. DENSE AND ERECT PANICLES 1 (DEP1), encoding a G protein that regulates nitrogen signaling and modulation, was previously reported to have been subjected to artificial selection during O. sativa spp. japonica domestication (Sun et al. 2014). The DEP1 alleles from Oryza rufipogon, the wild ancestor of cultivated rice, confer robust resistance to nitrogen limitation, such that the wild rice shows less sensitivity than japonica cultivars (Sun et al. 2014). These data in rice also point to a weakening of the nutrient uptake system in domesticated crop species. New solutions are urgently needed to simultaneously increase yields while maintaining, or preferably decreasing, fertilization use to maximize the efficiency of nutrient use in crops. Otherwise, under conditions of fertilizer shortage, crop yields could be severely impacted in the future. Restoring favorable alleles from the wild ancestor, teosinte, might provide a strategy for improving the nutrient uptake system of cultivated maize and maintaining environmentally sustainable increases in grain yield by avoiding the excessive application of fertilizer.
Materials and methods
Plant materials and growth conditions
The transgenic maize (Z. mays) plants of miR399-OE, miR399-STTM, and CRISPR-Cas9 mutant Zmpho2 were generated by the China National Seed Group Co. Ltd., China, with the inbred line ZZC01 as the receptor line, while the CRISPR-Cas9 mutant id1-c1 was generated by Functional Genomics and Molecular Breeding of Crops, China Agricultural University, Beijing, with the inbred line LH244 as the receptor line. The heterogeneous inbred family (MR1451) for developing the near isogenic lines (NILs) around ZmPHO2 locus was originally generated from a maize–teosinte BC2S3 recombinant inbred line (RIL) population derived from a cross between maize inbred line W22 and teosinte accession CIMMYT 8759 (Z. mays ssp. parviglumis) (Liang et al. 2019). NILs homozygous for W22 and CIMMYT 8759 around the ZmPHO2 locus were designated ZmPHO2-NILmaize and ZmPHO2-NILteosinte and were used for assays. Transgenic miR399-OE and miR399-STTM lines were grown in experimental fields from 2018 to 2021 in the summer in Tieling (Liaoning, 41.8° N, 123.4° E), China, and in the winter in Sanya (Hainan, 18.4° N, 109.2° E), China, for the screening of homozygous plants and bulking up of seeds. Homozygous miR399-OE, miR399-STTM, and the corresponding wild-type plants were grown in the field for phenotypic investigation and in a greenhouse at 25°C, 16-h light/8-h dark photoperiod with light intensity at 130 µmol m−2 s−1 (metal halide bulb 400 w/d) for tissue collection. Phenotypic investigation was performed with mature plants in the field, while measurements of P content and RNA gel blot assays were performed with seedlings grown in pots (30 cm × 25 cm × 12 cm, length × width × depth) containing vermiculite and organic soil mix (1:1) with full nutrients (N + P2O5 + K2O, 110–1200 mg/kg; PH 5.5–7.5) in the greenhouse.
The id1-c1 heterozygous mutant, created by CRISPR/Cas9-mediated gene editing, was grown in the experimental field in Tieling (Liaoning, 41.8° N, 123.4° E) or Sanya (Hainan, 18.4° N, 109.2° E) for positive mutant identification and generation of seed stocks, and in a greenhouse for genotyping and tissue collection.
Plasmid construction and genetic transformation
For the construction of the miR399-OE binary vector, 2 stem-loop structures harboring different mature miR399 sequences (i.e. miR399a/c/h and miR399e/i/j) were first inserted into a primary miRNA backbone under the control of the maize ubiquitin (ZmUBI) promoter in the pOT2-poly-cis-UN cloning vector by PCR amplification. After verification by Sanger sequencing and digestion with restriction enzymes, the UBIpro:miR399:NOS fragment from the pOT2-poly-cis-UN-miR399OE construct was cloned between the PacI and MluI restriction sites in the destination vector pZZ00026-PM, which contains the Streptomyces hygroscopicus phosphinothricin acetyltransferase (BAR) gene as a selectable marker driven by the cauliflower mosaic virus (CaMV) 35S promoter, via T4 ligase-mediated ligation (Supplemental Fig. S2A).
The miR399-STTM binary vector was prepared following previously described procedures (Tang and Tang 2013; Peng et al. 2018) with some modifications. In brief, the miR399-STTM fragment was first introduced into the pOT2-poly-cis-UN cloning vector to be placed under the control of the ZmUBI promoter by PCR amplification, SwaI digestion, and T4 ligase-mediated ligation. After verification by Sanger sequencing and restriction enzyme digestion, the UBIpro:miR399-STTM:NOS fragment from pOT2-poly-cis-UN-miR399STTM was cloned between the PacI and MluI sites in the binary vector pZZ00026-PM using T4 ligase (Supplemental Fig. S2B).
To prepare the CRISPR/Cas9 knockout vector for ID1 or ZmPHO2, 2 target sites located in the coding region near the start codon of each gene were selected with the aid of the online tool CRISPR-P (http://crispr.hzau.edu.cn/CRISPR2/) and verified by manual alignment to the maize “B73' reference genome (AGPv5; https://www.maizegdb.org/) (Hufford et al. 2021). After restriction enzyme digestion, the 2 single-guide RNAs (sgRNAs) were integrated into the BtgZ1 site to be placed under the control of the maize U6.1 promoter and the BsaI site to be placed under the control of the maize U6.2 promoter in the pENTR4-gRNA1 cloning vector, respectively (Fig. 2; Supplemental Fig. S16). After verification by Sanger sequencing, the U6pro-driven gRNA cassette was cloned into the destination vector pZmCas9 by LR recombination.
After verification by Sanger sequencing, the resulting binary construct containing miR399-OE, miR399-STTM, or the sgRNAs with Cas9 was introduced into Agrobacterium (Agrobacterium tumefaciens) strain EHA105 for genetic transformation. To identify mutation events, genomic fragments covering both target sites were amplified from T0 CRISPR/Cas9 plants by PCR. After gel purification, the resulting PCR products were cloned into the pEASY-Blunt cloning vector (TransGen, Beijing, China) and at least ten clones per PCR product were analyzed by Sanger sequencing. Finally, 1 and 3 independent T0 events with large fragment deletions at the target sites were identified for ID1 and ZmPHO2, respectively (Fig. 2; Supplemental Fig. S16).
Quantification of total phosphorus content
Shoots and roots of transgenic miR399-OE and miR399-STTM plants were collected separately as 3 biological replicates at the seedling stage. For each replicate, tissues from more than 5 seedlings were pooled and incubated at 105°C for 30 min. The samples were then dried at 65°C for 3 d, weighed, and milled to a fine powder. Portions (∼100 µg) were then digested in 5 mL 65% HNO3-H2O2 at 300°C until the solution became clear. Total phosphorus content was determined by the vanadomolybdophosphoric acid colorimetric method (Westerman 1991). The same protocol was also used to determine total phosphorus content in mature leaves, immature leaves, roots, stems, and leaf sheaths in the id1-c1 mutant and wild-type plants.
Measurement of relative chlorophyll content
At 15 d after pollination, when the premature senescence syndrome appeared, relative chlorophyll contents were measured from ear leaves with a mobile device, SPAD 502 Plus Chlorophyll Meter (Speactrum Technologies Inc.), which is widely used for the rapid, accurate, and nondestructive measurement of leaf chlorophyll concentrations. Measurements with the SPAD 502 Plus meter produce relative SPAD (soil plant analysis development) values that are proportional to the amount of chlorophyll present. Three different regions of the leaf (base, middle, and tip) were chosen for measurements. More than 30 plants were used for analysis within an experiment.
Gene expression analysis with RT-qPCR
Total RNA was extracted from mature or immature leaves of the id1-c1 mutant, miR399-OE transgenic plants, miR399-STTM transgenic plants, and their corresponding wild-type controls using TRIzol reagent (Invitrogen, USA). Genomic DNA removal and first-strand cDNA synthesis were carried out using a TaKaRa first-strand cDNA synthesis kit (TaKaRa, Japan). Quantitative PCR (qPCR) was performed using SYBR Green detection and relative gene expression was determined with the 2−△△Ct method (Livak and Schmittgen 2001) from 3 biological replicates. ZmUBI was used as the internal control for quantification of transcript levels. Primers used for qPCR are listed in Supplemental Data Set S5.
Small RNA gel blot
Total RNA was isolated by TRIzol reagent from mature and immature leaves of the id1-c1 mutant and its wild type. Three biological replicates were collected for each genotype with at least 5 leaves pooled per sample. Small RNA was first enriched with a high concentration of NaCl (for each reaction, 50 µL 5 M NaCl and 50 µL 50% PEG8000 were added into 400 µL total RNA), and then, the precipitated RNA was used for RNA gel blot analysis to assess the accumulation of mature miR399 in the id1-c1 mutant. In brief, a 15% polyacrylamide gel was prepared in 15 mL of gel mixture including 6.3 g urea, 1.5 mL 5×TBE solution and 5.63 mL 40% acrylamide/Bis (29:1) solution, supplied with 100 µL 10% ammonium persulfate (APS), and 10 µL TEMED. A minimum of 10 µg RNA per sample was loaded onto a 15% polyacrylamide gel and resolved at 120 V for 1 h. RNA was transferred from the gel to a nitrocellulose membrane (Bio-Rad) using a semidry transfer apparatus (Bio-Rad). Specific probes labeled with 5′-biotin were hybridized to the membrane. A probe complementary to U6 was used as an internal control. Hybridization was performed at 55°C for 16 h, followed by 3 washes in 20 mL 20×SSC buffer suppled with 0.1% SDS. Signals were detected using a Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher, 89880) with a chemiluminescence imaging system (Clinx Science Instruments Co. Ltd., China). Probe sequences can be found in Supplemental Data Set S5.
Recombinant protein expression and purification, electrophoretic mobility shift assay, and in vitro DNA pull-down assay
To construct the MBP-ID1 vector, the coding sequence (CDS) of ID1 was first amplified with cDNA prepared from the developing leaves of maize as template, using primers ID1-MBP-F and ID1-MBP-R (Supplemental Data Set S5). The PCR product was then cloned into a version of the pMSCG7 vector with the sequence encoding maltose-binding protein (MBP) at the 5′ end of the cloning site using Gibson assembly; the resulting construct was validated by Sanger sequencing. The empty vector and the version containing the ID1 CDS were introduced separately into RosettaTM DE3 E. coli (Sigma-Aldrich, 70954) for protein expression and purification. Transformed E. coli colonies were first cultured to a final OD at 600 nm of 0.7 and then induced with 0.1 mM IPTG (Thermo Fisher Scientific, 15529019) at 16°C for 16 h. The bacterial pellets were collected by centrifugation at 3,972 g with a JLA-16.250 rotor for 10 min at 4°C and resuspended in lysis buffer [100 mM Tris-HCl pH 7.5; 500 mM NaCl; 5 mM 2-mercaptoethanol; 5% (v/v) glycerol; 20 mM imidazole; 0.1% (v/v) CA630; 1 mM PMSF; 1×EDTA-free protease inhibitor cocktail]. After sonication (10 s sonication followed with 20 s cooling for 30∼60 times), the recombinant protein was purified with Profinity IMAC Resin, Ni charged (Bio-Rad, 1560133). The protein was eluted with elution buffer [50 mM Tris-HCl, pH 7.5; 500 mM NaCl; 5 mM 2-mercaptoethanol; 5% (v/v) glycerol; 400 mM imidazole; 0.1% (v/v) CA630], and the concentration was estimated using a Bradford assay (Quick Start Bradford 1× Dye Reagent #5000205; Bio-Rad).
For EMSA, a 20-µL reaction mixture containing 10× binding buffer (100 mM Tris-HCl, pH 7.5; 500 mM KCl; 10 mM DTT), 20 fmol biotin-labeled probe (Supplemental Data Set S5), recombinant proteins (∼1 µg), and varying amounts of unlabeled probes as competitors was incubated at room temperature for 1 h. DNA–protein complexes were resolved on a 6% polyacrylamide gel and transferred to a charged Hybond-N+ membrane (GE Healthcare). The membrane was crosslinked by UV light (1,200 mJ), probed with stabilized streptavidin–horseradish peroxidase (HRP), and subsequently detected using a Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher). Membranes were imaged using a ChemiDoc XRS+ camera (BioRad).
For the in vitro DNA pull-down assay, ∼10 pmol of the biotin-labeled probes used in the EMSAs mentioned above was first incubated with 10 µL Dynabeads M-280 streptavidin magnetic beads (Thermo Fisher Scientific) for 15 min at room temperature in 1× B&W buffer (5 mM Tris-HCl pH 7.4, 0.5 mM EDTA, 1 M NaCl). Probe-bound beads were then incubated with the same amount of MBP or MBP-ID1 protein (∼100 ng) in 100 µL binding buffer [10 mM Tris-HCl pH 7.5, 50 mM KCl, 5 mM MgCl2, 2.5% (v/v) glycerol, 0.05% (v/v) CA630] containing 10 µg BSA for 1 h at 4°C. The precipitates and 1/5 of the supernatant were boiled in SDS loading buffer and subjected to immunoblot analysis using an anti-MBP monoclonal antibody (New England Biolabs, E8032S, 1:200 dilution), followed by anti-mouse IgG-HRP conjugated antibody (Sigma, A4416, 1:5,000 dilution).
RNA-seq library preparation, sequencing, and data processing
The transgenic lines miR399-OE#2 and miR399-STTM#2, together with the corresponding wild type, were used for RNA-seq with 3 biological replicates. Mature leaves were collected at the floral transition stage and at least 5 leaves were pooled for each replicate. Total RNA extraction was performed using TRIzol reagent and genomic DNA was removed with recombinant DNase I (Roche) treatment (10 U DNase I per 20 µg total RNA) at 37°C for 1 h. Total RNA quality was verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA) before library preparation. Sequencing libraries were prepared with a NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB) and sequenced on an Illumina HiSeqX10 platform using a 150-bp paired-end sequencing strategy at Novogene Co. Ltd. (Beijing, China). Approximately 25 million high-quality 150-bp paired-end reads were generated from each library.
The raw data were first examined for library quality using the FastQC program (https://github.com/s-andrews/FastQC), which provides a modular set of analyses for a quick impression of the data, including “Per base sequence quality,” “Per sequence quality scores,” “Per base sequence content,” “Per base GC content,” “Per sequence GC content,” “Per base N content,” “Sequence Length Distribution,” “Sequence Duplication Levels,” “Overrepresented sequences,” and “Kmer Content.” Raw data after the quality check of FastQC were analyzed using the “pRNASeqTools mrna” pipeline (https://github.com/grubbybio/pRNASeqTools/), which integrates the trimming of low-quality nucleotides, reads alignment, reads/fragments counting, and differential gene expression analysis. In brief, high-quality trimmed reads were mapped to the B73 reference genome (AGPv5; https://www.maizegdb.org/) using STAR 2.7.3a (Dobin et al. 2013) with the parameters “–alignIntronMax 5000 –outSAMmultNmax 1 –outFilterMultimapNmax 50 –outFilterMismatchNoverLmax 0.1”. Then, featureCounts (v2.0.0) was employed to count reads mapped to exon regions for each gene with the parameters “-p -B -C -O -s 0” (Liao et al. 2014). Differentially expressed genes (DEGs) were identified with fold change [reads per million (RPM)] ≥ 2 and false discovery rate (FDR) < 0.05 using DESeq2 (Love et al. 2014). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the DEGs were performed using g:Profiler, a free online platform (https://biit.cs.ut.ee/gprofiler/gost) (Raudvere et al. 2019). GO terms with FDR ≤ 0.05 were retained and deemed significantly enriched.
Small RNA-seq data analysis
The raw small RNA-seq data for the id1-m1 mutant (accession number PRJNA439244) were downloaded from the National Center for Biotechnology Information Sequence Read Archive (Minow et al. 2018). The 3′ adaptor sequence was detected and removed, and then, size selection (from 18 to 42 nts) was carried out for adaptor-trimmed reads using Cutadapt v1.15 (Martin 2011). The retained reads were mapped to the AGPv5 reference genome for B73 (https://www.maizegdb.org/) using ShortStack v3.8.5 (Johnson et al. 2016). Information on annotated maize miRNAs was obtained from both miRBase v21 (http://www.mirbase.org/) and miRNEST 2.0 (http://rhesus.amu.edu.pl/mirnest/copy/). Adaptor-trimmed reads mapped to specific miRNAs were counted and summarized separately for each size class ranging from 18 to 27 nts. Normalization was performed by calculating the RPM value for each size class, and comparison was carried out using the R package DESeq2 (Love et al. 2014).
Dual-luciferase transient expression assay in maize protoplasts
To examine the regulation of ZmMIR399 genes by ID1, 2-kb promoter fragments for the ZmMIR399c and ZmMIR399j genes were cloned into the pGreenII 0800-LUC vector, which carries a minimal CaMV promoter (mpCaMV) upstream of the firefly luciferase (LUC) coding sequence, to drive LUC reporter transcription; the ID1 coding sequence was cloned into the pGreenII 62-SK vector as effector. Similarly, to validate the relationship between miR399s and ZmPHO2, different miR399 precursors were cloned into pGreenII 62-SK as effectors and a ∼1.6-kb 5′ UTR fragment (indicated by the magenta line segment in Fig. 2A) of ZmPHO2 from B73 was cloned into pGreenII 0800-LUC as the reporter (Supplemental Fig. S7A). To test whether the SNPs around the miR399-binding sites affect miR399-mediated repression of ZmPHO2, ∼1.6-kb 5′ UTR fragments (indicated by the magenta line segment in Fig. 2A) of ZmPHO2 from maize and teosinte were cloned into pGreenII 0800-LUC as reporters, and the miR399c precursor was cloned into pGreenII 62-SK as the effector. The Renilla luciferase gene (REN) under the control of the 35S promoter in the pGreenII 0800-LUC vector was used as an internal control to correct for differences in protoplast transfection efficiency.
Mesophyll protoplasts were isolated from leaves of 12-d-old etiolated B73 seedlings following a method described previously (Yoo et al. 2007). Each effector/reporter combination was cotransfected into mesophyll protoplasts, using a previously described polyethylene glycol method (Hellens et al. 2005; Yoo et al. 2007). After incubation in the dark for 12 to 16 h, LUC and REN activities were measured using a dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions. Relative LUC activity was calculated by normalizing LUC activity against REN activity. At least 3 technical replicates were performed per construct combination: the same batch of transfected protoplasts was split into different tubes as technical replicates. All experiments were independently performed 3 times. Primers used for plasmid construction are shown in Supplemental Data Set S5.
Agrobacterium-mediated infiltration and fluorescence microscopy in Nicotiana benthamiana leaves
To construct 35S:ID1-EYFP, the ID1 coding region was PCR-amplified from genomic DNA using primers ID1-TSK-F and ID1-TSK-R (Supplemental Data Set S5). The PCR product was then cloned into TSK108, a pENTRY-D-topo-based Gateway entry vector. The sequence of the pENTR clone was validated by Sanger sequencing and recombined with the binary vector pGWB641, which contains an EYFP sequence at the 3′ end of the recombination site, by Gateway cloning. Similarly, the vector 35S:FIB2-RFP was prepared in pGWB654 as a nucleolus marker.
For subcellular localization in N. benthamiana leaf epidermal cells, Agrobacterium strain GV3101 individually containing the constructs 35S:ID1-EYFP and 35S:FIB2-RFP was grown overnight at 28 °C with constant shaking at 200 rpm. The Agrobacterium cells were collected by centrifugation at 3,972 g at room temperature for 10 min. The supernatant was discarded, and the pellet was resuspended in infiltration buffer (10 mM MES pH 5.6, 10 mM MgCl2, 150 mM acetosyringone) to a final OD at 600 nm of 1.0. Subsequently, the Agrobacterium resuspensions of 35S:ID1-EYFP and 35S:FIB2-RFP were mixed in equal volume and coinfiltrated into 4-wk-old leaves of N. benthamiana. After infiltration, plants were placed under long-day conditions (16-h light/8-h dark, 22 °C) for ∼48 h, and EYFP and RFP fluorescence was observed via confocal microscopy using a Zeiss 710 laser scanning microscope equipped with a Plan-Apochromat 100×/1.4 oil-immersion objective and an Axiocam 506 mono camera (Carl Zeiss, Jena, Germany). To stain the nucleus, the fluorescent stain, 4′ 6-diamidino-2-phenylindole (DAPI), was infiltrated into the leaves, and DAPI fluorescence was observed at 20 min after infiltration via confocal microscopy. The following Zeiss filter sets were used: YFP, exciter 500/25 nm/nm, emitter 535/40 nm/nm; DAPI, exciter 365 nm, emitter 445/50 nm/nm; and RFP, exciter 558 nm, emitter 583/40 nm/nm. Fluorescence images were collected using Zeiss ZEN software.
Chromatin immunoprecipitation (ChIP) assay
The full-length ID1 coding region was amplified from B73 genomic DNA and subcloned into TSK108. The validated clone was recombined with the binary vector pGWB614, which contains a 3×HA tag at the 3′ end of the recombination site, by Gateway cloning. The fusion construct, 35S:ID1-3×HA, was introduced into Agrobacterium strain EHA105 for genetic transformation with the maize inbred line ZZC01 as receptor. The leaf tissues of the 35S:ID1-3×HA transgene-positive line (T1 generation) at the seedling stage were collected to perform chromatin immunoprecipitation (ChIP) assays, with a previously described protocol (Kaufmann et al. 2010) as reference. Sonicated chromatin fragments were immunoprecipitated using anti-HA antibody (Sigma, H6908), and the resulting precipitated chromatin was quantified by qPCR. The percentage input was calculated with the following formula: 2−ΔCt times 100 and divided by the dilution of the input, in which Δcycle threshold (ΔCt) = (Ct [input]—Ct [ChIP]) (Li et al. 2016). The wild-type (WT) ZZC01 was used as control sample to calculate the fold enrichment.
Nucleotide diversity analysis and test for selection
The third-generation haplotype map data of Z. mays (HapMap 3: https://www.panzea.org/) (Bukowski et al. 2018) was downloaded and used to calculate nucleotide diversity along the 10-kb upstream and downstream regions of ZmMIR399 family genes and ZmPHO2 in maize, landraces, and teosinte. The population genomic analysis was done with the R package PopGenome (Pfeifer et al. 2014). Sliding windows (window size = 1,000 bp, step size = 100 bp) across the target regions were generated using the function “sliding.window.transform”. Nucleotide diversity (π) in each sliding window was calculated for the 3 groups, tropical maize, temperate maize, and teosinte, using the function “diversity.stats.” For the analysis of the resequenced ∼1.6-kb region of ZmPHO2, multiple sequence alignments were performed using BioEdit v.7.1.3.0 with some manual edits when necessary. Nucleotide diversity was calculated using DnaSP v.5.10.00 with a sliding window size of 100 bp. Coalescent simulations were employed to evaluate whether the loss of nucleotide diversity observed in maize relative to that in teosinte could be explained by a domestication bottleneck alone using Hudson's ms simulator (Hudson 2002). All parameters in the model were set according to previously established values (Wright et al. 2005; Tian et al. 2009; Xu et al. 2017; Liang et al. 2019). The population mutation and population recombination parameters were estimated from the teosinte sequences. A total of 10,000 coalescent simulations were performed.
Acknowledgments
We thank the Instrumental Analysis Center of Shenzhen University for technical assistance.
Author contributions
X.W., X.C., and L.L. designed and supervised the study; X.W., D.Y., YC.L., YM.L., J.H., X.Y., R.H., and Q.X. conducted laboratory experiments, field work, and data analysis; YM.L., H. J., and F.T. created the id1-c1 mutant by CRISPR/Cas9 technology, generated the near-isogenic lines and provided the DIBOA and DIMBOA standards; X.W. performed the RNA-seq and small RNA-seq data analysis and interpretation; X.W., X.C., and L.L. wrote the manuscript. All authors read and revised the manuscript.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Alignment of mature miR399 sequences in maize and the genomic locations of ZmMIR399 genes.
Supplemental Figure S2. Schematic diagram of the miR399-OE and miR399-STTM vectors used for genetic transformation.
Supplemental Figure S3. Phenotype of field-grown miR399-overexpressing transgenic plants.
Supplemental Figure S4. Phenotype of leaf necrosis at the seedling stage.
Supplemental Figure S5. Analysis of agronomic traits of 3 independent miR399-overexpressing transgenic lines.
Supplemental Figure S6. Properties of miR399-STTM transgenic plants.
Supplemental Figure S7. Transient coexpression assays in maize protoplasts for validating the relationships between miR399s and ZmPHO2.
Supplemental Figure S8. Gene structures of ZmPHO2 in different versions of maize genome annotations (GRMZM2G381709 in AGPv3; Zm00001d038972 in AGPv4; Zm0001eb295490 in AGPv5).
Supplemental Figure S9. Clustering and principal component analysis (PCA) of experimental samples miR399-OE#3 and miR399-STTM#2 used in RNA-seq.
Supplemental Figure S10. Gene ontology (GO) and KEGG pathway enrichment analyses.
Supplemental Figure S11. The genes involved in the benzoxazinoid biosynthesis pathway and their expression differences between miR399-OEP#3 and miR399-OEN#3.
Supplemental Figure S12. RT-qPCR validation of expression changes of all genes in the benzoxazinoid biosynthesis pathway between the miR399-OE or miR399-STTM transgene-positive lines and the corresponding transgene-negative lines.
Supplemental Figure S13. The relative metabolite content of both DIBOA and DIMBOA between the miR399-OE or miR399-STTM transgene-positive lines and the corresponding transgene-negative lines.
Supplemental Figure S14. Reanalysis of small RNA-seq data downloaded from (Minow et al. 2018).
Supplemental Figure S15. ZmPHO2 expression in mature leaves (ML) and immature leaves (IL) in the id1-m1 mutant and wild type (WT).
Supplemental Figure S16. The id1 knockout line (id1-c1) created by CRISPR/Cas9.
Supplemental Figure S17. ID1 expression levels in transgene-positive (miR399-OEP#3 or miR399-STTMP#2) and the corresponding transgene-negative (miR399-OEN#3 or miR399-STTMN#2) plants.
Supplemental Figure S18. Total phosphorus (P) concentration in the id1-c1 mutant and wild type (WT) at the floral transition stage.
Supplemental Figure S19. Subcellular localization of the ID1 protein in Nicotiana benthamiana leaf epidermal cells.
Supplemental Figure S20. Diagram showing the distribution of ID1-binding motifs in the promoter regions of ten ZmMIR399 genes.
Supplemental Figure S21. Purification of recombinant ID1 protein.
Supplemental Figure S22. Immunoblot analysis of 35S:ID1-3xHA transgenic lines.
Supplemental Figure S23. Determination of the levels of miR399 and the long noncoding RNA PILNCR1, both of which were known to be upregulated under low-Pi (LP) conditions.
Supplemental Figure S24. Relative ID1 expression under low-Pi stress conditions.
Supplemental Figure S25. Nucleotide diversity (π) analysis of the genomic regions surrounding ZmMIR399 family genes in maize, landraces, and teosinte groups with maize HapMap 3 data.
Supplemental Figure S26. Nucleotide diversity (π) analysis of the 5′ regulatory region of ZmPHO2 in 26 diverse maize inbred lines and 14 teosinte accessions.
Supplemental Figure S27. Expression levels of ID1, ZmPHR1, ZmMYB78, and PILNCR genes in mature leaves (ML) and immature leaves (IL) of the id1-c1 mutant.
Supplemental Table S1. List of 26 diverse maize inbred lines and 14 teosinte accessions used for resequencing the 5′ regulatory region of ZmPHO2.
Supplemental Data Set S1. Differentially expressed genes identified between miR399-OE-positive and negative lines.
Supplemental Data Set S2. Differentially expressed genes identified between miR399-STTM-positive and negative lines.
Supplemental Data Set S3. GO and KEGG enrichments for DEGs between miR399-OE-positive and negative lines.
Supplemental Data Set S4. GO and KEGG enrichments for DEGs between miR399-STTM-positive and negative lines.
Supplemental Data Set S5. Primers and probes used in this study.
Supplemental Data Set S6. Results of statistical analysis.
Funding
This research was supported by the National Natural Science Foundation of China (31900446 to X.W.), National Key Research and Development Program of China (2019YFA0903900 to L.L.), the Guangdong Innovation Research Team Foundation (2014ZT05S078 to X.C.), the China Postdoctoral Science Foundation (2018M640823 to X.W.), and a Shenzhen Basic Research General Project (JCYJ20190808112207542 to L.L).
Data availability
The raw RNA-seq data in this study have been submitted to the GenBank database under accession number PRJNA819017.
References
Author notes
These authors contributed equally.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plcell/pages/General-Instructions) is: Lin Liu ([email protected]).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}