





Abstract
The U.S. Food and Drug Administration (FDA) granted accelerated approval to rucaparib in May 2020 for the treatment of adult patients with deleterious BRCA mutation (germline and/or somatic)‐associated metastatic castrate‐resistant prostate cancer (mCRPC) who have been treated with androgen receptor‐directed therapy and a taxane. This approval was based on data from the ongoing multicenter, open‐label single‐arm trial TRITON2. The primary endpoint, confirmed objective response rate, in the 62 patients who met the above criteria, was 44% (95% confidence interval [CI]: 31%–57%). The median duration of response was not estimable (95% CI: 6.4 to not estimable). Fifty‐six percent of patients had a response duration of >6 months and 15% >12 months. The safety profile of rucaparib was generally consistent with that of the class of poly‐(ADP‐ribose) polymerase enzyme inhibitors and other trials of rucaparib in the treatment of ovarian cancer. Deaths due to adverse events (AEs) occurred in 1.7% of patients, and 8% discontinued rucaparib because of an AE. Grade 3–4 AEs occurred in 59% of patients. No patients with prostate cancer developed myelodysplastic syndrome or acute myeloid leukemia. The trial TRITON3 in patients with mCRPC is ongoing and is planned to verify the clinical benefit of rucaparib in mCRPC. This article summarizes the FDA thought process and data supporting this accelerated approval.
The accelerated approval of rucaparib for the treatment of adult patients with deleterious BRCA mutation (germline and/or somatic)‐associated metastatic castrate‐resistant prostate cancer who have been treated with androgen receptor‐directed therapy and a taxane represents the first approved therapy for this selected patient population. This approval was based on a single‐arm trial demonstrating a confirmed objective response rate greater than that of available therapy with a favorable duration of response and an acceptable toxicity profile. The ongoing trial TRITON3 is verifying the clinical benefit of this drug.
Introduction
Prostate cancer is the most common cancer and the second leading cause of cancer death among U.S. men, with more than 174,000 new cases and more than 31,000 deaths in the U.S. in 2019 [1]. Abiraterone and enzalutamide have demonstrated an overall survival (OS) benefit versus placebo in metastatic castrate‐resistant prostate cancer (mCRPC) and are approved for treatment in this setting either before or after docetaxel chemotherapy [2–5]. The taxanes docetaxel [6] and cabazitaxel [7] have demonstrated a statistically significant OS improvement of approximately 3 months versus mitoxantrone plus prednisone/prednisolone in the second‐ or third‐line setting in mCRPC. These agents can be toxic, and in this patient population, many are not candidates for these therapies. Sipuleucel‐T produced a 4‐month overall survival advantage versus placebo in asymptomatic or minimally symptomatic mCRPC. In men with mCRPC involving bone without visceral metastases, the calcium mimetic alpha particle emitter Radium‐223 demonstrated a 3‐month OS benefit versus placebo. However, for many patients with mCRPC who have progressed after androgen receptor (AR)‐directed therapy and a taxane, there are limited therapies available and an unmet need for safe and effective therapies.
Genomic studies of metastatic prostate cancer have identified loss‐of‐function mutations in DNA repair pathways as a frequent occurrence in mCRPC (∼20%–25%) [8], with BRCA2 being the most commonly mutated gene. The presence of these DNA repair pathway mutations is associated with a more aggressive phenotype and a poorer prognosis compared with the absence of these mutations. Preliminary clinical evidence supported the potential for benefit from PARP inhibitors in mCRPC with either germline or somatic DNA repair defects [9]. Given the frequency of occurrence of genetic alterations (both germline and somatic) in metastatic prostate cancer, germline testing and consideration of tumor testing for somatic mutations in homologous recombination repair (HRR) genes for all patients with metastatic prostate cancer is recommended [10], although until May 2020, there were no approved therapies specific for patients with metastatic prostate cancer and mutations in DNA repair genes.
On May 15, 2020, the U.S. Food and Drug Administration (FDA) granted accelerated approval to rucaparib (RUBRACA; Clovis Oncology, Inc., Boulder, CO) (Table 1), an inhibitor of the poly‐(ADP‐ribose) polymerase (PARP) enzyme, for the treatment of adults with deleterious BRCA mutation (germline and/or somatic)‐associated mCRPC who have been treated with AR‐directed therapy and a taxane [11]. This approval was based on efficacy and safety data evaluated in the ongoing multicenter, open‐label single‐arm trial TRITON2, which demonstrated an improved objective response rate compared with historical data for patients who had progressed after taxane‐based chemotherapy and AR‐directed therapy, along with a favorable duration of response of >6 months. Herein, we summarize key review findings that supported this approval in this novel patient population.
Clinical Trial Design
TRITON2 (ClinicalTrials.gov Identifier NCT02952534) was a multicenter, open‐label, phase II trial of rucaparib that enrolled men with mCRPC associated with HRR deficiency [12]. The trial enrolled patients with mCRPC with deleterious mutations in BRCA (1 and/or 2), ATM, or other HRR genes associated with potential sensitivity to PARP inhibitors into various cohorts.
These mutations were identified by central or local testing. Central testing was provided during prescreening. The tumor tissue–based central assay was the FoundationOne Laboratory Developed Test (Foundation Medicine, Inc., Cambridge, MA), a next‐generation sequencing (NGS)‐based test performed on DNA extracted from formalin‐fixed paraffin embedded (FFPE) tissue. The plasma‐based central test was the FoundationOne Liquid Laboratory Developed Test (Foundation Medicine, Inc.), an NGS‐based test performed on cell‐free DNA extracted from plasma. The central germline assay test used was the Color Hereditary Cancer Test (Color Genomics, Inc., Burlingame, CA). Central testing allowed the simultaneous submission of both a plasma sample for cell‐free DNA and an FFPE tissue sample for DNA analyses. A patient would be eligible for the trial if either the plasma or tissue sample was positive.
In addition to identifying patients eligible for trial participation, the additional pretreatment plasma samples were obtained to support development in parallel of an NGS‐based liquid assay in collaboration with Foundation Medicine, Inc., as a companion diagnostic to identify patients with prostate cancer with BRCA1/BRCA2 mutations in cell‐free tumor DNA isolated from plasma for treatment with rucaparib. This assay, FoundationOne Liquid CDx (F1 Liquid CDx), was subsequently approved by FDA on August 26, 2020, to identify patients with prostate cancer harboring BRCA1 and/or BRCA2 alterations (i.e., mutations) for treatment with rucaparib.
Patients were required to be medically or surgically castrated with a serum testosterone level ≤ 50 ng/dL with disease progression after at least one but not more than two AR‐directed therapies and disease progression after one prior line of taxane‐based chemotherapy. Disease progression for eligibility purposes was defined as any one of the following: a rise in prostate‐specific antigen (PSA) on two consecutive measurements separated by at least 1 week and with the most recent determination ≥2 ng/mL; new or progressive soft tissue masses on computed tomography or magnetic resonance imaging scans per RECIST version 1.1; or at least two new metastatic lesions on radionuclide bone scan.
Cohort A enrolled patients with measurable visceral and/or nodal disease and either a BRCA or ATM mutation. Cohort B enrolled patients without measurable visceral and/or nodal disease and either a BRCA or ATM mutation. Cohort C enrolled patients with or without measurable visceral and/or nodal disease and an HRR mutation other than BRCA or ATM.
All patients received rucaparib 600 mg twice daily orally and continuously in 28‐day cycles. Dose reductions due to unacceptable toxicity were permitted. Patients treated with a luteinizing hormone–releasing hormone (LHRH) analog were required to continue this therapy.
Soft tissue (visceral and nodal) disease was evaluated for evidence of radiographic response based on modified RECIST version 1.1 (mRECIST v1.1). Bone lesions were followed and evaluated for evidence of radiographic progression based on Prostate Cancer Working Group 3 (PCWG3) criteria. Tumor assessments were performed at baseline, at the end of every 8 weeks relative to study day 1 for up to 24 weeks, and then every 12 weeks, until confirmed radiographic disease progression by mRECIST v1.1 and/or PCWG3 criteria, per the investigator, loss to follow‐up, withdrawal, or study closure. Toxicity was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‐CTCAE) version 4.03. Clinical laboratory evaluations were graded according to NCI‐CTCAE version 4.03 or version 5.0 when this version became available.
The FDA efficacy review focused on the BRCA mutant population from Cohort A with measurable disease by independent radiologic review (IRR; n = 62; Fig. 1), with supportive evidence from the BRCA mutant population from Cohort B without measurable disease by IRR (n = 53). For Cohort A, the primary efficacy endpoint was confirmed radiographic objective response rate (ORR), defined as complete response (CR) or partial response (PR) by mRECIST v1.1 and no confirmed bone progression per PCWG3 prior to PR or CR. Confirmation of radiographic response was by blinded central IRR. The key secondary endpoint considered in the FDA review was duration of response (DOR) by mRECIST v1.1/PCWG3. For Cohort B, the primary efficacy endpoint was PSA response (≥50% decrease) as assessed by a local laboratory and analyzed separately for patients with deleterious BRCA 1/2 mutations and ATM mutations. The proportion of patients with ≥50% decrease in PSA from baseline was reported. As in Cohort A, the key secondary endpoint considered in the FDA review was DOR.
Rucaparib background information
| Structure | The chemical name is 8‐fluoro‐2‐{4‐[(methylamino)methyl] phenyl}‐1,3,4,5‐tetrahydro‐6H‐azepino[5,4,3‐cd] indol‐6‐one ((1S,4R)‐7,7‐dimethyl‐2‐oxobicyclo [2.2.1] hept‐1‐yl) methanesulfonic acid salt. The chemical formula of rucaparib camsylate is C19H18FN3O•C10H16O4S and the relative molecular mass is 555.67 Daltons. The chemical structure of rucaparib camsylate is shown below:
|
| Mechanism of action | Rucaparib is an inhibitor of the mammalian PARP enzyme, including PARP‐1, PARP‐2, and PARP‐3, which play a role in DNA repair. |
| Pharmacokinetics | The pharmacokinetic profile of rucaparib was characterized in patients with cancer. Rucaparib demonstrated linear pharmacokinetics over a dose range from 240 to 840 mg twice daily with time‐independence and dose‐proportionality. The mean steady‐state rucaparib Cmax was 1,940 ng/mL (54% CV) and AUC0–12h was 16,900 hours⋅ng/mL (54% CV) at the approved recommended dose. Accumulation was 3.5‐ to 6.2‐fold. Absorption The median Tmax was 1.9 hours at the approved recommended dose. The mean absolute bioavailability of rucaparib immediate‐release tablet was 36% with a range from 30% to 45%. Following a high‐fat meal, the Cmax was increased by 20% and AUC0–24h was increased by 38%, and Tmax was delayed by 2.5 hours, as compared with dosing under fasted conditions. Distribution Rucaparib had a steady‐state volume of distribution of 113–262 L following a single intravenous dose of 12–40 mg rucaparib. In vitro, the protein binding of rucaparib was 70% in human plasma at therapeutic concentrations. Rucaparib preferentially distributed to red blood cells with a blood‐to‐plasma concentration ratio of 1.83. Elimination The apparent clearance ranged from 15.3 to 79.2 L/hour, following rucaparib 600 mg orally twice daily. The clearance ranged from 13.9 to 18.4 L/hour, following a single intravenous dose of rucaparib 12–40 mg. The mean terminal T1/2 of rucaparib was 25.9 hours following a single oral dose of 600 mg rucaparib. Metabolism In vitro, rucaparib had a low metabolic turnover rate and was metabolized primarily by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. Following administration of a single oral dose of [14C]‐rucaparib to patients with solid tumors, unchanged rucaparib accounted for 64.0% of the radioactivity in plasma. Oxidation, N‐demethylation, N‐methylation, and glucuronidation were the major metabolic pathways for rucaparib. Rucaparib accounted for 44.9% and 94.9% of radioactivity in urine and feces, respectively. |
| Prior approvals | Rucaparib is also approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum‐based chemotherapy. Rucaparib is also approved for the treatment of adult patients with a deleterious BRCA mutation (germline and/or somatic)‐associated epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more chemotherapies. Select patients for therapy based on an FDA‐approved companion diagnostic for rucaparib. |

| Structure | The chemical name is 8‐fluoro‐2‐{4‐[(methylamino)methyl] phenyl}‐1,3,4,5‐tetrahydro‐6H‐azepino[5,4,3‐cd] indol‐6‐one ((1S,4R)‐7,7‐dimethyl‐2‐oxobicyclo [2.2.1] hept‐1‐yl) methanesulfonic acid salt. The chemical formula of rucaparib camsylate is C19H18FN3O•C10H16O4S and the relative molecular mass is 555.67 Daltons. The chemical structure of rucaparib camsylate is shown below:
|
| Mechanism of action | Rucaparib is an inhibitor of the mammalian PARP enzyme, including PARP‐1, PARP‐2, and PARP‐3, which play a role in DNA repair. |
| Pharmacokinetics | The pharmacokinetic profile of rucaparib was characterized in patients with cancer. Rucaparib demonstrated linear pharmacokinetics over a dose range from 240 to 840 mg twice daily with time‐independence and dose‐proportionality. The mean steady‐state rucaparib Cmax was 1,940 ng/mL (54% CV) and AUC0–12h was 16,900 hours⋅ng/mL (54% CV) at the approved recommended dose. Accumulation was 3.5‐ to 6.2‐fold. Absorption The median Tmax was 1.9 hours at the approved recommended dose. The mean absolute bioavailability of rucaparib immediate‐release tablet was 36% with a range from 30% to 45%. Following a high‐fat meal, the Cmax was increased by 20% and AUC0–24h was increased by 38%, and Tmax was delayed by 2.5 hours, as compared with dosing under fasted conditions. Distribution Rucaparib had a steady‐state volume of distribution of 113–262 L following a single intravenous dose of 12–40 mg rucaparib. In vitro, the protein binding of rucaparib was 70% in human plasma at therapeutic concentrations. Rucaparib preferentially distributed to red blood cells with a blood‐to‐plasma concentration ratio of 1.83. Elimination The apparent clearance ranged from 15.3 to 79.2 L/hour, following rucaparib 600 mg orally twice daily. The clearance ranged from 13.9 to 18.4 L/hour, following a single intravenous dose of rucaparib 12–40 mg. The mean terminal T1/2 of rucaparib was 25.9 hours following a single oral dose of 600 mg rucaparib. Metabolism In vitro, rucaparib had a low metabolic turnover rate and was metabolized primarily by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. Following administration of a single oral dose of [14C]‐rucaparib to patients with solid tumors, unchanged rucaparib accounted for 64.0% of the radioactivity in plasma. Oxidation, N‐demethylation, N‐methylation, and glucuronidation were the major metabolic pathways for rucaparib. Rucaparib accounted for 44.9% and 94.9% of radioactivity in urine and feces, respectively. |
| Prior approvals | Rucaparib is also approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum‐based chemotherapy. Rucaparib is also approved for the treatment of adult patients with a deleterious BRCA mutation (germline and/or somatic)‐associated epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more chemotherapies. Select patients for therapy based on an FDA‐approved companion diagnostic for rucaparib. |
Abbreviations: CV, coefficient of variation; FDA, Food and Drug Administration; PARP, polyadenosine 5’‐diphosphoribose polymerase.
Taken from rucaparib U.S. prescribing information [11].
Rucaparib background information
| Structure | The chemical name is 8‐fluoro‐2‐{4‐[(methylamino)methyl] phenyl}‐1,3,4,5‐tetrahydro‐6H‐azepino[5,4,3‐cd] indol‐6‐one ((1S,4R)‐7,7‐dimethyl‐2‐oxobicyclo [2.2.1] hept‐1‐yl) methanesulfonic acid salt. The chemical formula of rucaparib camsylate is C19H18FN3O•C10H16O4S and the relative molecular mass is 555.67 Daltons. The chemical structure of rucaparib camsylate is shown below:
|
| Mechanism of action | Rucaparib is an inhibitor of the mammalian PARP enzyme, including PARP‐1, PARP‐2, and PARP‐3, which play a role in DNA repair. |
| Pharmacokinetics | The pharmacokinetic profile of rucaparib was characterized in patients with cancer. Rucaparib demonstrated linear pharmacokinetics over a dose range from 240 to 840 mg twice daily with time‐independence and dose‐proportionality. The mean steady‐state rucaparib Cmax was 1,940 ng/mL (54% CV) and AUC0–12h was 16,900 hours⋅ng/mL (54% CV) at the approved recommended dose. Accumulation was 3.5‐ to 6.2‐fold. Absorption The median Tmax was 1.9 hours at the approved recommended dose. The mean absolute bioavailability of rucaparib immediate‐release tablet was 36% with a range from 30% to 45%. Following a high‐fat meal, the Cmax was increased by 20% and AUC0–24h was increased by 38%, and Tmax was delayed by 2.5 hours, as compared with dosing under fasted conditions. Distribution Rucaparib had a steady‐state volume of distribution of 113–262 L following a single intravenous dose of 12–40 mg rucaparib. In vitro, the protein binding of rucaparib was 70% in human plasma at therapeutic concentrations. Rucaparib preferentially distributed to red blood cells with a blood‐to‐plasma concentration ratio of 1.83. Elimination The apparent clearance ranged from 15.3 to 79.2 L/hour, following rucaparib 600 mg orally twice daily. The clearance ranged from 13.9 to 18.4 L/hour, following a single intravenous dose of rucaparib 12–40 mg. The mean terminal T1/2 of rucaparib was 25.9 hours following a single oral dose of 600 mg rucaparib. Metabolism In vitro, rucaparib had a low metabolic turnover rate and was metabolized primarily by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. Following administration of a single oral dose of [14C]‐rucaparib to patients with solid tumors, unchanged rucaparib accounted for 64.0% of the radioactivity in plasma. Oxidation, N‐demethylation, N‐methylation, and glucuronidation were the major metabolic pathways for rucaparib. Rucaparib accounted for 44.9% and 94.9% of radioactivity in urine and feces, respectively. |
| Prior approvals | Rucaparib is also approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum‐based chemotherapy. Rucaparib is also approved for the treatment of adult patients with a deleterious BRCA mutation (germline and/or somatic)‐associated epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more chemotherapies. Select patients for therapy based on an FDA‐approved companion diagnostic for rucaparib. |
| Structure | The chemical name is 8‐fluoro‐2‐{4‐[(methylamino)methyl] phenyl}‐1,3,4,5‐tetrahydro‐6H‐azepino[5,4,3‐cd] indol‐6‐one ((1S,4R)‐7,7‐dimethyl‐2‐oxobicyclo [2.2.1] hept‐1‐yl) methanesulfonic acid salt. The chemical formula of rucaparib camsylate is C19H18FN3O•C10H16O4S and the relative molecular mass is 555.67 Daltons. The chemical structure of rucaparib camsylate is shown below:
|
| Mechanism of action | Rucaparib is an inhibitor of the mammalian PARP enzyme, including PARP‐1, PARP‐2, and PARP‐3, which play a role in DNA repair. |
| Pharmacokinetics | The pharmacokinetic profile of rucaparib was characterized in patients with cancer. Rucaparib demonstrated linear pharmacokinetics over a dose range from 240 to 840 mg twice daily with time‐independence and dose‐proportionality. The mean steady‐state rucaparib Cmax was 1,940 ng/mL (54% CV) and AUC0–12h was 16,900 hours⋅ng/mL (54% CV) at the approved recommended dose. Accumulation was 3.5‐ to 6.2‐fold. Absorption The median Tmax was 1.9 hours at the approved recommended dose. The mean absolute bioavailability of rucaparib immediate‐release tablet was 36% with a range from 30% to 45%. Following a high‐fat meal, the Cmax was increased by 20% and AUC0–24h was increased by 38%, and Tmax was delayed by 2.5 hours, as compared with dosing under fasted conditions. Distribution Rucaparib had a steady‐state volume of distribution of 113–262 L following a single intravenous dose of 12–40 mg rucaparib. In vitro, the protein binding of rucaparib was 70% in human plasma at therapeutic concentrations. Rucaparib preferentially distributed to red blood cells with a blood‐to‐plasma concentration ratio of 1.83. Elimination The apparent clearance ranged from 15.3 to 79.2 L/hour, following rucaparib 600 mg orally twice daily. The clearance ranged from 13.9 to 18.4 L/hour, following a single intravenous dose of rucaparib 12–40 mg. The mean terminal T1/2 of rucaparib was 25.9 hours following a single oral dose of 600 mg rucaparib. Metabolism In vitro, rucaparib had a low metabolic turnover rate and was metabolized primarily by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. Following administration of a single oral dose of [14C]‐rucaparib to patients with solid tumors, unchanged rucaparib accounted for 64.0% of the radioactivity in plasma. Oxidation, N‐demethylation, N‐methylation, and glucuronidation were the major metabolic pathways for rucaparib. Rucaparib accounted for 44.9% and 94.9% of radioactivity in urine and feces, respectively. |
| Prior approvals | Rucaparib is also approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum‐based chemotherapy. Rucaparib is also approved for the treatment of adult patients with a deleterious BRCA mutation (germline and/or somatic)‐associated epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more chemotherapies. Select patients for therapy based on an FDA‐approved companion diagnostic for rucaparib. |
Abbreviations: CV, coefficient of variation; FDA, Food and Drug Administration; PARP, polyadenosine 5’‐diphosphoribose polymerase.
Taken from rucaparib U.S. prescribing information [11].
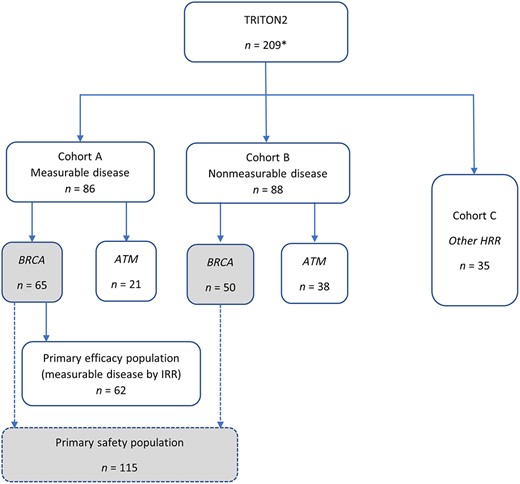
Analysis populations for regulatory review. Italicized font denotes mutation‐defined subgroups. Gray‐filled boxes denote primary safety population. *n = 209 represents the extended safety population.
TRITON2 planned to enroll 360 patients, with the following approximate numbers in each cohort: A = 150, B = 150, and C = 60. For Cohort A, which was divided into two subgroups (BRCA and ATM), the estimated enrollment for patients with BRCA mutations was 100.
A Simon two‐stage design was used to evaluate antitumor activity of rucaparib for futility and efficacy. The design set a 5% probability of accepting a minimally effective drug and a 90% probability of accepting an effective drug, with a target ORR of 20% for a minimally effective drug and an ORR of 35% for an effective drug. Stage 1 evaluation occurred after enrollment of 37 patients. At the time of the analysis cutoff date for this New Drug Application submission, Cohort A was not fully enrolled.
Results
Efficacy
The primary efficacy population submitted by the applicant consisted of all patients with BRCA‐mutated mCRPC evaluable for radiographic response by IRR in Cohort A enrolled as of May 3, 2019, with a data cutoff date of December 23, 2019 (i.e., those with measurable disease at baseline; n = 62; Fig. 1). A supportive efficacy population consisted of all patients evaluable for radiographic response by the investigator (n = 65). An additional 53 patients from Cohort B with BRCA‐mutated mCRPC, but with nonmeasurable disease per IRR, were included in an exploratory analysis based on PSA response. This exploratory supportive analysis was performed as PSA response was a prespecified endpoint and PSA is widely used to clinically assess response in men with prostate cancer.
The demographic characteristics and disease characteristic of the primary efficacy population are presented in Table 2. The median age was 73 years, and 82% were ≥ 65 years of age. Most were white. Patients were equally distributed between U.S. and non‐U.S. sites. Patients tended to present with locally advanced or metastatic high‐grade disease. Forty‐five percent had T3/4 tumors, 40% had lymph node involvement, 42% presented with distant metastases, and 63% had Gleason score 8–10 tumors at diagnosis. Twenty‐four percent had metastases only to lymph nodes and 34% had visceral disease. The most common HRR mutation was in the BRCA2 gene (85%), and 2/3 of patients had somatic mutations. Prior therapy included at least one prior AR‐directed therapy for all patients, and two prior AR‐directed therapies in 34% of patients.
Demographic and disease characteristics of the primary efficacy analysis population
| Characteristics | TRITON 2 (n = 62), n (%) |
|---|---|
| Sex | |
| Male | 62 |
| Female | 0 |
| Age, years | |
| Mean (SD) | 71.2 (7.9) |
| Median | 73.0 |
| Minimum, maximum | 52, 88 |
| Age group, years | |
| <65 | 11 (17.7) |
| 65–74 | 25 (40.3) |
| ≥75 | 26 (42.0) |
| Race | |
| White | 45 (72.5) |
| Black | 6 (9.7) |
| Asian | 1 (1.6) |
| Missing | 10 (16.1) |
| Ethnicity | |
| Hispanic or Latino | 2 (3.2) |
| Not Hispanic or Latino | 52 (83.9) |
| Not reported | 8 (12.9) |
| Geographic region | |
| U.S. | 30 (48.4) |
| Rest of the world | 32 (51.6) |
| ECOG | |
| 0 | 16 (25.8) |
| 1 | 45 (72.6) |
| ≥2 | 1 (1.6) |
| Time since cancer diagnosis, years | |
| Mean (SD) | 6.4 (4.7) |
| Median | 4.6 |
| Range | 0.8–21.2 |
| Clinical tumor stage at diagnosis | |
| T1 | 6 (9.7) |
| T2 | 19 (30.6) |
| T3 | 16 (25.8) |
| T4 | 12 (19.4) |
| Missing | 9 (14.5) |
| Clinical lymph node stage at diagnosis | |
| N0 | 18 (29.0) |
| N1 | 25 (40.3) |
| NX | 16 (25.8) |
| Missing | 3 (4.8) |
| Distant metastases at diagnosis | |
| M0 | 23 (37.1) |
| M1 | 26 (41.9) |
| MX | 11 (17.7) |
| Missing | 2 (3.2) |
| Gleason score | |
| <7 | 5 (8.1) |
| 7 | 13 (21.0) |
| 8 | 16 (25.8) |
| 9 | 19 (30.7) |
| 10 | 4 (6.5) |
| Not reported | 5 (8.1) |
| Baseline PSA, ng/mL | |
| Mean (SD) | 349 (871) |
| Median | 71 |
| Range | 1.8–4,782 |
| Tumor burden per IRR, mm | |
| ≤50 | 31 (50) |
| >50 | 31 (50) |
| Metastatic sites per IRR | |
| Bone | 44 (71) |
| Nodal | 51 (82.3) |
| Nodal only | 15 (24.2) |
| Visceral | 21 (33.9) |
| Liver | 13 (21.0) |
| Lung | 11 (17.7) |
| Number of bone lesions per IRR | |
| <10 | 37 (59.7) |
| ≥10 | 25 (40.3) |
| Gene mutationa | |
| BRCA1 | 9 (14.5) |
| BRCA2 | 53 (85.5) |
| Mutation testing sourcea | |
| Central | 36 (58.1) |
| Blood | 16 (25.8) |
| Tumor | 20 (32.3) |
| Local | 26 (41.9) |
| Blood | 6 (9.7) |
| Tumor | 19 (30.6) |
| Other | 1 (1.6) |
| Germline vs. somatic (all centrally reviewed)a | |
| Germline | 21 (33.9) |
| Somatic | 41 (66.1) |
| Prior systemic treatments | |
| Abiraterone | 38 (62.3) |
| Enzalutamide | 43 (70.5) |
| Apalutamide | 1 (1.6) |
| Darolutamide | 0 |
| ≥ 2 AR‐directed therapies | 21 (34.4) |
| Docetaxel (hormone‐sensitive) | 8 (13.1) |
| Docetaxel (castrate‐resistant) | 58 (95.1) |
| Cabazitaxel | 4 (6.6) |
| Sipuleucel‐T | 7 (11.5) |
| Radium‐223 | 7 (11.5) |
| Number of prior CRPC therapies | |
| 1 | 0 |
| 2 | 32 (51.6) |
| 3 or more | 30 (48.4) |
| Characteristics | TRITON 2 (n = 62), n (%) |
|---|---|
| Sex | |
| Male | 62 |
| Female | 0 |
| Age, years | |
| Mean (SD) | 71.2 (7.9) |
| Median | 73.0 |
| Minimum, maximum | 52, 88 |
| Age group, years | |
| <65 | 11 (17.7) |
| 65–74 | 25 (40.3) |
| ≥75 | 26 (42.0) |
| Race | |
| White | 45 (72.5) |
| Black | 6 (9.7) |
| Asian | 1 (1.6) |
| Missing | 10 (16.1) |
| Ethnicity | |
| Hispanic or Latino | 2 (3.2) |
| Not Hispanic or Latino | 52 (83.9) |
| Not reported | 8 (12.9) |
| Geographic region | |
| U.S. | 30 (48.4) |
| Rest of the world | 32 (51.6) |
| ECOG | |
| 0 | 16 (25.8) |
| 1 | 45 (72.6) |
| ≥2 | 1 (1.6) |
| Time since cancer diagnosis, years | |
| Mean (SD) | 6.4 (4.7) |
| Median | 4.6 |
| Range | 0.8–21.2 |
| Clinical tumor stage at diagnosis | |
| T1 | 6 (9.7) |
| T2 | 19 (30.6) |
| T3 | 16 (25.8) |
| T4 | 12 (19.4) |
| Missing | 9 (14.5) |
| Clinical lymph node stage at diagnosis | |
| N0 | 18 (29.0) |
| N1 | 25 (40.3) |
| NX | 16 (25.8) |
| Missing | 3 (4.8) |
| Distant metastases at diagnosis | |
| M0 | 23 (37.1) |
| M1 | 26 (41.9) |
| MX | 11 (17.7) |
| Missing | 2 (3.2) |
| Gleason score | |
| <7 | 5 (8.1) |
| 7 | 13 (21.0) |
| 8 | 16 (25.8) |
| 9 | 19 (30.7) |
| 10 | 4 (6.5) |
| Not reported | 5 (8.1) |
| Baseline PSA, ng/mL | |
| Mean (SD) | 349 (871) |
| Median | 71 |
| Range | 1.8–4,782 |
| Tumor burden per IRR, mm | |
| ≤50 | 31 (50) |
| >50 | 31 (50) |
| Metastatic sites per IRR | |
| Bone | 44 (71) |
| Nodal | 51 (82.3) |
| Nodal only | 15 (24.2) |
| Visceral | 21 (33.9) |
| Liver | 13 (21.0) |
| Lung | 11 (17.7) |
| Number of bone lesions per IRR | |
| <10 | 37 (59.7) |
| ≥10 | 25 (40.3) |
| Gene mutationa | |
| BRCA1 | 9 (14.5) |
| BRCA2 | 53 (85.5) |
| Mutation testing sourcea | |
| Central | 36 (58.1) |
| Blood | 16 (25.8) |
| Tumor | 20 (32.3) |
| Local | 26 (41.9) |
| Blood | 6 (9.7) |
| Tumor | 19 (30.6) |
| Other | 1 (1.6) |
| Germline vs. somatic (all centrally reviewed)a | |
| Germline | 21 (33.9) |
| Somatic | 41 (66.1) |
| Prior systemic treatments | |
| Abiraterone | 38 (62.3) |
| Enzalutamide | 43 (70.5) |
| Apalutamide | 1 (1.6) |
| Darolutamide | 0 |
| ≥ 2 AR‐directed therapies | 21 (34.4) |
| Docetaxel (hormone‐sensitive) | 8 (13.1) |
| Docetaxel (castrate‐resistant) | 58 (95.1) |
| Cabazitaxel | 4 (6.6) |
| Sipuleucel‐T | 7 (11.5) |
| Radium‐223 | 7 (11.5) |
| Number of prior CRPC therapies | |
| 1 | 0 |
| 2 | 32 (51.6) |
| 3 or more | 30 (48.4) |
Abbreviations: AR, androgen receptor; CRPC, castrate‐resistant prostate cancer; ECOG, Eastern Cooperative Oncology Group; IRR, independent radiologic review; PSA, prostate‐specific antigen.
aAll 62 patients had a deleterious somatic or germline BRCA mutation detected from central plasma (26%), central tissue (32%), or local (42%) testing. Plasma BRCA mutation status was verified retrospectively by FoundationOne Liquid CDx in 66% of the patients.
Demographic and disease characteristics of the primary efficacy analysis population
| Characteristics | TRITON 2 (n = 62), n (%) |
|---|---|
| Sex | |
| Male | 62 |
| Female | 0 |
| Age, years | |
| Mean (SD) | 71.2 (7.9) |
| Median | 73.0 |
| Minimum, maximum | 52, 88 |
| Age group, years | |
| <65 | 11 (17.7) |
| 65–74 | 25 (40.3) |
| ≥75 | 26 (42.0) |
| Race | |
| White | 45 (72.5) |
| Black | 6 (9.7) |
| Asian | 1 (1.6) |
| Missing | 10 (16.1) |
| Ethnicity | |
| Hispanic or Latino | 2 (3.2) |
| Not Hispanic or Latino | 52 (83.9) |
| Not reported | 8 (12.9) |
| Geographic region | |
| U.S. | 30 (48.4) |
| Rest of the world | 32 (51.6) |
| ECOG | |
| 0 | 16 (25.8) |
| 1 | 45 (72.6) |
| ≥2 | 1 (1.6) |
| Time since cancer diagnosis, years | |
| Mean (SD) | 6.4 (4.7) |
| Median | 4.6 |
| Range | 0.8–21.2 |
| Clinical tumor stage at diagnosis | |
| T1 | 6 (9.7) |
| T2 | 19 (30.6) |
| T3 | 16 (25.8) |
| T4 | 12 (19.4) |
| Missing | 9 (14.5) |
| Clinical lymph node stage at diagnosis | |
| N0 | 18 (29.0) |
| N1 | 25 (40.3) |
| NX | 16 (25.8) |
| Missing | 3 (4.8) |
| Distant metastases at diagnosis | |
| M0 | 23 (37.1) |
| M1 | 26 (41.9) |
| MX | 11 (17.7) |
| Missing | 2 (3.2) |
| Gleason score | |
| <7 | 5 (8.1) |
| 7 | 13 (21.0) |
| 8 | 16 (25.8) |
| 9 | 19 (30.7) |
| 10 | 4 (6.5) |
| Not reported | 5 (8.1) |
| Baseline PSA, ng/mL | |
| Mean (SD) | 349 (871) |
| Median | 71 |
| Range | 1.8–4,782 |
| Tumor burden per IRR, mm | |
| ≤50 | 31 (50) |
| >50 | 31 (50) |
| Metastatic sites per IRR | |
| Bone | 44 (71) |
| Nodal | 51 (82.3) |
| Nodal only | 15 (24.2) |
| Visceral | 21 (33.9) |
| Liver | 13 (21.0) |
| Lung | 11 (17.7) |
| Number of bone lesions per IRR | |
| <10 | 37 (59.7) |
| ≥10 | 25 (40.3) |
| Gene mutationa | |
| BRCA1 | 9 (14.5) |
| BRCA2 | 53 (85.5) |
| Mutation testing sourcea | |
| Central | 36 (58.1) |
| Blood | 16 (25.8) |
| Tumor | 20 (32.3) |
| Local | 26 (41.9) |
| Blood | 6 (9.7) |
| Tumor | 19 (30.6) |
| Other | 1 (1.6) |
| Germline vs. somatic (all centrally reviewed)a | |
| Germline | 21 (33.9) |
| Somatic | 41 (66.1) |
| Prior systemic treatments | |
| Abiraterone | 38 (62.3) |
| Enzalutamide | 43 (70.5) |
| Apalutamide | 1 (1.6) |
| Darolutamide | 0 |
| ≥ 2 AR‐directed therapies | 21 (34.4) |
| Docetaxel (hormone‐sensitive) | 8 (13.1) |
| Docetaxel (castrate‐resistant) | 58 (95.1) |
| Cabazitaxel | 4 (6.6) |
| Sipuleucel‐T | 7 (11.5) |
| Radium‐223 | 7 (11.5) |
| Number of prior CRPC therapies | |
| 1 | 0 |
| 2 | 32 (51.6) |
| 3 or more | 30 (48.4) |
| Characteristics | TRITON 2 (n = 62), n (%) |
|---|---|
| Sex | |
| Male | 62 |
| Female | 0 |
| Age, years | |
| Mean (SD) | 71.2 (7.9) |
| Median | 73.0 |
| Minimum, maximum | 52, 88 |
| Age group, years | |
| <65 | 11 (17.7) |
| 65–74 | 25 (40.3) |
| ≥75 | 26 (42.0) |
| Race | |
| White | 45 (72.5) |
| Black | 6 (9.7) |
| Asian | 1 (1.6) |
| Missing | 10 (16.1) |
| Ethnicity | |
| Hispanic or Latino | 2 (3.2) |
| Not Hispanic or Latino | 52 (83.9) |
| Not reported | 8 (12.9) |
| Geographic region | |
| U.S. | 30 (48.4) |
| Rest of the world | 32 (51.6) |
| ECOG | |
| 0 | 16 (25.8) |
| 1 | 45 (72.6) |
| ≥2 | 1 (1.6) |
| Time since cancer diagnosis, years | |
| Mean (SD) | 6.4 (4.7) |
| Median | 4.6 |
| Range | 0.8–21.2 |
| Clinical tumor stage at diagnosis | |
| T1 | 6 (9.7) |
| T2 | 19 (30.6) |
| T3 | 16 (25.8) |
| T4 | 12 (19.4) |
| Missing | 9 (14.5) |
| Clinical lymph node stage at diagnosis | |
| N0 | 18 (29.0) |
| N1 | 25 (40.3) |
| NX | 16 (25.8) |
| Missing | 3 (4.8) |
| Distant metastases at diagnosis | |
| M0 | 23 (37.1) |
| M1 | 26 (41.9) |
| MX | 11 (17.7) |
| Missing | 2 (3.2) |
| Gleason score | |
| <7 | 5 (8.1) |
| 7 | 13 (21.0) |
| 8 | 16 (25.8) |
| 9 | 19 (30.7) |
| 10 | 4 (6.5) |
| Not reported | 5 (8.1) |
| Baseline PSA, ng/mL | |
| Mean (SD) | 349 (871) |
| Median | 71 |
| Range | 1.8–4,782 |
| Tumor burden per IRR, mm | |
| ≤50 | 31 (50) |
| >50 | 31 (50) |
| Metastatic sites per IRR | |
| Bone | 44 (71) |
| Nodal | 51 (82.3) |
| Nodal only | 15 (24.2) |
| Visceral | 21 (33.9) |
| Liver | 13 (21.0) |
| Lung | 11 (17.7) |
| Number of bone lesions per IRR | |
| <10 | 37 (59.7) |
| ≥10 | 25 (40.3) |
| Gene mutationa | |
| BRCA1 | 9 (14.5) |
| BRCA2 | 53 (85.5) |
| Mutation testing sourcea | |
| Central | 36 (58.1) |
| Blood | 16 (25.8) |
| Tumor | 20 (32.3) |
| Local | 26 (41.9) |
| Blood | 6 (9.7) |
| Tumor | 19 (30.6) |
| Other | 1 (1.6) |
| Germline vs. somatic (all centrally reviewed)a | |
| Germline | 21 (33.9) |
| Somatic | 41 (66.1) |
| Prior systemic treatments | |
| Abiraterone | 38 (62.3) |
| Enzalutamide | 43 (70.5) |
| Apalutamide | 1 (1.6) |
| Darolutamide | 0 |
| ≥ 2 AR‐directed therapies | 21 (34.4) |
| Docetaxel (hormone‐sensitive) | 8 (13.1) |
| Docetaxel (castrate‐resistant) | 58 (95.1) |
| Cabazitaxel | 4 (6.6) |
| Sipuleucel‐T | 7 (11.5) |
| Radium‐223 | 7 (11.5) |
| Number of prior CRPC therapies | |
| 1 | 0 |
| 2 | 32 (51.6) |
| 3 or more | 30 (48.4) |
Abbreviations: AR, androgen receptor; CRPC, castrate‐resistant prostate cancer; ECOG, Eastern Cooperative Oncology Group; IRR, independent radiologic review; PSA, prostate‐specific antigen.
aAll 62 patients had a deleterious somatic or germline BRCA mutation detected from central plasma (26%), central tissue (32%), or local (42%) testing. Plasma BRCA mutation status was verified retrospectively by FoundationOne Liquid CDx in 66% of the patients.
The primary efficacy results are as follows. The confirmed ORR by IRR was 44% (95% confidence interval [CI]: 31%–57%). All seven patients who achieved complete responses had somatic BRCA mutation at baseline, six of seven had BRCA2 mutation, and five of seven had metastatic involvement of lymph nodes only at baseline. For the 20 patients with partial response, 9 had germline BRCA mutations and the other 11 had somatic mutations. Of the nine patients with BRCA1‐mutated mCRPC, three patients had tumor response. Overall, (60%) of the partial responses were in patients with metastases to lymph nodes only at baseline.
The median DOR for the primary efficacy population has not been reached, and the lower bound of the 95% CI is about 6 months. There is reasonable concordance between the independent radiologic review and investigator assessment of duration of response.
Exploratory analyses were performed on the TRITON2 patients with nonmeasurable disease at baseline from Cohort B. As most patients with mCRPC have nonmeasurable disease, this analysis explored whether there was evidence of durable benefit in these patients. Patients in Cohort B consisted of those with BRCA mutations and nonmeasurable disease by IRR (n = 53).
The demographics and disease characteristics between patients with measurable or nonmeasurable disease are summarized in supplemental online Table 1. Patients with measurable disease were older and had a lower performance status than patients with nonmeasurable disease. They also had a higher frequency of nodal involvement and were more likely to have a larger tumor burden and visceral disease at baseline, and a higher baseline PSA. Patients with nonmeasurable disease had a higher frequency of higher‐grade tumors and a higher frequency of bone metastases. Overall, there were adverse features observed in both groups.
Exploratory analyses in patients with nonmeasurable disease suggested the PSA percentage change from baseline was similar between the patients with measurable versus nonmeasurable disease at baseline and larger decreases in PSA trended favorably with outcome. Despite the difficulties with interpreting PSA response, these additional post hoc analyses added supportive evidence that patients with nonmeasurable disease were seeing benefit of rucaparib, with no evidence of a detriment.
Safety
The safety of rucaparib was primarily evaluated in the patients with BRCA‐mutated mCRPC in TRITON2 Cohorts A and B (Fig. 1). The safety population comprised 115 men with mCRPC who received at least one dose of rucaparib 600 mg b.i.d. as of the data cutoff of September 13, 2019. The primary safety population was supplemented with an additional 94 patients treated in TRITON2 with other forms of HRR‐deficient mCRPC, including 59 with ATM mutations, 14 with CDK12 mutations, 7 with CHEK2 mutations, and 14 with mutations in “other” HRR genes (extended safety population, n = 209). Baseline demographic and characteristics for the 115 BRCA‐mutated patients from TRITON2 versus all enrolled TRITON2 patients were similar.
Fatal adverse reactions occurred in two (1.7%) patients in the primary safety population of TRITON2, one each attributed to acute respiratory distress syndrome and pneumonia. In the extended safety population, an additional two patients had fatal adverse reactions, one each attributed to intestinal ischemia and pulmonary embolism.
In the primary safety population, nine (8%) patients required permanent discontinuation because of treatment‐emergent adverse events (TEAEs). TEAEs leading to discontinuation (one patient each) were QT prolongation, acute respiratory distress syndrome, anemia, balance disorder, cardiac failure, decreased appetite/fatigue/weight decrease, leukopenia/neutropenia, alanine aminotransferase (ALT)/aspartate aminotransferase (AST) increase, and pneumonia.
Rucaparib was interrupted in 65 (57%) patients. The most common TEAEs leading to treatment interruption were anemia (22%), thrombocytopenia (14%), and fatigue (10%).
Rucaparib dose was reduced owing to TEAEs in 47 (41%) patients. The most common TEAEs leading to dose reduction were anemia (14%), fatigue (10%), and thrombocytopenia (7%).
The most common any‐grade TEAE was fatigue (62%). The incidence of grade 3–4 adverse events was consistent with the experience of rucaparib in ovarian cancer. Patients with germline BRCA mutations had higher rates of bone marrow suppression versus those with somatic BRCA mutations (grade 3–4: 41% vs. 30%; grade 1–5: 59% vs. 51%). Myelodysplastic syndrome/acute myeloid leukemia was not observed in patients with mCRPC. During the review, FDA also identified a postmarket safety signal for hypersensitivity. Based on causal association, hypersensitivity was added to the safety section of labeling.
The most common laboratory abnormalities worsening from baseline to grade 3–4 were decrease in hemoglobin (25%), decrease in lymphocytes (17%), decrease in phosphate (15%), decrease in absolute neutrophil count (10%), and decrease in platelets (10%). Five percent developed grade 3–4 ALT/AST elevations; no cases met Hy's law criteria. Although 10% of patients had a shift to grade 3–4 neutropenia during the course of treatment, most patients did not have infections while neutropenic. Similarly, infection was a serious adverse event in 11% of patients, most of whom were not neutropenic. Twenty‐nine percent of patients required at least one red blood cell transfusion (∼10% per cycle).
Discussion
FDA review of the application found that treatment with rucaparib had a favorable risk–benefit profile (Table 3) in adults with mCRPC who have progressed on an AR‐directed therapy and a taxane. Treatment resulted in a confirmed ORR of 44% in this single‐arm trial, with a median DOR >6 months and an acceptable toxicity profile. This ORR and DOR are improved over those of available therapies and are reasonably likely to predict clinical benefit and thus met requirements for accelerated approval. Although further trial(s) may be required to confirm clinical benefit, this confirmation does not need to occur in the same exact population as the accelerated approval. Thus, TRITON3 is a randomized phase III trial of rucaparib versus physician's choice of either docetaxel or other AR‐directed therapy in patients with mCRPC associated with HRR deficiency progressing after treatment with one prior AR‐directed therapy that could confirm benefit.
Risk–benefit assessment
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
| Patients with mCRPC who have progressed on AR‐directed therapies and a taxane have a serious and life‐threatening condition. |
| Current treatment options |
| Current options for patients with mCRPC who have progressed on AR‐directed therapy and a taxane are limited. |
| Benefit |
| Rucaparib has demonstrated a substantial response rate with a median duration of response of more than 6 months and an acceptable safety profile. |
| Risk and risk management |
| The risk–benefit profile of rucaparib is acceptable in the approved patient population. The randomized trial TRITON3 is ongoing and will provide further data on the efficacy and safety of rucaparib in mCRPC. |
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
| Patients with mCRPC who have progressed on AR‐directed therapies and a taxane have a serious and life‐threatening condition. |
| Current treatment options |
| Current options for patients with mCRPC who have progressed on AR‐directed therapy and a taxane are limited. |
| Benefit |
| Rucaparib has demonstrated a substantial response rate with a median duration of response of more than 6 months and an acceptable safety profile. |
| Risk and risk management |
| The risk–benefit profile of rucaparib is acceptable in the approved patient population. The randomized trial TRITON3 is ongoing and will provide further data on the efficacy and safety of rucaparib in mCRPC. |
Abbreviations: ALT, alanine aminotransferase; AML, acute myeloid leukemia; AR, androgen receptor; AST, aspartate aminotransferase; CI, confidence interval; mCRPC, metastatic castrate‐resistant prostate cancer; MDS, myelodysplastic syndrome; ORR, objective response rate.
Risk–benefit assessment
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
| Patients with mCRPC who have progressed on AR‐directed therapies and a taxane have a serious and life‐threatening condition. |
| Current treatment options |
| Current options for patients with mCRPC who have progressed on AR‐directed therapy and a taxane are limited. |
| Benefit |
| Rucaparib has demonstrated a substantial response rate with a median duration of response of more than 6 months and an acceptable safety profile. |
| Risk and risk management |
| The risk–benefit profile of rucaparib is acceptable in the approved patient population. The randomized trial TRITON3 is ongoing and will provide further data on the efficacy and safety of rucaparib in mCRPC. |
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
| Patients with mCRPC who have progressed on AR‐directed therapies and a taxane have a serious and life‐threatening condition. |
| Current treatment options |
| Current options for patients with mCRPC who have progressed on AR‐directed therapy and a taxane are limited. |
| Benefit |
| Rucaparib has demonstrated a substantial response rate with a median duration of response of more than 6 months and an acceptable safety profile. |
| Risk and risk management |
| The risk–benefit profile of rucaparib is acceptable in the approved patient population. The randomized trial TRITON3 is ongoing and will provide further data on the efficacy and safety of rucaparib in mCRPC. |
Abbreviations: ALT, alanine aminotransferase; AML, acute myeloid leukemia; AR, androgen receptor; AST, aspartate aminotransferase; CI, confidence interval; mCRPC, metastatic castrate‐resistant prostate cancer; MDS, myelodysplastic syndrome; ORR, objective response rate.
Because TRITON2 was a single‐arm trial, the analysis of the primary efficacy population focused on ORR and DOR per mRECIST v1.1/PCWG3 and confirmed by blinded independent review in patients with measurable disease at baseline, as the measurable effect on tumor would be attributable to the study drug and not the natural history of the disease. Time‐to‐event endpoints, such as progression‐free survival, are not appropriate for a regulatory decision in this case, as this is a single‐arm study and the results based on these endpoints would not be interpretable. Similarly, PSA‐based endpoints have not been validated as surrogates of clinical endpoints, such as survival, and therefore were not used as the basis of the regulatory decision. PSA is an especially unreliable endpoint when evaluating therapies not targeted against the androgen receptor that controls PSA expression. In addition, symptomatic and/or radiographic progression may occur in more than 50% of men with mCRPC before PSA progression is detected [13], which could result in overestimation of the efficacy of the study agent.
This regulatory decision therefore included patients from Cohort A with BRCA mutations and measurable disease at baseline established by blinded independent radiologic review in the primary efficacy population during FDA's review, with efficacy evaluation looking for IRR‐evaluated ORR of adequate magnitude, supported by a clinically relevant duration of response. Efficacy in the measurable disease population along with exploratory analysis in the nonmeasurable disease group and an understanding that the mechanism of action of rucaparib would not suggest a differential response between patients with measurable versus nonmeasurable disease supported an ultimate indication inclusive of all patients regardless of measurable disease status. In addition, unpublished analyses reviewed by FDA from trials that enrolled patients with mCRPC showed consistent treatment effects comparing those with and without measurable disease.
At the time of FDA approval of rucaparib, the companion diagnostic assay for detection of BRCA1 or BRCA2 mutations was still under FDA review. The benefits from the use of rucaparib in mCRPC are sufficiently pronounced to outweigh the risks from the lack of an approved or cleared in vitro diagnostic companion diagnostic device. FDA may approve a therapeutic product in these circumstances if the therapeutic product is intended to treat a serious or life‐threatening condition for which no satisfactory alternative treatment exists [14]. The completion of adequate analytical and clinical validation studies using specimens from TRITON2 to support appropriate identification of patients through an in vitro diagnostic device as a companion diagnostic that detects deleterious BRCA1 and BRCA2 alterations in plasma and tissue were postmarketing commitments for this indication. The companion diagnostic, FoundationOne Liquid CDx, was subsequently approved by FDA on August 26, 2020. ORR appeared consistent in germline versus somatic BRCA mutation subgroups, which was an important regulatory consideration because rucaparib was approved without a diagnostic device, and eligibility for rucaparib in real‐world clinical practice may require germline BRCA mutation until fulfillment of the postmarketing commitment.
In TRITON2, ARIEL2, and ARIEL3 (the trials supporting the ovarian indications), most hypersensitivity events manifested in the form of rash, which was already a recognized adverse drug reaction in association with rucaparib. However, even excluding skin reactions resulted in an increased rate of hypersensitivity events, and thus, hypersensitivity was added as a unique adverse reaction in addition to rash in product labeling. Overall, frequency of grade 3–4 TEAEs, TEAEs leading to death, and AEs requiring dose interruption/reduction/permanent discontinuation are acceptable in this patient population and risks are appropriately conveyed in labeling.
Conclusion
Taken together, the evidence as summarized in our risk–benefit assessment in Table 3 was considered sufficient for accelerated approval of rucaparib for the intended clinical use. This provides the first non‐AR‐directed targeted agent in this disease setting and addresses an unmet clinical need for this patient population. Based on the regulatory requirement for accelerated approval, the continued approval of rucaparib for this indication may be contingent upon verification of clinical benefit in a confirmatory trial. The ongoing phase III trial TRITON3 will serve this purpose.
Author Contributions
Conception/design: Mitchell S. Anscher, Elaine Chang, Xin Gao, Yutao Gong, Chana Weinstock, Erik Bloomquist, Oluseyi Adeniyi, Rosane Charlab, Sarah Zimmerman, Maritsa Serlemitsos‐Day, Yang Min Ning, Ruth Mayrosh, Barbara Fuller, Ann Marie Trentacosti, Pamela Gallagher, Karen Bijwaard, Frances Fahnbulleh, Felicia Diggs, Shaily Arora, Kirsten B. Goldberg, Shenghui Tang, Laleh Amiri‐Kordestani, Richard Pazdur, Amna Ibrahim, Julia A. Beaver
Collection and/or assembly of data: Mitchell S. Anscher, Elaine Chang, Xin Gao, Yutao Gong, Chana Weinstock, Erik Bloomquist, Oluseyi Adeniyi, Rosane Charlab, Sarah Zimmerman, Maritsa Serlemitsos‐Day, Yang Min Ning, Ruth Mayrosh, Barbara Fuller, Ann Marie Trentacosti, Pamela Gallagher, Karen Bijwaard, Frances Fahnbulleh, Felicia Diggs, Shaily Arora, Kirsten B. Goldberg, Shenghui Tang, Laleh Amiri‐Kordestani, Richard Pazdur, Amna Ibrahim, Julia A. Beaver
Data analysis and interpretation: Mitchell S. Anscher, Elaine Chang, Xin Gao, Yutao Gong, Chana Weinstock, Erik Bloomquist, Oluseyi Adeniyi, Rosane Charlab, Sarah Zimmerman, Maritsa Serlemitsos‐Day, Yang Min Ning, Ruth Mayrosh, Barbara Fuller, Ann Marie Trentacosti, Pamela Gallagher, Karen Bijwaard, Frances Fahnbulleh, Felicia Diggs, Shaily Arora, Kirsten B. Goldberg, Shenghui Tang, Laleh Amiri‐Kordestani, Richard Pazdur, Amna Ibrahim, Julia A. Beaver
Manuscript writing: Mitchell S. Anscher, Elaine Chang, Xin Gao, Yutao Gong, Chana Weinstock, Erik Bloomquist, Oluseyi Adeniyi, Rosane Charlab, Sarah Zimmerman, Maritsa Serlemitsos‐Day, Yang Min Ning, Ruth Mayrosh, Barbara Fuller, Ann Marie Trentacosti, Pamela Gallagher, Karen Bijwaard, Frances Fahnbulleh, Felicia Diggs, Shaily Arora, Kirsten B. Goldberg, Shenghui Tang, Laleh Amiri‐Kordestani, Richard Pazdur, Amna Ibrahim, Julia A. Beaver
Final approval of manuscript: Mitchell S. Anscher, Elaine Chang, Xin Gao, Yutao Gong, Chana Weinstock, Erik Bloomquist, Oluseyi Adeniyi, Rosane Charlab, Sarah Zimmerman, Maritsa Serlemitsos‐Day, Yang Min Ning, Ruth Mayrosh, Barbara Fuller, Ann Marie Trentacosti, Pamela Gallagher, Karen Bijwaard, Frances Fahnbulleh, Felicia Diggs, Shaily Arora, Kirsten B. Goldberg, Shenghui Tang, Laleh Amiri‐Kordestani, Richard Pazdur, Amna Ibrahim, Julia A. Beaver
Disclosures
The authors indicated no financial relationships.
References
Author notes
Disclosures of potential conflicts of interest may be found at the end of this article.

{kind=link}