





Abstract
Astroblastoma (ABM) is a rare glial brain tumor. Recurrent meningioma 1 (MN1) alterations have been recently identified in most pediatric cases. Adolescent and adult cases, however, remain molecularly poorly defined.
We performed clinical and molecular characterization of a retrospective cohort of 14 adult and 1 adolescent ABM.
Strikingly, we found that MN1 fusions are a rare event in this age group (1/15). Using methylation profiling and targeted sequencing, most cases were reclassified as either pleomorphic xanthoastrocytomas (PXA)‐like or high‐grade glioma (HGG)‐like. PXA‐like ABM show BRAF mutation (6/7 with V600E mutation and 1/7 with G466E mutation) and CD34 expression. Conversely, HGG‐like ABM harbored specific alterations of diffuse midline glioma (2/5) or glioblastoma (GBM; 3/5). These latter patients showed an unfavorable clinical course with significantly shorter overall survival (p = .021). Mitogen‐activated protein kinase pathway alterations (including FGFR fusion, BRAF and NF1 mutations) were present in 10 of 15 patients and overrepresented in the HGG‐like group (3/5) compared with previously reported prevalence of these alterations in GBM and diffuse midline glioma.
We suggest that gliomas with astroblastic features include a variety of molecularly sharply defined entities. Adult ABM harboring molecular features of PXA and HGG should be reclassified. Central nervous system high‐grade neuroepithelial tumors with MN1 alterations and histology of ABM appear to be uncommon in adults. Astroblastic morphology in adults should thus prompt thorough molecular investigation aiming at a clear histomolecular diagnosis and identifying actionable drug targets, especially in the mitogen‐activated protein kinase pathway.
Astroblastoma (ABM) remains a poorly defined and controversial entity. Although meningioma 1 alterations seem to define a large subset of pediatric cases, adult cases remain molecularly poorly defined. This comprehensive molecular characterization of 1 adolescent and 14 adult ABM revealed that adult ABM histology comprises several molecularly defined entities, which explains clinical diversity and identifies actionable targets. Namely, pleomorphic xanthoastrocytoma‐like ABM cases show a favorable prognosis whereas high‐grade glioma (glioblastoma and diffuse midline gliome)‐like ABM show significantly worse clinical courses. These results call for in‐depth molecular analysis of adult gliomas with astroblastic features for diagnostic and therapeutic purposes.
Introduction
Astroblastoma (ABM) is a rare neuroepithelial tumor and has been recognized by the World Health Organization as a distinct tumor entity [1]. It was first described in 1926 by Bailey and Cushing [2] and further characterized by Bailey and Bucy [3] in 1930. ABM is generally regarded as an entity occurring in children and young adults [4–6]. However, in a recent epidemiological survey, 56% of cases were diagnosed after 30 years of age [7]. The diagnosis of ABM is based on a typical histomorphology with perivascular astroblastic pseudorosettes and vascular hyalinization [2,3,8]. However, perivascular arrangement of tumor cells is not specific for ABM and may also be seen in other central nervous system tumors such as glioblastoma (GBM), primitive neuroectodermal tumors, angiocentric glioma, and ependymomas 9,10. The lack of specific neuropathological features explains why the validity of ABM as a distinct entity is still a matter of debate [11].
Recently, efforts have been made to identify specific molecular alterations in ABM. In 2016, Sturm et al. [12] described a new molecularly defined tumor entity characterized by meningioma 1 (MN1) and BEN domain containing 2 (BEND2) fusion genes that histologically frequently corresponds to ABM. Several reports have confirmed the existence of recurrent MN1 alterations in pediatric as well as adult ABM [13–15]. In contrast, Lehman and colleagues [16] showed that tumors with the histological diagnosis of ABM frequently harbor BRAF V600E mutations.
Here, we report a comprehensive clinical, radiological, histological, and molecular characterization of a retrospective series of adolescent and adult ABM and show that they share common clinical and molecular features with other glial tumor entities.
Materials and Methods
Patient Cohort and Histological Review
Patients were retrospectively identified using systematic archival review for the term “astroblastoma” between 1990 and 2017 at the Pitié Salpêtrière University Hospital (i.e., cases with a final diagnosis of astroblastoma or astroblastoma being mentioned in the pathology report as a differential diagnosis). Tumors samples were stored with signed consent form in the tumor tissue bank OncoNeuroTek. All identified cases were centrally reviewed by experienced neuropathologists (K.M. and/or F.B.). Required histologic features for inclusion in the study were (a) the presence of perivascular astroblastic pseudorosettes; (b) the presence of hyalinized vessels; (c) cuboidal, columnar, or tapered perivascular cellular processes, occasionally ending in broad endfeet; and (d) absence of definitive criteria for other central nervous system (CNS) tumors. Hematoxylin and eosin staining as well as immunohistochemistry for Ki67, GFAP, IDH1 R132H, ATRX, EMA, OLIG2, vimentin, FGFR3, H3K27M, p53, and NFkB (commonly found in supratentorial ependymomas [17]) were performed. Additional immunohistochemical assessment of CD34 was done post hoc on all cases with a confirmed diagnosis of ABM.
Clinical and Radiological Data
For all patients, age at diagnosis, sex, clinical presentation, treatments, and clinical outcome were retrospectively collected. From initial diagnosis, progression‐free survival and overall survival were compared using a log‐rank test and plotted according to the Kaplan‐Meier method.
When available, brain magnetic resonance imaging (MRI) data (T1w, T2w, and contrast‐enhanced T1w sequences) were reviewed and annotated for supra‐ versus infratentorial location, perilesional edema, contrast enhancement, cystic component, and type of tumor boundary (well defined or diffuse).
Panel DNA Sequencing
All exons of genes frequently mutated in brain tumors were sequenced using a custom next‐generation sequencing panel (details in supplemental online Table 1). Briefly, tumor DNA was extracted from formalin‐fixed paraffin‐embedded or fresh‐frozen tumor specimens using the QIAamp DNA mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. DNA was quantified using a QuantiFluor dsDNA assay (Promega, Madison, WI). Target regions were captured from fragmented genomic DNA samples using a custom SeqCap EZ choice kit (Roche, Basel, Switzerland), and paired‐end 75‐bp massively parallel sequencing was carried out on a NextSeq500 sequencer (Illumina, San Diego, CA) according to the manufacturer's protocols.
Patient characteristics
| Patient no. | Age at diagnosis, years | Sex | Tumor location | PFS1, days | Treatment(s) after first surgery | Death | Overall survival, months |
|---|---|---|---|---|---|---|---|
| PXA‐like ABM | |||||||
| 1 | 66 | M | Parietal | 1,291 | Surgery | No | 62.7 (SD) |
| 2 | 18 | M | Occipital | 594 | RT, CT (CCNU, bevacizumab, vemurafenib) | Yes | 58.5 |
| 3 | 23 | F | Temporal | 509 | Multiple surgery | No | 91.1 (SD) |
| 4 | 24 | M | Temporal | 1,604 | RT‐CT then CT alone (TMZ) | No | 52.7 (SD) |
| 5 | 50 | F | Frontal | 178 | CT (TMZ) | No | 5.9 (SD) |
| 6 | 70 | F | Temporal | 249 | RT‐CT (carmustine) | No | 8.2 (LTFU) |
| 7 | 25 | F | Frontal | 30 | RT‐CT (TMZ) | No | 1 |
| HGG‐like ABM | |||||||
| 8 | 45 | F | Third ventricle | 278 | RT, CT (carboplatin, VP16) | Yes | 10.4 |
| 9 | 33 | M | Parietal parasagittal | 405 | RT‐CT (TMZ), surgery, CT (Campto, bevacizumab, carboplatin) | Yes | 26.6 |
| 10 | 76 | F | Parietal and occipital | 250 | RT‐CT (TMZ), CT (bevacizumab, carmustine) | Yes | 19.9 |
| 11 | 67 | M | Temporal | 780 | No | No | 25.6 (SD) |
| 12 | 59 | F | Frontal | 17 | RT‐CT (carmustine) | No | 0.6 (LTFU) |
| MN1‐BEND2 fusion | |||||||
| 13 | 15 | F | Parietal and occipital | 3,600 | Surgery | No | 229 (SD) |
| Unclassifiable patients | |||||||
| 14 | 69 | F | Parietal and occipital | 193 | RT‐CT (VP‐16, carboplatin) | No | 6.3 (LTFU) |
| 15 | 44 | F | Temporal | 467 | RT | No | 15.4 (SD) |
| Patient no. | Age at diagnosis, years | Sex | Tumor location | PFS1, days | Treatment(s) after first surgery | Death | Overall survival, months |
|---|---|---|---|---|---|---|---|
| PXA‐like ABM | |||||||
| 1 | 66 | M | Parietal | 1,291 | Surgery | No | 62.7 (SD) |
| 2 | 18 | M | Occipital | 594 | RT, CT (CCNU, bevacizumab, vemurafenib) | Yes | 58.5 |
| 3 | 23 | F | Temporal | 509 | Multiple surgery | No | 91.1 (SD) |
| 4 | 24 | M | Temporal | 1,604 | RT‐CT then CT alone (TMZ) | No | 52.7 (SD) |
| 5 | 50 | F | Frontal | 178 | CT (TMZ) | No | 5.9 (SD) |
| 6 | 70 | F | Temporal | 249 | RT‐CT (carmustine) | No | 8.2 (LTFU) |
| 7 | 25 | F | Frontal | 30 | RT‐CT (TMZ) | No | 1 |
| HGG‐like ABM | |||||||
| 8 | 45 | F | Third ventricle | 278 | RT, CT (carboplatin, VP16) | Yes | 10.4 |
| 9 | 33 | M | Parietal parasagittal | 405 | RT‐CT (TMZ), surgery, CT (Campto, bevacizumab, carboplatin) | Yes | 26.6 |
| 10 | 76 | F | Parietal and occipital | 250 | RT‐CT (TMZ), CT (bevacizumab, carmustine) | Yes | 19.9 |
| 11 | 67 | M | Temporal | 780 | No | No | 25.6 (SD) |
| 12 | 59 | F | Frontal | 17 | RT‐CT (carmustine) | No | 0.6 (LTFU) |
| MN1‐BEND2 fusion | |||||||
| 13 | 15 | F | Parietal and occipital | 3,600 | Surgery | No | 229 (SD) |
| Unclassifiable patients | |||||||
| 14 | 69 | F | Parietal and occipital | 193 | RT‐CT (VP‐16, carboplatin) | No | 6.3 (LTFU) |
| 15 | 44 | F | Temporal | 467 | RT | No | 15.4 (SD) |
Abbreviations: ABM, astroblastoma; CT, chemotherapy; F, female; HGG, high‐grade glioma; LTFU, lost to follow‐up; M, male; MN1, meningioma 1; N/A, not available; PFS1, progression‐free survival until first recurrence; PXA, pleomorphic xanthoastrocytoma; RT, radiotherapy; SD, stable disease; TMZ, temozolomide.
Patient characteristics
| Patient no. | Age at diagnosis, years | Sex | Tumor location | PFS1, days | Treatment(s) after first surgery | Death | Overall survival, months |
|---|---|---|---|---|---|---|---|
| PXA‐like ABM | |||||||
| 1 | 66 | M | Parietal | 1,291 | Surgery | No | 62.7 (SD) |
| 2 | 18 | M | Occipital | 594 | RT, CT (CCNU, bevacizumab, vemurafenib) | Yes | 58.5 |
| 3 | 23 | F | Temporal | 509 | Multiple surgery | No | 91.1 (SD) |
| 4 | 24 | M | Temporal | 1,604 | RT‐CT then CT alone (TMZ) | No | 52.7 (SD) |
| 5 | 50 | F | Frontal | 178 | CT (TMZ) | No | 5.9 (SD) |
| 6 | 70 | F | Temporal | 249 | RT‐CT (carmustine) | No | 8.2 (LTFU) |
| 7 | 25 | F | Frontal | 30 | RT‐CT (TMZ) | No | 1 |
| HGG‐like ABM | |||||||
| 8 | 45 | F | Third ventricle | 278 | RT, CT (carboplatin, VP16) | Yes | 10.4 |
| 9 | 33 | M | Parietal parasagittal | 405 | RT‐CT (TMZ), surgery, CT (Campto, bevacizumab, carboplatin) | Yes | 26.6 |
| 10 | 76 | F | Parietal and occipital | 250 | RT‐CT (TMZ), CT (bevacizumab, carmustine) | Yes | 19.9 |
| 11 | 67 | M | Temporal | 780 | No | No | 25.6 (SD) |
| 12 | 59 | F | Frontal | 17 | RT‐CT (carmustine) | No | 0.6 (LTFU) |
| MN1‐BEND2 fusion | |||||||
| 13 | 15 | F | Parietal and occipital | 3,600 | Surgery | No | 229 (SD) |
| Unclassifiable patients | |||||||
| 14 | 69 | F | Parietal and occipital | 193 | RT‐CT (VP‐16, carboplatin) | No | 6.3 (LTFU) |
| 15 | 44 | F | Temporal | 467 | RT | No | 15.4 (SD) |
| Patient no. | Age at diagnosis, years | Sex | Tumor location | PFS1, days | Treatment(s) after first surgery | Death | Overall survival, months |
|---|---|---|---|---|---|---|---|
| PXA‐like ABM | |||||||
| 1 | 66 | M | Parietal | 1,291 | Surgery | No | 62.7 (SD) |
| 2 | 18 | M | Occipital | 594 | RT, CT (CCNU, bevacizumab, vemurafenib) | Yes | 58.5 |
| 3 | 23 | F | Temporal | 509 | Multiple surgery | No | 91.1 (SD) |
| 4 | 24 | M | Temporal | 1,604 | RT‐CT then CT alone (TMZ) | No | 52.7 (SD) |
| 5 | 50 | F | Frontal | 178 | CT (TMZ) | No | 5.9 (SD) |
| 6 | 70 | F | Temporal | 249 | RT‐CT (carmustine) | No | 8.2 (LTFU) |
| 7 | 25 | F | Frontal | 30 | RT‐CT (TMZ) | No | 1 |
| HGG‐like ABM | |||||||
| 8 | 45 | F | Third ventricle | 278 | RT, CT (carboplatin, VP16) | Yes | 10.4 |
| 9 | 33 | M | Parietal parasagittal | 405 | RT‐CT (TMZ), surgery, CT (Campto, bevacizumab, carboplatin) | Yes | 26.6 |
| 10 | 76 | F | Parietal and occipital | 250 | RT‐CT (TMZ), CT (bevacizumab, carmustine) | Yes | 19.9 |
| 11 | 67 | M | Temporal | 780 | No | No | 25.6 (SD) |
| 12 | 59 | F | Frontal | 17 | RT‐CT (carmustine) | No | 0.6 (LTFU) |
| MN1‐BEND2 fusion | |||||||
| 13 | 15 | F | Parietal and occipital | 3,600 | Surgery | No | 229 (SD) |
| Unclassifiable patients | |||||||
| 14 | 69 | F | Parietal and occipital | 193 | RT‐CT (VP‐16, carboplatin) | No | 6.3 (LTFU) |
| 15 | 44 | F | Temporal | 467 | RT | No | 15.4 (SD) |
Abbreviations: ABM, astroblastoma; CT, chemotherapy; F, female; HGG, high‐grade glioma; LTFU, lost to follow‐up; M, male; MN1, meningioma 1; N/A, not available; PFS1, progression‐free survival until first recurrence; PXA, pleomorphic xanthoastrocytoma; RT, radiotherapy; SD, stable disease; TMZ, temozolomide.
Mutation and Copy Number Profiling
Quality control of the reads was performed with FastQC on demultiplexed data. Trimmomatic [18] was used to remove low‐quality segments (phred base quality <20) of the reads at 3′ and 5′ ends. Reads smaller than 40 base pairs after trimming were discarded. Reads were aligned against the hg19 assembly of the human genome using BWA‐MEM (version 0.7.15) [19]. We then applied Genome Analysis Toolkit (GATK) [20] for base quality score recalibration and indel realignment. PatternCNV [21] was used to estimate copy number variation based on read depth. Mutations and indels were called using GATK4 MuTect2 (beta version). Single nucleotide variants were annotated using Variant Effect Predictor [22]. Putative somatic variants were selected by filtering out all Single nucleotide polymorphisms in the gnomAD release 2.0.1 with an overall population allele frequency > 0.01 [23]. Variants were filtered for missense and nonsense mutations, and a minimum variant allele frequency > 0.1 was required. Disease‐causing variants were annotated using known cancer hotspots [24] and ClinVar [25].
RT‐PCR
First strand complementary DNA (cDNA) was generated from 500 ng total RNA using Maxima First Strand Synthesis Kit (Thermo Fisher Scientific, Illkirch, France) and diluted 1:10 in molecular biology‐grade water. Reverse transcription polymerase chain reaction (RT‐PCR) with primers specific for MN1‐BEND2 fusions was performed using 0.02 U/μL Q5 polymerase (New England Biolabs, Ipswich, MA), 200 μM dNTPs, 500 nM forward and reverse primers, Q5 reaction buffer with High GC Enhancer, and 5 μL template cDNA in total reaction volume of 20 μL. Thermal cycling was performed as follows: 98°C initial denaturation for 2 minutes, followed by 30 cycles of denaturation at 98°C for 10 seconds, annealing at 65°C for 20 seconds, and extension at 72°C for 90 seconds, as well as a final extension at 72°C for 2 minutes. Amplicons were analyzed using a Caliper LabChip GX DNA 5K assay (Perkin Elmer, Waltham, MA).
qPCR
Quantitative real‐time polymerase chain reaction (qPCR) of BEND2 expression was performed in 20 μL reactions using 10 μL 2X LightCycler 480 SYBR Green master mix (Roche, Basel, Switzerland), 1 μL of a 10 μM forward and reverse primer mix, 4 μL H2O, and 5 μL of 1:10 prediluted cDNA. The following PCR program was used: 10 minutes of preincubation at 95°C, 45 cycles of 10 seconds of denaturation at 95°C, 10 seconds of annealing at 60°C, and 10 seconds of extension at 72°C. All reactions were run in duplicates and normalized to a housekeeping gene (PPIA) using the ddCt method.
RNA Sequencing and Fusion Gene Discovery
Five hundred nanograms of total RNA was used for library preparation using NextSeq High Output Kit v2.5 kits (Illumina) and subjected to 2 × 75 bp paired‐end sequencing on a NextSeq 500 device (Illumina). Quality control of raw sequencing data (e.g., for potential ribosomal contamination) and insert size estimation were performed using FastQC, Picard tools, samtools, and rseqc [26]. Reads were mapped using STAR v2.4.0 [27] to the hg19 human genome assembly. Gene expression analysis was performed as described previously [28]. Briefly, for each gene present in the Human FAST DB v2016_1 annotation, reads aligning on constitutive regions (that are not prone to alternative splicing) were counted. Based on these read counts, normalization was performed using DESeq2 [29] and R (v.3.2.5).
Fusion detection was performed using five different tools: Defuse v0.6.0 [30], FusionCatcher v0.99.5a [31] without BLAT, JAFFA v1.06 [32], SoapFuse v1.27 [33], TopHat fusion v2.06.13 [34]. Comparison of results between algorithms was done by FuMa v2 [35] with FASTD DB v2016_1 annotations. High‐confidence fusion candidates identified by >3 tools were reviewed manually.
Methylation Profiling
Genome‐wide methylation profiling was performed as previously reported [36]. Briefly, 500 ng of DNA was subjected to bisulfite conversion and hybridized to Infinium MethylationEPIC BeadChip microarrays (Illumina). Raw IDAT files were used as input for online methylation‐based random forest classification (www.molecularneuropathology.org) using classifier version v11b4.
Data Availability
All raw sequencing data have been deposited at the European Genome‐phenome Archive (accession no. EGAS00001003604). Microarray‐based methylome data have been made available at ArrayExpress (accession no. E‐MTAB‐7490).
Results
Patient and Tumor Characteristics
We initially identified 25 patients with an archival diagnosis of ABM. After centralized neuropathological review, a histological diagnosis of ABM was confirmed in 15 cases: 14 adults and 1 adolescent. Ten cases were excluded because of a morphological diagnosis of glioblastoma in seven cases, no availability of tumor DNA and RNA in two cases, and co‐occurrence of astroblastoma and IDH‐mutant glioblastoma within the same tumor in one patient. Typical ABM histology with astroblastic pseudorosettes, perivascular pattern, vascular hyalinization, and immunoreactivity for GFAP, OLIG2, and vimentin were seen in all included cases. Positive immunostaining for EMA and p53 was found in only three cases and four cases, respectively. Of 15 ABMs, 10 were classified as “high grade” based on hypercellular zones with increased mitotic index, vascular proliferation, and necrosis.
The 15 cases included in the current study were investigated further (10 females and 5 males, sex ratio 2:1). Median age at diagnosis was 44.5 years (range: 24‐66) with a bimodal age distribution: four patients were <25 years of age and seven were >50 years of age. The most common initial symptom was neurological deficit (8/15 patients), followed by headaches (6/15) and seizure (4/15).
All patients underwent brain computed tomography or MRI scan. All cases were supratentorial, including one intraventricular tumor (third ventricle). Location was temporal, frontal, parietal, occipital, and parieto‐occipital in six, three, two, one, and three cases, respectively. Brain MRI was available for review in 11/15 patients. All tumors appeared well circumscribed and demarcated from normal brain on MRI. Contrast enhancement was detected in all patients, and a cystic component was observed in 6/11 patients. All patients underwent surgical resection (13 patients with gross total resection, 2 with partial resection).
After surgery, 11 of 15 patients received adjuvant treatment (6 with combined radio‐ and chemotherapy, 4 with radiotherapy alone, and 1 with chemotherapy alone) because of high‐grade morphology (10 cases) or partial resection (1 case).
After first‐line treatment, eight patients did not experience tumor relapse. In contrast, two, four, and one patients experienced one, two, and three or more tumors relapses, respectively. Four patients died during the follow‐up period (26.7%). The median progression‐free survival and overall survival were 1.6 and 4.9 years, respectively (Table 1).
Molecular Profiling
Depending on DNA and RNA availability, quantity, and quality, molecular profiling was performed. Targeted next‐generation panel sequencing was performed in all 15 patients, transcriptome sequencing in 4 samples, and methylation profiling in 6 cases.
Together with immunohistochemistry, molecular profiling allowed the reclassification of cases into three main groups (Fig. 1A).
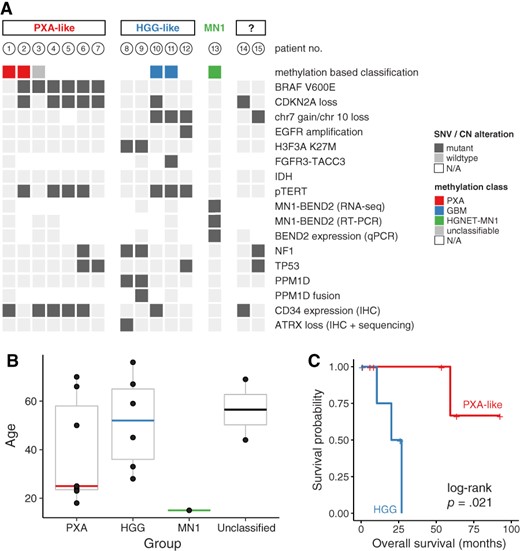
Summary of genetic alterations (A), age distribution (B), and overall survival (C) by subgroups.
Abbreviations: CN, copy number; EGFR, epidermal growth factor receptor; GBM, glioblastoma; HGG, high‐grade glioma; HGNET, high grade neuroepithelial tumor; IHC, immunohistochemistry; MN1, meningioma 1; N/A, not available; PXA, pleomorphic xanthoastrocytoma; qPCR, quantitative real‐time polymerase chain reaction; RT‐PCR, reverse transcription polymerase chain reaction; SNV, single nucleotid variant.
ABM with Molecular Features of PXA
Of 15 tumors, 7 (47%) were either classified as pleomorphic xanthoastrocytoma (PXA) using methylation‐based classification or showed BRAF mutation with concomitant CDKN2A loss and/or immunohistochemical CD34 expression. Six tumors harbored a BRAFV600E mutation, which was associated with CDKN2A homozygous deletion in five cases and TERT C228T promoter mutations in four cases.
One patient's tumor harbored a BRAF G466E alteration, a class III mutation resulting in a kinase‐dead form of BRAF [37]. This alteration was accompanied by a truncating NF1 mutation, a combination that is frequently observed in melanoma and known to result in a mechanistically different, yet functionally similar, ERK pathway activation [38].
None of these cases harbored typical histological features of PXA (i.e., cellular pleomorphism with spindle cells, mononucleated and multinucleated giant cells, granular bodies, or positive reticulin staining) [39]. Using immunohistochemistry, these cases harbored nuclear OLIG2 expression in all cases and cytoplasmic CD34 expression in 5/7 patients (whereas only 2/8 cases without BRAF mutation expressed CD34). Mean age at diagnosis was 25 years (Fig. 1B). In this subgroup, one patient out of seven died during follow‐up (Fig. 1C). The median progression‐free survival was 2.6 years.
ABM with Molecular Patterns of HGG
Of 15 patients, 5 were classified as high‐grade glioma because of the presence of H3.3 K27M mutations or glioblastoma molecular features (methylation class GBM or any two of the following criteria: combined chr7 gain/chr10 loss, epidermal growth factor receptor (EGFR) amplification, and/or TERT promoter mutation) [40]. Three patients had TERT C228T promoter mutations, one of which harbored a FGFR3:TACC3 fusion. Interestingly, the two cases with molecular pattern of diffuse midline glioma (i.e., combination of H3F3A, PPM1D, and NF1 mutations) did not arise within classic midline location (i.e., thalamus, pons, and spinal cord [41]): one case was located in the third ventricle and the other one in frontoparietal parasagittal localization. Of note, none of the H3F3A‐wildtype cases in this group showed loss of histone H3 lysine 27 trimethylation.
Mean age at diagnosis in this group was 52 years (Fig. 1B). Strikingly, this subgroup was associated with poor clinical outcome compared with PXA‐like cases: three out of five patients (60%) died during the follow‐up. The median progression‐free survival and overall survival were 0.9 and 1.9 years, respectively (Fig. 1C).
In contrast to PXA‐like cases, MRI of high‐grade glioma (HGG)‐like cases showed moderate‐to‐diffuse peripheral edema with a median volume of 23.5 mL (18.8–90) versus 2.7 mL (0.7–129) in PXA‐like subgroup (p < .001; Fig. 2).
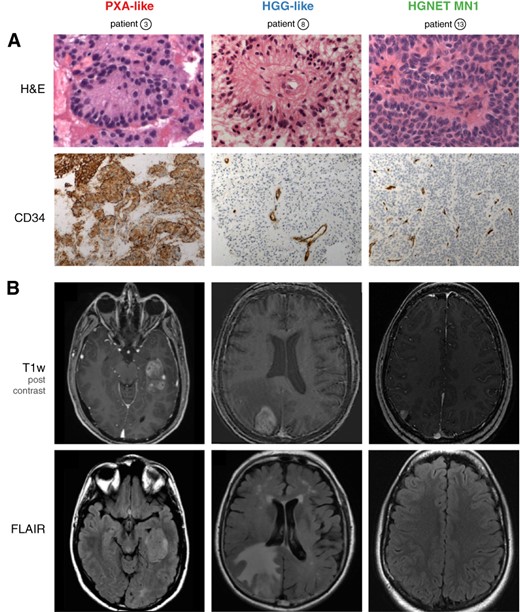
Representative histological (A) and magnetic resonance imaging (MRI) (B) features of subgroups. (A): H&E staining shows large astroblastic pseudorosettes in all cases (upper panel). Unlike HGG and HGNET‐MN1, PXA‐like astroblastoma (ABM) demonstrate an intense cytoplasmic and pericellular expression of CD34 (lower panel). (B): Representative MRI features of subgroups. Axial post‐contrast T1‐weighted magnetic resonance images demonstrate a solid component in all cases (upper panel). Unlike the other subgroups, HGG‐like ABM show moderate‐to‐extensive perifocal edema on FLAIR sequences (lower panel).
Abbreviations: FLAIR, T2‐weighted fluid‐attenuated inversion recovery; H&E, hematoxylin and eosin; HGG, high‐grade glioma; HGNET, high grade neuroepithelial tumor; MN1, meningioma 1; PXA, pleomorphic xanthoastrocytoma; T1w, T1‐weighted.
ABM with MN1‐BEND2 Fusion
Only one young patient (15 years of age) was epigenetically classified as high‐grade neuroepithelial tumor with MN1 alteration (CNS HGNET‐MN1) [12]. RNA sequencing identified an MN1:BEND2 fusion, which could be confirmed by RT‐PCR and BEND2 expression by real‐time PCR, whereas all others cases did not express BEND2 (supplemental online Fig. 1). The patient has had stable disease since tumor resection over 19 years of follow‐up.
Finally, two patients could not be assigned to either subgroup given the available molecular data. One patient (lost during follow‐up) harbored an NF1 mutation and combined chr7 gain/chr10 loss. However, because this patient did not harbor EGFR amplification, this case may correspond to PXA or GBM‐like ABM [42]. The other patient harbored CDKN2A homozygous deletion and was positive for CD34 expression immunohistochemically.
Discussion
In this study, we retrospectively characterized a series of 14 adults and 1 adolescent with centrally reviewed ABM. The present cohort shares characteristics with previous published studies [4,6,9,43–46] with ABM being a supratentorial tumor with a clear female predominance and heterogeneous clinical courses. Comprehensive interrogation of genetic alterations in these 15 patients (including methylation profiling, targeted DNA sequencing, RNA sequencing, RT‐PCR, and qPCR) allowed reclassification of cases in three subgroups (PXA‐like ABM, HGG‐like ABM, and ABM with MN1‐BEND2 fusion) with distinct radiological and histological features as well as clinical outcomes.
ABM has been controversial in recent years, as some literature supports it as true entity with frequent MN1 alterations [12,13,15] whereas other studies suggest it does not exist as an established and unique tumor entity but could overlap with other well‐known tumors [11,14,16,47].
Especially, in accordance with our study, gliomas with astroblastic features may harbor molecular signatures of PXA [14–16] and HGG [11,14,47]. Moreover, reclassification of ABM into a more specific molecularly defined entity could explain the clinical unpredictability and difficulty in grading these tumors.
The largest group was ABM with a molecular signature of PXA. Previously, methylation‐based reclassification of an ABM case into PXA 14,15 and high prevalence of BRAF mutations 15,16 have been reported. BRAF mutations are also frequently observed in ganglioglioma (GG) and pilocytic astrocytoma (PA). However, combined BRAF V600E mutation with (a) CDKN2A loss, usually only seen in PXA [48], (b) TERT promoter mutations, common in anaplastic PXA but virtually never seen in PA and rare in GG 49,50, and (c) positive CD34 staining demonstrates compelling evidence that indeed many ABM share the molecular identity of PXA.
The second group includes ABM with molecular features of HGGs (i.e., GBM and diffuse midline glioma, K27M‐mutant). As expected, these patients experienced an unfavorable clinical course. These tumors were accompanied by larger perifocal edema, a typical feature of high‐grade glioma. It is interesting to note that mitogen‐activated protein kinase (MAPK) pathway alterations including BRAF mutation, NF1 mutation, or FGFR fusion were present in 67% of cases (10/15) in the entire cohort. Even though MAPK pathway alterations are frequent in GBM (mostly affecting EGFR), they rarely involve BRAF, NF1, and FGFR 51,52. Rather, the mutational spectrum resembles that of pilocytic astrocytomas [53]. Strikingly, these alterations are actionable drug targets, potentially subject to BRAF inhibition (e.g., vemurafenib), MEK inhibition (e.g., trametinib), or FGFR inhibition (e.g., NCT02052778). In addition to BRAF mutations, a NRAS mutation has previously been reported in a case of ABM [14], extending the spectrum of observed MAPK/RAS pathway mutations. Because all of these alterations were identified retrospectively, none of the reported patients received targeted therapy.
Despite thorough multimodal screening for MN1 alterations, only one adolescent patient was assigned the diagnosis of CNS high‐grade neuroepithelial tumor with MN1 alteration. This result differs from three previous studies in adult ABM [13–15] in which MN1 rearrangements were frequent. Of note, we cannot entirely rule out MN1 alterations with fusion partners other than BEND2 in tumors that only were screened by RT‐PCR and for BEND2 expression and did not undergo methylation profiling (six cases). However, all of these cases harbored glioblastoma molecular features or a BRAF or H3.3 K27M mutation and could thus reliably be assigned to one of the other subgroups. This finding is in line with evidence of previous studies, in which MN1 alterations, BRAF mutation, and HGG molecular features were mutually exclusive [12–15,47].
Our results differ partly from those of five other retrospective studies [12–15,47]. Comparing our results directly with these studies is not possible as we encounter major differences in study population (majority of adult patients in our study vs. a significant proportion of pediatric cases in others) [12–15], required histologic features, number of patients, and molecular analyses performed. However, analysis of available data (summarized in Table 2) suggests that ABM with MN1 alterations appears to be frequent in children (found in 29/41 [70.7%] pediatric ABM cases) and uncommon in adults (found in 6/44 [13,6%]). In contrast, ABM with molecular features of PXA (i.e., BRAF mutation, CD34 expression, corresponding methylation class) appears to be recurrent in adolescents and adults (found in 14/44 [31.8%]) and rare in children (1/41 [2.4%]). Finally, most of these studies did not specifically investigate molecular features of high‐grade glioma (chr7 gain/chr 10 loss, histone mutation, methylation‐based classification) and could explain why the majority of these cases (5/7) are found in our study. Additional studies with a focus on this issue would be necessary to further explore this entity.
Review of literature
| Reference | No. of patients with ABM | No. of adult cases | No. of pediatric cases | No. of cases with PXA features (BRAF mutation) | No. of cases with MN1 rearrangement | No. of cases with HGG features |
|---|---|---|---|---|---|---|
| This study | 15 | 14 | 1 | 7 | 1 (none in adult case) | 5 |
| Sturm et al. [12] | 23 | 0 | 23 | — | 16 (none in adult cases) | — |
| Bale et al. [47] | 4 | 4 | 0 | 0 | — | 1 |
| Hirose et al. [13] | 8 | 4 | 4 | 0 | 4 (2 in adult cases) | — |
| Wood et al. [14] | 8 | 6 | 2 | 1 | 4 (2 in adult cases) | 1 |
| Lehman et al. [15] | 27 | 16 | 11 | 7 (1 in pediatric case) | 10 (2 in adult cases) | — |
| Total | 85 | 44 | 41 | 15 (1 in pediatric case) | 35 (6 in adult cases) | 7 |
| Reference | No. of patients with ABM | No. of adult cases | No. of pediatric cases | No. of cases with PXA features (BRAF mutation) | No. of cases with MN1 rearrangement | No. of cases with HGG features |
|---|---|---|---|---|---|---|
| This study | 15 | 14 | 1 | 7 | 1 (none in adult case) | 5 |
| Sturm et al. [12] | 23 | 0 | 23 | — | 16 (none in adult cases) | — |
| Bale et al. [47] | 4 | 4 | 0 | 0 | — | 1 |
| Hirose et al. [13] | 8 | 4 | 4 | 0 | 4 (2 in adult cases) | — |
| Wood et al. [14] | 8 | 6 | 2 | 1 | 4 (2 in adult cases) | 1 |
| Lehman et al. [15] | 27 | 16 | 11 | 7 (1 in pediatric case) | 10 (2 in adult cases) | — |
| Total | 85 | 44 | 41 | 15 (1 in pediatric case) | 35 (6 in adult cases) | 7 |
Four of four cases were tested for BRAF mutation.
Three of eight cases were tested for BRAF mutation.
Abbreviations: —, not specified; ABM, astroblastoma; HGG, high‐grade glioma; MN1, meningioma 1; PXA, pleomorphic xanthoastrocytoma.
Review of literature
| Reference | No. of patients with ABM | No. of adult cases | No. of pediatric cases | No. of cases with PXA features (BRAF mutation) | No. of cases with MN1 rearrangement | No. of cases with HGG features |
|---|---|---|---|---|---|---|
| This study | 15 | 14 | 1 | 7 | 1 (none in adult case) | 5 |
| Sturm et al. [12] | 23 | 0 | 23 | — | 16 (none in adult cases) | — |
| Bale et al. [47] | 4 | 4 | 0 | 0 | — | 1 |
| Hirose et al. [13] | 8 | 4 | 4 | 0 | 4 (2 in adult cases) | — |
| Wood et al. [14] | 8 | 6 | 2 | 1 | 4 (2 in adult cases) | 1 |
| Lehman et al. [15] | 27 | 16 | 11 | 7 (1 in pediatric case) | 10 (2 in adult cases) | — |
| Total | 85 | 44 | 41 | 15 (1 in pediatric case) | 35 (6 in adult cases) | 7 |
| Reference | No. of patients with ABM | No. of adult cases | No. of pediatric cases | No. of cases with PXA features (BRAF mutation) | No. of cases with MN1 rearrangement | No. of cases with HGG features |
|---|---|---|---|---|---|---|
| This study | 15 | 14 | 1 | 7 | 1 (none in adult case) | 5 |
| Sturm et al. [12] | 23 | 0 | 23 | — | 16 (none in adult cases) | — |
| Bale et al. [47] | 4 | 4 | 0 | 0 | — | 1 |
| Hirose et al. [13] | 8 | 4 | 4 | 0 | 4 (2 in adult cases) | — |
| Wood et al. [14] | 8 | 6 | 2 | 1 | 4 (2 in adult cases) | 1 |
| Lehman et al. [15] | 27 | 16 | 11 | 7 (1 in pediatric case) | 10 (2 in adult cases) | — |
| Total | 85 | 44 | 41 | 15 (1 in pediatric case) | 35 (6 in adult cases) | 7 |
Four of four cases were tested for BRAF mutation.
Three of eight cases were tested for BRAF mutation.
Abbreviations: —, not specified; ABM, astroblastoma; HGG, high‐grade glioma; MN1, meningioma 1; PXA, pleomorphic xanthoastrocytoma.
Our study has several limitations inherent to its retrospective observational design. The diagnosis of ABM is based on fairly exclusive histologic features. This could lead to differences in patient selection and also explain differences in results between studies. Survival data should be interpreted with caution considering the three patients lost to follow‐up and the study inclusion period (from 1990 to 2017): we cannot exclude the possibility that the evolution of imaging and histopathology techniques along with treatments of patients (including surgery techniques and chemotherapy and radiotherapy protocols) during the study period may have influenced our results. Finally, because of lack of material, methylation profiling could not be performed in all cases. Thus, we cannot prove that all BRAF‐mutant, CD34‐positive cases are indeed PXAs. Ganglioglioma, for example, remains a differential diagnosis.
Conclusion
Our data indicate that adult ABM comprises several molecularly defined entities. CNS high‐grade neuroepithelial tumor with MN1 alteration appears to be uncommon in adults. Adults’ ABM frequently harbor molecular features of PXA and HGG. Astroblastic morphology in adults should prompt thorough molecular investigation aiming at a clear histomolecular diagnosis and identifying actionable drug targets, especially in MAPK pathway genes.
Acknowledgments
We thank Amithys Rahimian‐Aghda, Marine Giry, Inès Detrait, and Yohann Schmitt for biobanking, sequencing studies, and expert technical assistance. We also thank Noémie Robil and Pierre de la Grange for fusion gene analysis, David Capper for methylation profiling, and Jean‐François Côté for providing human testis tissue. P.E. is a participant of the BIH‐Charité Clinician Scientist Program funded by the Charité ‐ Universitätsmedizin Berlin and the Berlin Institute of Health. P.E. has been supported by the Deutsche Forschungsgemeinschaft (EU 162/1). This research was supported by a grant of the Ligue Contre le Cancer and the Institut Universitaire de Cancérologie (RS17/75‐130 to A.I.). INCA‐DGOS‐Inserm_12560 SiRIC CURAMUS is financially supported by the French National Cancer Institute, the French Ministry of Solidarity and Health, and Inserm. The research leading to these results has received funding from the program Investissements d'avenir (ANR‐10‐IAIHU‐06).
Author Contributions
Conception/design: William Boisseau, Philipp Euskirchen, Karima Mokhtari, Ahmed Idbaih
Provision of study material or patients: Philipp Euskirchen, Karima Mokhtari, Caroline Dehais, Khê Hoang‐Xuan, Marc Sanson, Laurent Capelle, Aurélien Nouet, Carine Karachi, Franck Bielle, Justine Guégan, Yannick Marie, Nadine Martin‐Duverneuil, Luc Taillandier, Audrey Rousseau, Jean‐Yves Delattre, Ahmed Idbaih,
Collection and/or assembly of the data: William Boisseau, Philipp Euskirchen, Karima Mokhtari, Ahmed Idbaih
Data analysis and interpretation: William Boisseau, Philipp Euskirchen, Karima Mokhtari, Ahmed Idbaih
Final approval of manuscript: William Boisseau, Philipp Euskirchen, Karima Mokhtari, Caroline Dehais, Khê Hoang‐Xuan, Marc Sanson, Laurent Capelle, Aurélien Nouet, Carine Karachi, Franck Bielle, Justine Guégan, Yannick Marie, Nadine Martin‐Duverneuil, Luc Taillandier, Audrey Rousseau, Jean‐Yves Delattre, Ahmed Idbaih
Disclosures
Mehdi Touat: Taiho Oncology, Integragen (C/A), Agios Pharmaceuticals (SAB); Franck Bielle: Abbvie, Sanofi (RF [family member]), Celgene (E [family member]), Crossject (OI [family member]); Ahmed Idbaih: CarThera (travel), Transgene, Sanofi, Air Liquide, Gecko, Beta Innov (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
For Further Reading: Priscilla K. Brastianos, Franziska Maria Ippen et al. Emerging Gene Fusion Drivers in Primary and Metastatic Central Nervous System Malignancies: A Review of Available Evidence for Systemic Targeted Therapies. The Oncologist 2018;23:1063–1075.
Implications for Practice: Driver gene fusions involving receptor tyrosine kinases have been identified across a wide range of tumor types, including primary central nervous system (CNS) tumors and extracranial solid tumors that are associated with high rates of metastasis to the CNS (e.g., lung, breast, melanoma). This review discusses the systemic therapies that target emerging gene fusions, with a focus on brain‐penetrant agents that will target the intracranial disease and, where present, also extracranial disease.
References
Author notes
Contributed equally.
Disclosures of potential conflicts of interest may be found at the end of this article.

{kind=link}
{kind=link}