





Abstract
The progress in the development of systemic treatment for advanced pancreatic cancer (APC) has been slow. The mainstream treatment remains using chemotherapy including gemcitabine, FOLFIRINOX, and nab-paclitaxel. Erlotinib is the only approved biological therapy with marginal benefit. Studies of agents targeting epidermal growth factor receptor, angiogenesis, and RAS signaling have not been satisfying, and the usefulness of targeted therapy in APC is uncertain. Understanding in molecular processes and tumor biology has opened the door for new treatment strategies such as targeting insulin-like growth factor 1 receptor, transforming growth factor β, phosphoinositide 3-kinase/AKT/mammalian target of rapamycin pathway, and Notch pathway. New directions also include the upcoming immunotherapy and many novel agents that act on the microenvironment. The practice of personalized medicine using predictive biomarkers and pharmacogenomics signatures may also enhance the effectiveness of existing treatment. Future treatment approaches may involve comprehensive genomic assessment of tumor and integrated combinations of multiple agents to overcome treatment resistance.
摘要
晚期胰腺癌 (APC) 全身治疗的发展进展缓慢。主流治疗仍然是使用吉西他滨、FOLFIRINOX 和纳米白蛋白结合型紫杉醇 (nab-paclitaxel) 之类的化疗。厄罗替尼是唯一获批且具有边际效益的生物治疗。对表皮生长因子受体、血管生成和 RAS 信号通道之靶向药物的研究一直无法令人满意,并且 APC 靶向疗法的效果不明确。对分子过程和肿瘤生物学的了解为发展新的治疗策略(如以雷帕霉素通路和 Notch 信号通路的胰岛素样生长因子 1 受体、转化生长因子 β、磷脂酰肌醇 3 激酶/AKT/哺乳动物靶向为目标的疗法)开启了大门。新的发展方向还包括即将面世的免疫治疗和许多作用于肿瘤微环境的新药。利用预测性生物标志物和药物基因组学标识的个体化医学实践也有可能增强现有治疗的效果。进一步的治疗方法可能涉及全面的肿瘤基因组评估和综合使用多种药物以克服抗治疗性。The Oncologist 2014;19:937–950
Implications for Practice:
Pancreatic cancer is a dismal disease with an unmet need for novel treatment approaches. This paper provides a comprehensive review in the current development of new treatment strategies, supported with concise scientific background and brief descriptions of ongoing phase II/III clinical trials. It provides both nonspecialists and specialists in pancreatic cancer care an overview of the topic that they can easily reference in their clinical practice or research.
Introduction
Advanced pancreatic cancer (APC) is one of the most dismal human cancers. Conventional approaches such as radiation, chemotherapy, or a combination of both have little impact in the disease course. Gemcitabine (GEM) replaced fluorouracil (5FU) as the standard treatment based on its effect on alleviating disease-related symptoms and modest improvement of 1-year survival [1]. There was no breakthrough until the PRODIGE 4/ACCORD11 trial demonstrated superior survival benefit of FOLFIRINOX (a combination regimen consisting of oxaliplatin, irinotecan, 5FU, and leucovorin) over GEM [2]. However, this increase in median overall survival (OS) from 6.9 to 10.5 months is at a cost of significant toxicities, and this regimen is only indicated for patients with good performance status. Recently the Food and Drug Administration (FDA) has approved another cytotoxic agent, nab-paclitaxel, in combination with GEM for treatment of APC based on results from the MPACT trial [3]. This combination merely increased the median OS from 6.7 to 8.5 months when compared with single-agent GEM. There is an unmet need for more effective treatment. Although the development of biological agents has revolutionized the management of many cancers, the only targeted therapy shown to have antitumor activity is erlotinib. However, the addition of erlotinib to GEM only showed a statistically significant but not clinical meaningful survival benefit of 10 days at the median [4]. Years of research have suggested APC is a complex disease, and the usual targeting approach based on single gene aberration might be inadequate. Many novel treatment strategies are in development. This review summarized previous development of APC treatment and described recent findings. Table 1 provides a summary of major completed phase III trials in advanced pancreatic cancer, and Table 2 lists the drugs in development in major phase II and III trials.
Summary of major completed phase III trials in advanced pancreatic cancer
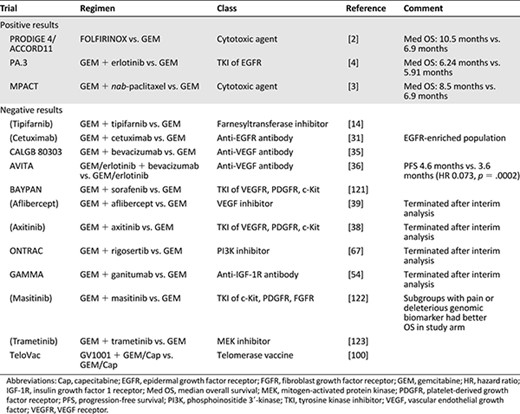
Summary of major completed phase III trials in advanced pancreatic cancer
Summary of drugs in development in major phase II and III trials
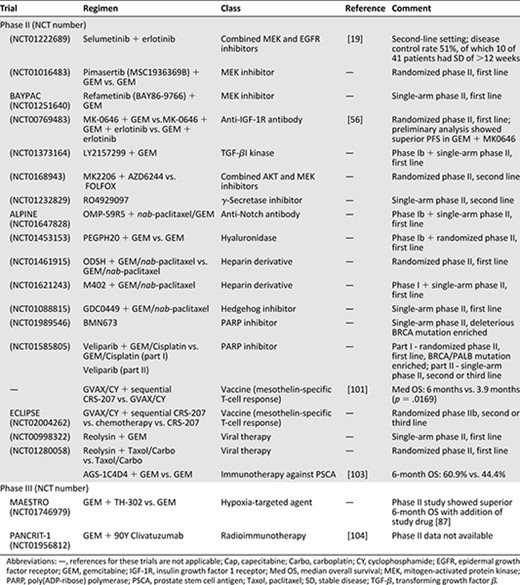
Summary of drugs in development in major phase II and III trials
Overview in Genetic Basis of Pancreatic Cancer
The pancreatic cancer genome project sequenced >750,000,000 base pairs of DNA from pancreatic adenocarcinoma and identified >1,500 somatic mutations in 1,007 out of 20,661 protein-coding genes [5]. It showed pancreatic cancer to be a heterogeneous disease and that its carcinogenesis could not be explained by single oncogene addiction. Each pancreatic cancer contained on average 63 genetic alterations, the majority of which were point mutations. The constitutively active KRASG12D mutation on chromosome 12p is the most common mutation and is reported in 70%–90% of cases [5–7]. Activated oncogenes including FRAF on chromosome 7q and AKT2 on chromosome 19q are also present in up to 20% of cases. Tumor suppressor gene mutations frequently found include p16/CDKN2A (75%–80%) [5, 8, 9], p53 (50%–75%), SMAD4 (50%–60%), and BRCA2 (10%) [10]. Notably, the BRCA2 mutation was initially believed to be only associated with familial cases, but 10% of sporadic cases were also reported to harbor this mutation.
These genetic alterations defined a core set of 12 cellular signaling processes that were each genetically altered in 67%–100% of the tumors [5]. They included apoptosis, DNA damage repair, regulation of G1/S phase transition, hedgehog signaling, hemophilic cell adhesion, integrin signaling, K-ras signaling, JNP signaling, regulation of invasion, small GTPase-dependent signaling, transforming growth factor β (TGF-β) signaling, and Wnt/notch signaling. These processes have been the main focus of drug development in this cancer.
Signaling Pathways in Pancreatic Cancer and Potential Therapeutic Targets
RAS
Although KRAS is the most common mutation in APC, this gene is difficult to target directly. RAS is active when bound to GTP. Inactivation is achieved by hydrolysis of the γ-phosphate of GTP to GDP with GTPase-activating proteins (GAPs) acting as the catalyst [11]. The catalytic domain contains a nucleotide-binding protein. Codon 12 of KRAS encodes for the phosphate-binding loop and the two switch regions that bind the nucleotide. KRASG12D mutation renders RAS catalytically insensitive to GAP-mediated GTP hydrolysis, and RAS becomes constitutively active. Direct blockade of these functional sites of RAS protein with small molecules is challenging because so far no accessible active-site pocket could be identified.
Post-translational modification of RAS involves four steps: isoprenylation, proteolysis, methylation, and palmitoylation. Isoprenylation requires transferring a farnesyl group to the pre-RAS protein by the farnesyltransferase (FTase) and transferring a geranylgeranyl group to the K- and N-Ras by geranylgeranyltransferase I (GGTase I) [11]. FTase was considered to be the dominant enzyme. Tipifarnib (R115777) is an orally active FTase inhibitor (FTI) that demonstrated acceptable toxicity profile but no appreciable antitumor activity as monotherapy [12, 13]. When combined with GEM in a randomized phase III double-blind placebo-controlled trial, this drug did not derive additional survival benefit over single-agent GEM [14]. Another FTI, lonafarnib (SCH66336), was also compared with GEM in a phase II trial. It showed modest clinical activity but no clinical advantage over GEM in initial presentation of data [15]. The combination of lonafarnib with GEM was not pursued further. The failure of these trials suggested that FTase inhibition might lead to diversion to the alternative pathways through GGTase.
The idea of gene therapy by silencing of activated KRAS is gaining popularity, and preclinical studies are underway. Treatment of cultured pancreatic cancer cells with antisense oligonucleotides resulted in KRAS mutation-matched suppression of tumor invasion [16]. K-ras small interfering RNA (siRNA) also led to significant inhibition of KRAS endogenous expression and cell proliferation in tumor cell cultures [17]. In animal models, combination treatment of KRAS siRNA with GEM led to growth inhibition of orthotopic pancreatic tumor and prolongation of animal survival compared with single-agent GEM [17]. Nevertheless, gene-silencing strategy is limited by its transient nature, unpredictable intratumoral bioavailability, and lack of reliable delivering system. Technology for clinical application of gene therapy is still under development.
The RAS/RAF/mitogen-activated protein kinase (MEK)/extracellular signal-related kinase (ERK) pathway is the dominant effector of KRAS activation. A number of MEK inhibitors are being tested in clinical trials. For instance, selumetinib (AZD6244) was compared with capecitabine in a phase II open-label randomized study in APC patients who have failed first-line gemcitabine therapy and demonstrated similar efficacy as capecitabine [18]. Based on preclinical evidence of synergistic activity between epidermal growth factor receptor (EGFR) and MEK inhibitors, selumetinib is also being tested in combination with erlotinib in gemcitabine-resistant APC in an ongoing phase II trial and demonstrated preliminary antitumor activity (01222689) [19]. Another MEK inhibitor, trametinib (GSK1120212), demonstrated a tolerable toxicity profile when combined with GEM and there were evidence of clinical activity in pancreatic cancer from the phase I trial [20]. This combination was further tested in a randomized phase II trial but did not demonstrate clinical benefit [21]. Other MEK inhibitors by various sponsors are in clinical development, and results from phase II studies are expected to be available in the next 2 years. For instance, both pimasertib (MSC1936369B) and refametinib (BAY86-9766) are in phase II studies in combinations with GEM at the moment (01016483 and 01251640, respectively).
Targeting KRAS and its downstream signaling will remain a central focus of research in pancreatic cancer. In the transgenic KRASG12D mouse model, inactivation of references for these trials are not applicable during cancer progression led to tumor regression, suggesting that this mutation is also required for tumor maintenance and survival [22]. Moreover, it was associated with the up-regulation of Hedgehog signaling and other pathways known to mediate the maintenance of the protumorigenesis stroma and microenvironment. The significance of tumor microenvironment in APC will be further discussed later.
Epidermal Growth Factor Receptor
The EGFR family consists of four tyrosine kinase receptors including ErB-1 (EGFR), ErbB-2 receptor (HER-2/neu), ErbB-3 (HER-3), and ErbB-4 (HER-4). ErbB-1 and ErbB-2 expression has been found in 90% and 21% of pancreatic cancer, respectively [23, 24]. Increased coexpression of EGFR and its ligand in pancreatic cancer was associated with more liver metastasis and poorer prognosis [25–27]. EGFR targeting therapy has been studied extensively in APC, but the results were disappointing.
Tyrosine Kinase Inhibitors
Erlotinib is an orally active small molecule tyrosine kinase inhibitor (TKI) of EGFR. In the PA.3 trial, which was a phase III randomized double-blind clinical study of erlotinib in combination with GEM in APC, a total of 569 patients were given GEM plus either a daily dose of 100 mg of erlotinib (GE) or a placebo as first-line treatment [4]. A small improvement in median OS was observed in the combination arm (6.24 versus 5.91 months, hazard ratio [HR] 0.82, p = .038). Patients who received GE had toxicities such as rashes, diarrhea, infection, and stomatitis. The presence of rash was associated with a higher disease control rate (p = .05) and longer survival (p = .037; HR 0.74) after adjusting for other prognostic factors. Molecular analyses of K-ras mutation status and EGFR gene copy number were performed in 26% of the tumor samples, and they were not associated with survival benefit of the erlotinib/GEM combination [28]. Although erlotinib has proven survival benefit, the GE combination is limited by its cost-effectiveness and thus generally not supported by funding agencies.
In a second-line setting, a phase II trial by Kulke et al. [29] evaluated the combination of erlotinib (150 mg daily) and capecitabine (1,000 mg/m2 twice daily for 2 weeks every 21-day cycle) in GEM-refractory APC and showed only 10% radiological response and median OS of 6.5 months. The erlotinib combination with cytotoxic agents is not favored in general for its limited efficacy. Further phase III data are not available.
Monoclonal Antibodies
Cetuximab is a chimeric monoclonal antibody with high specificity against ErbB-1 receptors. The combination of cetuximab and GEM as first-line treatment in EGFR-enriched APC showed initial encouraging results in a phase II study with stable disease (SD) and partial response shown in 63% and 12% of patients [30]. Nevertheless, in a phase III study, this combination did not show any survival benefit over single-agent GEM [31]. Approximately 90% of tumor expressed EGFR, and no treatment benefit was detected in the analysis of this subgroup. EGFR expression does not confer response to anti-EGFR therapy in APC.
In preclinical models of pancreatic cancer, dual inhibition of the EGFR pathway by TKIs and monoclonal antibodies demonstrated promising antitumor activity. A randomized phase II study of panitumumab, erlotinib, and GEM in APC showed a trend in OS benefit when compared with GEM plus erlotinib at a median follow-up of 6 months [32]. However, this three-drug combination resulted in severe toxicities, especially skin rashes, and the trial was terminated.
Other Anti-EGFR Strategy
Trastuzumab is a monoclonal antibody targeting HER2. In a small phase II trial, 34 APC patients with tumors Her2/neu overexpression 2+/3+ by immunohistochemistry were given trastuzumab in combination with GEM. The survival results were very similar to single-agent GEM with an objective response rate (ORR) of only 6% and a median OS of 7 months [33]. Notably, only 12% of these patients were HER2 3+. The rest were HER2 2+, and in situ hybridization was not performed. Previous experience from breast cancer and gastric cancer suggested that trastuzumab is predominantly effective in Her2 3+ or amplified tumors. Given the relatively low Her2 expression in APC, anti-HER2 therapy is unlikely to be developed into a mainstream treatment in this tumor.
Angiogenesis
Angiogenesis is believed to play an important role in sustaining tumor growth and targeting the vascular endothelial growth factor (VEGF) pathway has shown promising results in the treatment of many solid tumors. In pancreatic cancer, high expression of VEGF is associated with increased microvessel density, liver metastasis and poor prognosis [34]. Unfortunately, anti-VEGF therapy has not shown much success in APC.
Anti-VEGF Antibody
Bevacizumab is a humanized monoclonal antibody against VEGF that has been extensively studied in APC. In the Cancer and Leukemia Group B (CALGB) 80303 phase III clinical trial, 602 patients with locally advanced/metastatic pancreatic cancer were randomized to receive GEM with or without bevacizumab [35]. Patients in both groups had similar median OS and progression-free survival (PFS), whereas the addition of bevacizumab gave more toxicity. AVITA is another large phase III trial of bevacizumab in APC. In this study, 301 patients with metastatic pancreatic cancer were randomly assigned to receive GEM plus erlotinib with either bevacizumab or placebo. Although the addition of bevacizumab improved median PFS (4.6 vs. 3.6 months, HR 0.73, p = .0002), it did not translate into OS benefit (7.1 vs. 6.0 months, HR 0.89, p = .2) [36].
Other Antiangiogenic Agents
Axitinib is a potent and selective oral inhibitor of VEGF receptors 1, 2, and 3. In an initial randomized phase II trial, the addition of axitinib to GEM in APC patients had apparent survival advantage [37]. This beneficial effect, however, was not confirmed upon interim analysis of a large phase III trial, which was subsequently terminated [38]. Aflibercept is a recombinant fusion protein with high VEGF affinity that functions as a VEGF inhibitor. It has also been combined with GEM in a phase III trial against single-agent GEM. The study was stopped for futility after an interim analysis of OS showing no difference between the two arms [39].
Sorafenib inhibits VEGFR, platelet-derived growth factor receptor (PDGFR) and c-Kit. Apart from its antiangiogenic property, it also modulates the RAS-RAF-MAPK signaling pathway. Nonetheless, the phase III BAYPAN study showed addition of sorafenib to GEM did not improve PFS in APC patients [40]. Another interesting agent is masitinib, This drug has been approved to be used in the treatment of mast cell tumors in animals. It is a TKI of c-Kit, PDGFR, and fibroblast growth factor receptor. In a phase III study of APC, the combination of GEM plus masitinib did not show improved OS [41]. However, subgroup analysis found patients with significant pain yielded superior survival in the combination arm with median OS 8.1 months versus 5.4 months. Another subgroup analysis of patients with a specific deleterious genomic biomarker also had superior survival receiving combination treatment with median OS of 11.0 months versus 5.0 months (HR 0.29, p = .000038).
The angiogenic and EGFR pathways are closely related: VEGF signaling is up-regulated by EGFR expression, whereas VEGF up-regulation independent of EGFR signaling may contribute to resistance to EGFR inhibition [42]. Thus, simultaneous inhibition of both pathways may theoretically improve the efficacy and overcome resistance. However, such results were not found in the AVITA study. Nevertheless, patients with more aggressive disease, as suggested by higher c-reactive protein and lactate dehydrogenase, derived greater benefit from bevacizumab [36]. In another study, 139 APC patients were randomized to either GEM, bevacizumab plus cetuximab (n = 68) or GEM, or bevacizumab plus erlotinib (n = 71) [43]. Despite the slightly higher ORR when combining the dual targeted agents with GEM, the median OS (7 months) was not better than the historical results treated by GEM alone. Notably, Starling et al. [44] reported an interesting phase I study in combining GEM, capecitabine, erlotinib, and bevacizumab in the treatment of APC patients. The ORR was surprising high (50%), with a median OS of 12.5 months. Further clinical development of this regimen is not available, and there is not sufficient evidence to support its use in clinical practice.
Suffice it to say, antiangiogenesis is ineffective clinically in treating APC patients. Although most preclinical models of pancreatic cancer suggested potential activity of many antiangiogenic agents, they failed to simulate human tumor microenvironment where dense stromal tissue with decreased vascular density is now known to be the main obstacle for effective drug delivery. Moreover, the withdrawal of antiangiogenic agents after therapy might associate with increased tumor aggressiveness and invasion, offsetting the potential therapeutic benefits offered by the antiangiogenic agents. Interestingly, it has also been postulated that potent angiogenesis inhibition might alter the natural history of tumors by paradoxically increasing tumor invasion and metastasis [45].
Although most preclinical models of pancreatic cancer suggested potential activity of many antiangiogenic agents, they failed to simulate human tumor microenvironment where dense stromal tissue with decreased vascular density are now known to be the main obstacle for effective drug delivery. Moreover, the withdrawal of antiangiogenic agents after therapy might associate with increased tumor aggressiveness and invasion, offsetting the potential therapeutic benefits offered by the antiangiogenic agents.
Future Perspectives
Although attempts on targeting the RAS, EGFR, and VEGF signaling were disappointing, new targets and therapeutic strategies are emerging. Figure 1 shows the major signaling pathways and potential actionable targets in advanced pancreatic cancer.
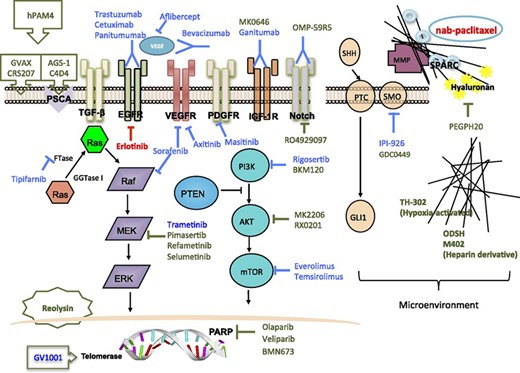
Major signaling pathways and potential actionable targets in advanced pancreatic cancer. Red lines and text indicate Food and Drug Administration-approved agents. Blue lines and text indicate agents that showed negative results in previous clinical trials. Olive green lines and text indicate agents in clinical trials.
Abbreviations: EGFR, epidermal growth factor receptor; ERK, extracellular signal-related kinase; FTase, farnesyltransferase; FTI, farnesyltransferase inhibitor; GGTase I, geranylgeranyltransferase I; HER, human epidermal growth factor receptor; IGF-1R, insulin-like growth factor 1 receptor; MEK, mitogen-activated protein kinase/ERK kinase; MMP, matrix metalloproteinases; MMPI, MMP inhibitor; mTOR, mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase; PDGFR, platelet-derived growth factor receptor; PI3K, phosphoinositide 3-kinase; PSCA, prostate stem cell antigen; PTC, patched-1 receptor; PTEN, phosphatase and tensin homolog deleted on chromosome ten; SHH, sonic hedgehog; SMO, smoothened; SPARC, secreted protein acidic and rich in cysteine; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
Insulin-Like Growth Factor 1 Receptor
Insulin-like growth factor (IGF) family plays a pivotal role in carcinogenesis [46]. In particular, IGF-1 receptor (IGF-1R) mRNA levels in pancreatic cancer were found to be 32-fold that of normal pancreas, and deviant regulation of an IGF-1 autocrine loop was associated with increased tumorigenicity in human pancreatic cancer cell lines [47, 48]. The binding of insulin to the IGF receptors is followed by tyrosine phosphorylation of insulin receptor substrates, which further propagate downstream signaling via phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway and results in tumor growth and progression [49]. The crosstalk between IGF and other pathways, for example EGFR pathway, has been implicated in resistance to anti-EGFR therapy [50]. It is scientifically plausible to use either IGF inhibitor alone or in combination with other agents in the treatment of APC.
Cixutumumab (IMC-A12) is a fully human monoclonal antibody against IGF-1R. Early phase I trial evaluating the triplet combination of gemcitabine, erlotinib, and cixutumumab showed no excessive toxicity, and the regimen was carried to a randomized phase II trial in APC (SWOG-S0727) [51]. Preliminary results were negative. Ganitumab (AMG479) is another monoclonal antibody against IGF-1R in clinical development (AMG479). The combination of ganitumab plus GEM in an randomized phase II trial had a trend toward favorable 6-month survival of 59% compared with 50% in single-agent GEM [52]. A large phase III study was initiated (GAMMA trial) [53]. Unfortunately, it was terminated after a preplanned interim analysis concluded that the addition of ganitumab to GEM was unlikely to demonstrate survival benefit [54].
Another humanized monoclonal antibody is dalotuzumab (MK-0646). It can bind to IGF-1R and induce receptor internalization and degradation. It also blocks IGF-1/2-mediated signal and has antibody-dependent cell-mediated cytotoxicity in vitro. Phase I/II trial of this agent in combination with GEM and erlotinib in APC demonstrated acceptable toxicity [55]. The phase II portion is ongoing (00769483). It is a crossover, randomized, three-arm study of combination therapies: GEM and MK0646 (arm A), GEM plus MK0646 and erlotinib (arm B), and GEM plus erlotinib (arm C). Preliminary analysis was encouraging because it showed superior PFS in arm A with tolerable toxicities [56].
Anti-IGF-1R antibodies are generally well tolerated. Common toxicities include hyperglycemia, hyperlipidemia, marrow toxicity, and fatigue. Because IGF-1R/IGF-1 signaling has been implicated in crosstalk with other pathways and resistance to chemotherapy, and it is ubiquitously expressed, we will continue to see anti-IGF-1R therapy being combined with other therapeutic agents in clinical development. A better understanding of these pathway interactions and investigation of predictive markers of response are needed.
Transforming Growth Factor β
TGF-β signaling has been implied in cancer cell proliferation, tumor angiogenesis, metastasis, and suppression of antitumor immunity [57, 58]. The intracellular mediator of TGF-β signaling is SMAD4. Allelic deletion of SMAD4 is found in 50% of human pancreatic cancers [59]; thus aberration of TGF-β-SMAD4 suppressive signal is believed to be an important step in pathogenesis of this cancer [5]. SMAD4 mutation leads to feedback overexpression of TGF-β1. In the absence of SMAD4 counteraction, the preferential activation of the alternative intracellular NF-κB signal leads to downregulation of tumor suppressor phosphatase and tensin homolog (PTEN) [60]. PTEN is a negative regulator of the oncogenic P13K/AKT signaling pathway. In pancreatic cancer, a high level of PTEN expression is associated with less aggressive tumor and confers better survival [61], implying that downregulation of PTEN leads to more aggressive disease. Although PTEN mutation is rare in pancreatic cancer, the altered downstream modeling of the TGF-β pathway provides an alternative mechanism, leading to PTEN downregulation and tumor progression.
The development of anti-TGF treatment in APC is still in the early clinical stage. For instance, trabedersen (AP12009) is a phosphorothioate antisense oligodeoxynucleotide specific for human TGF-β2 mRNA. A phase I/II study involving pancreatic cancer patients demonstrated good tolerability and encouraging clinical activity [62]. The company is preparing to launch a phase II trial for APC, but such a trial has not been registered. LY2157299 is a small molecule inhibitor of TGF-βI kinase. A phase I/IIb study combining this drug with GEM in APC is underway (01373164).
TGF-β signaling has pleiotropic effect in regulation of cell growth and tumor physiology. In normal epithelial tissue, it acts as tumor suppressor and mediator of growth arrest, whereas in tumor it processes both tumor-suppressing and tumor-promoting functions depending on cellular context [58, 63]. This context-dependent regulation of TGF-β activity has made studying this signaling pathway difficult.
PI3K/AKT/mTOR Pathway
The PI3K/AKT/mTOR pathway is one of the major downstream signaling pathways mediating the effect of K-ras. A number of mTOR inhibitors, including everolimus (RAD001) and temsirolimus, have been tested in phase II trial in patients with GEM-refractory APC but yielded negative results [64, 65]. Rigosertib is a small molecular inhibitor of PI3K originally found to have clinical activity in patients with myelodysplastic syndrome. It demonstrated a favorable profile in phase I trial [66] and was quickly moved into a phase II/III study of GEM plus rigosertib versus GEM (ONTRAC trial). Unfortunately, the trial was terminated after interim analysis, suggesting that the combination was unlikely to show survival benefit [67]. Early phase clinical trials of other inhibitors of P13K/AKT/mTOR pathway or combining these inhibitors with chemotherapy in APC are ongoing. For instance, XL147, buparlisib (BKM120) is a PI3K inhibitor in phase I trial in combination with chemotherapy FOLFOX6 (01571024). MK-2206 is an AKT inhibitor now in early clinical studies in combination with dinaciclib, a cyclin-dependent kinase inhibitor (phase I, 01783171) or with AZD6244, a MEK inhibitor (phase II, 0168943). Archexin (RX-0201) is another AKT inhibitor, and a phase II study for APC is being planned.
Notch Pathway
Notch signaling is known to have a critical role in organ development and cell differentiation. It mediates pancreatic cancer stem cell function, which is believed to contribute to resistance to chemotherapy, tumor recurrence, and metastasis. Upon activation of Notch receptor, Notch is cleaved by a cascade of proteolytic enzyme including metalloprotease, tumor necrosis factor-α-converting enzyme and γ-secretase [68]. The oral γ-secretase inhibitor RO4929097 has completed phase I trial and is now in phase II trial as a second-line treatment of APC (01232829). Recently preliminary results from two phase I clinical trials of anti-Notch antibodies, OMP-59R5 and demcizumab, have been presented [69, 70]. A phase II study of OMP-59R5 in combination with nab-paclitaxel and gemcitabine is now ongoing (ALPINE trial [01647828]). Given its unique role in cancer stem cells, this class of agent will become an active area of clinical research.
Extracellular Matrix, Microenvironment, and Stroma
Matrix Metalloproteinases Inhibitors
In the last few years scientists have grown to appreciate the importance of microenvironment in sustaining pancreatic tumor growth. The microenvironment of APC is characterized by extensive deposit of extracellular matrix (ECM) components and hypovascularity. These desmoplastic features are believed to impede drug delivery and contribute to primary resistance of drug therapy. Matrix metalloproteinase (MMP) inhibitors (MMPI) were the first class of drug treatment designed to act on the microenvironment. MMPs are a family of proteolytic enzymes responsible for breakdown of connective tissue proteins. They are crucial in maintaining the growth, differentiation, and repair of normal healthy tissue Aberrant MMP expression is associated with invasive activities of solid tumors [71]. Marimastat was the first MMPI, and it has been combined with GEM as the first-line treatment of APC [72]. Tanomastat (BAY12-9566) was another MMPI being tested against GEM as a single-agent first-line treatment in APC [73]. However, no clinical activity of these agents was observed.
The microenvironment of APC is characterized by extensive deposit of ECM components and hypovascularity. These desmoplastic features are believed to impede drug delivery and contribute to primary resistance of drug therapy.
Hyaluronidase
Recent study on hyaluronidase appeared to be promising. Hyaluronan (HA) is a nonsulfated glycosaminoglycan in the ECM. It has high abundance in pancreatic tumor and has been implicated in angiogenesis, epithelial mesenchymal transition, and chemoresistance [74]. Moreover, patients with high a HA level usually have poor prognosis. PEGPH20 is a PEGylated human recombinant PH20 hyaluronidase. In preclinical model of pancreatic cancer, PH20 depleted HA, induced re-expansion of intratumor blood vessels, and enhanced delivery of GEM [75]. A phase Ib study of GEM combined with PEGPH20 in APC patients demonstrated encouraging overall tumor response. Of 21 evaluable patients, 7 demonstrated partial response, whereas 9 had stable disease. Tissue analysis suggested that HA score might be a potential predictive marker [76]. A phase II study of this combination is underway (01453153), and results are eagerly awaited.
Heparin-Derivative Agents
Heparin is a tissue-derived glycosaminoglycan that has been used as standard anticoagulant for decades. It also possesses nonanticoagulant function in maintaining the integrity of ECM [77, 78]. In vitro, a heparin-derived agent devoid of anticoagulant effect has been found to inhibit tumor growth via disruption of heparanase activity. Nonanticoagulant heparin S-NACH was also found to inhibit pancreatic cancer cell adhesion and metastasis [79]. In an animal study, heparin derivate showed anticancer and antiangiogenic effects [80]. Given the important role of stromal microenvironment in maintenance of pancreatic cancer, these agents might have potential use in the management of this resistant disease. ODSH (2-0, 3-0 desulfated heparin) and M402 are agents of this class now in phase II study in combination with GEM and nab-paclitaxel for patients with APC (01461915 and 01621243, respectively).
Hedgehog Antagonists
The Hedgehog (Hh) pathway has been shown to be an important signaling system within the microenvironment of pancreatic cancer. It is involved in all three compartments of pancreatic cancers: the differentiated bulk of cancer cells that overexpress hedgehog ligands, pancreatic cancer stem cells, and pancreatic stromal cells that constitute the corresponding receptors [81]. Sonic hedgehog (SHH) is a secreted hedgehog ligand that binds to the membrane patched-1 receptor (PTC). This ligand-receptor interaction releases the inhibition of PTC on another transmembrane protein smoothened (SMO), which then activates the Hh pathway and leads to the expression of Hh-specific genes. These proteins are not found in normal pancreatic tissue but are markedly overexpressed in the abnormal pancreatic epithelium and the reactive stroma that surrounds the epithelium [82]. This unique feature makes it an attractive target for therapeutic intervention. Saridegib (IPI-926) is a small molecule semisynthetic derivative of cyclopamine that potently inhibits SMO. In a genetically engineered mouse model, IPI-926 in combination with GEM increased survival in these otherwise GEM-resistant animals. It was associated with elevated intratumoral concentration of GEM, reduction in the dense fibrotic reaction, and increase in tumor neo-vascularization [83]. The compound was combined with GEM and brought to phase I trial. The results were encouraging with favorable toxicity profile, and apparently 31% of the patients demonstrated partial response [84]. Nevertheless, when it was brought to the double-blind randomized placebo-controlled phase II study comparing IPI-926 plus GEM versus GEM alone in 122 metastatic pancreatic cancer patients, the new drug combination was associated with shorter survival in interim analysis, and the trial was terminated prematurely [85]. Vismodegib (GDC-0449) is another hedgehog antagonist now in phase II evaluation in combination with GEM and nab-paclitaxel (01088815).
Remodeling of microenvironment is a novel concept in systemic treatment of cancer, and it probably has implication on the efficiency of drug delivery, as well as homotypic and heterotypic signaling. IPI-926 is the first compound of this category, and its failure in clinical study discouraged the enthusiasm in the development of drug targeting microenvironment. When targeting the stromal tissue, we should beware of the difference in the microenvironment between local tumor and metastasis. Metastasis is characterized by the tendency to escape from the primary tumor, capability to survive in the circulation, invasion, and ability to establish colonies in distant sites. Unlike local disease, many cells involved in facilitating metastasis are derived from the bone marrow lineage; thus they entail a distinct entity and warrant special consideration in the design of clinical studies [86].
Hypoxia-Targeting Agent
The dense fibro-inflammatory microenvironment of pancreatic cancer results in hypoxia. Recently research found that tumor hypoxia leads to activation of hypoxia-inducible factor-1α that in turns activates secretion of SHH by tumor cells. In line with the concept of hypoxia-targeting therapy comes the development of TH-302. TH-302 is a prodrug activated only under hypoxic conditions. When activated, it releases a potent DNA alkylating agent, bromo isophosphoramide mustard. It is expected to have relative selective action in the hypoxic microenvironment of solid tumors and bypass the generalized cytotoxic effect of traditional systemic treatment. In a large randomized phase II study of GEM plus TH-302 versus GEM, the combination treatment showed a favorable 6-month survival of 73% compared with 57% in the control arm (p = .04) [87]. A phase III trial of this combination is ongoing (01746979).
Enhanced Drug Delivery to Microenvironment
Inefficient drug delivery might explain the lack of efficacy of systemic treatment. Novel drug delivery vehicles have reformed the clinical use of traditional cytotoxic agents. nab-Paclitaxel is an albumin bound formulation that increases tumor accumulation of paclitaxel via binding of albumin to the surrounding stroma rich in overexpression of secreted protein acidic and rich in cysteine (SPARC). In the animal model, intratumoral concentration of GEM was increased by 2.8-fold in mice receiving nab-paclitaxel/GEM combination versus GEM alone. In a recently published phase III study comparing nab-paclitaxel plus GEM versus GEM, the addition of nab-paclitaxel significant prolonged medial OS from 6.7 to 8.5 months, with a corresponding increase in response rate from 7% to 23% [3]. The nab-paclitaxel/GEM combination becomes the second regimen proven to be superior to GEM and has been approved by the FDA for treatment of APC. In a second-line setting, nab-paclitaxel monotherapy demonstrated preliminary evidence of clinical activity in GEM-refractory APC patients in a phase II trial [88]. nab-Paclitaxel and its combination with different agents is now one of the most popular areas of clinical research in APC. Another innovative approach to improve drug delivery using nanotechnology and cancer-specific liposomes [89] is under development.
Synthetic Lethality
Germline mutation in BRCA1 and BRCA2 has been reported in familiar cases of pancreatic cancer. It has also been suggested that the incidence of BRCA2 mutation in sporadic pancreatic cancer may be as high as that in breast or ovarian cancer [9]. Loss of heterozygosity at the BRCA1/2 locus leads to deficient DNA double-strand break repair. Proper function of BRCA1/2 also requires a gene called “partner and localizer of BRCA2” (PALB2). PALB2 binds directly to BRCA1 and serves a molecular scaffold in forming the BRCA1-PALB2-BRCA2 complex [90]. Defects in any of these components would lead to an unstable complex and impair DNA repairing function. Poly(ADP-ribose) polymerase (PARP) inhibitor targets the PARP and inhibits tumor cells from DNA repair, and in tumors that are already deficient in DNA repair, the damages so produced would not be compatible with cell survival. This action of synthetic lethality by PARP inhibitor has been attested by a proof-of-concept trial that showed initial encouraging results in using the PARP inhibitor olaparib (AZD2281) in BRCA2-deficient breast cancer patients who failed other traditional treatments [91] and set the stage for development of this class of agent in many solid tumors including APC. A number of early phase clinical trials are ongoing exploring the tolerability or clinical efficacy of PARP inhibitors in patients with APC. Among these agents are olaparib in combination with GEM (phase I, 00515866), BMN673 (phase II, 01989546), and veliparib alone or in combination with GEM/cisplatin (phase II, 01585805).
Immunotherapy
Tumor cells are capable of evading the patrol of immune system by tuning down effector activities or blinding the immune system from recognizing them. An effective immune response against tumor relies on fine orchestration of different components in both the innate and adaptive immune system. Immunotherapy in pancreatic cancer is a rapidly expanding field with many exciting breakthroughs in recent research. Many of these strategies set out to strengthen tumor detection and effector response. A few agents that have entered clinical development with early signs of antitumor activities will be discussed here.
Ras-Specific Immunotherapy
GI-4000 is whole, heat-killed recombinant Saccharomyces cerevisiae yeast that expresses mutated RAS proteins [92]. After administration, the yeast-expressed mutated Ras protein is digested into peptides for both major histocompatibility complex class I and II pathways of antigen presentation to produce a highly specific and potent T-cell response. The CD8 killer T cells are activated to provide systemic surveillance and to selectively eliminate tumor cells that express the mutated Ras. In preclinical testing using mice bearing Ras-mutated tumor, this immunotherapy demonstrated dose-dependent elimination of tumors [92]. This drug has been tested in phase I trial with a satisfactory safety profile [93]. A phase II trial studying the efficacy of GI-4000 plus adjuvant GEM (00300950) in preventing recurrence of pancreatic cancer after curative resection is underway [94]. Use of this agent in APC has not been planned.
Another RAS-specific immunotherapy is reovirus. Reovirus is an acronym for respiratory enteric orphan virus. These viruses are tumor-targeted replication-competent viruses with specificity for Ras-activated cells [95]. Early animal model study of pancreatic cancer showed intraperitoneal administration of this virus impeded peritoneal dissemination [96]. It has been demonstrated recently in human that the reovirus Reolysin processed a unique ability to evade the neutralizing effect of the innate immune system, transfect cancer cells, replicate in these cells, and then induce tumorcidal effect without affecting normal healthy tissue [97]. This agent is now being combined with chemotherapy in two phase II clinical trials for APC patients (00998322, 01280058).
Strategies Against Tumor Antigens
Much effort has been made to develop immunotherapy against tumor antigens. GV1001 vaccine, a reverse-transcriptase subunit of telomerase (human telomerase reverse transcriptase)-derived peptide, is a novel concept in immunotherapy. Telomerase is highly expressed in cancer cells and believed to play a crucial role in promoting tumor survival [98]. In the initial phase I/II study in patients with APC, the induction of immune response as measured by delayed-type hypersensitivity and T-cell proliferation was correlated with prolonged survival [99]. The phase III trial (TeloVac) is a large three-arm randomized controlled study comparing patients receiving GV1001 with GEM/capecitabine, in combination or sequentially, versus GEM/capecitabine alone. The results were announced at the ASCO meeting in June 2013 and disappointingly concluded that the combination therapy with this novel vaccine did not confer superior survival over background chemotherapy [100].
Nevertheless, another phase II study of vaccine therapy later that year announced encouraging results. This study combined two immunogenic agents, GVAX and CRS-207, in APC and suggested a synergistic effect [101]. GVAX is composed of pancreatic cancer cells that have been genetically modified to secrete granulocyte-macrophage colony-stimulating factor and is given with low dose cyclophosphamide (CY) to inhibit regulatory T cells. It can induce T-cell responses in mesothelin-expressing tumor like pancreatic cancer. CRS-207, on the other hand, is live-attenuated Listeria vaccines that express human mesothelin and can induce listeriolysin O and mesothelin-specific T-cell responses [102]. Patients with APC previously treated/refused prior chemotherapy were randomized to receive GVAX/CY only (arm B) or with sequential CRS-207 (arm A). Of all evaluated patients, SD was 34% in arm A vs. 19% in arm B, and OS was 6 months in arm A vs. 3.9 months in arm B (p = .0169). The study suggested that combining multiple immunotherapies could result in extended survival. It also showed that it might be possible to have an effective regimen in APC without using chemotherapy. The study met the criteria for early stopping for favorable results at primary analysis. A phase IIB trial (ECLIPSE) comparing this combination regimen with chemotherapy in APC patients previously treated is now recruiting (02004262).
Another tumor antigen is prostate stem cell antigen (PSCA), and interesting data on this topic have been presented. PSCA is a glycosyl-phosphatidylinositol-linked cell surface antigen and is expressed in prostate, pancreatic, bladder, and gastric cancers. AGS-1C4D4 is a fully human monoclonal antibody IgG1 against PSCA. Binding of AGS-1C4D4 to PSCA leads to complement-dependent cell lysis and antibody-dependent cell-mediated cytotoxicity in PSCA-expressing cells. In a phase II trial, AGS-1C4D4 plus GEM extended the 6-month survival from 44.4% to 60.9% compared with GEM alone in patients with metastatic pancreatic cancer [103]. The company is currently working on the potential use of PSCA expression as a biomarker to enrich further clinical trials.
Other Potential Immunotherapies
There are many potential targets for immunotherapy. (90)Y-clivatuzumab tetraxetan (hPAM4) antibody is an yttrium-90-labeled hPAM4 with demonstrated selectivity against pancreatic ductal carcinoma. When this radioimmunotherapy was combined with low-dose GEM in a phase I trial, observed median OS was 7.7 months in untreated patients [104]. The company recently announced the launch of a phase III trial (PANCRIT-1 [01956812]) in pretreated APC patients. It is the first-in-class radioimmunotherapy for APC. Beatty et al. [105] published an innovative approach of combining an agonist CD40 antibody with GEM in a small cohort of patients with APC and observe some tumor regressions. Preclinical study in a mouse model found that administration of CD40 agonist led to recruitment of macrophages, instead of T cells, and these cells rapidly infiltrated tumor stroma and became tumoricidal [106].
One of the most popular areas of study in immunotherapy nowadays is immune checkpoint inhibitor. T-cell activation is governed by inhibitory pathways mediated by receptors such as cytotoxic T-lymphocyte antigen 4 (CTLA4) or programmed cell death 1 (PD-1). The success of ipilimumab, a monoclonal antibody against CTLA4, in melanoma [107] has opened the door for this new concept of immunotherapy. Although in a phase 2 trial, single-agent ipilimumab failed to demonstrate appreciable antitumor activity in APC [108], the combination of ipilimumab with GEM is under phase I evaluation at the moment (01473940). PD-L1 is the ligand for PD-1. Likewise, in the phase I study of an anti-PD-L1 antibody nivolumab, 14 patients had APC, but none responded [109]. Preliminary data suggest a relationship between PD-L1 expression on tumor and objective response [110]. Anti-PD-1/L1 agent might be effective in a subset of APC patients, so the usefulness of this agent in pancreatic cancer is still uncertain.
Another new concept is adoptive cell transfer (ACT), which is a personalized approach to immunotherapy. In ACT, T cells are removed from the host tumor tissue, expanded ex vivo, manipulated, and then infused to the host. T cells can be manipulated by introduction of artificial T-cell receptors, also known as chimeric antigen receptors (CARs), which carry Fab fragments of an antibody that is specifically designed to recognize a tumor antigen of interest. By using ACT, multiple CARs can be engineered into T cells and probably increase treatment efficacy. This approach for treatment of pancreatic cancer is in the preclinical stage with preliminary success [111].
Personalized Medicine
Despite the marginal benefit of GEM, it remains the key player in the treatment of APC. A substantial body of research suggesting identification of predictive biomarkers might help design a more effective approach. For instance, deoxycytidine kinase and ribonucleoside reductases M1 and M2 (RRM1 and RRM2) are GEM metabolism gene products, and decreased levels were correlated with resistance to GEM [112–114]. The nucleoside transporter-1, hENT1, is another potential biomarker and it plays an important role in uptake of GEM in cells. Among APC patients treated with GEM, those with low hENT1 expression had significantly poorer prognosis than those with hENT1 expression [113]. CO-101 is a lipid drug conjugate of GEM designed to enter tumor cells independent of hENT1 expression and is expected to overcome GEM resistance. Nevertheless, in a recently published randomized phase II study of CO101 versus GEM by Poplin et al. [115] involving >300 APC patients, there was no difference in OS between the two treatments in the low hENT1 subgroup, which did not support the significance of hENT1 in predicting treatment outcome. This study dichotomized the hENT1 expression level of metastatic tissue into high/low using a cutoff parameter derived from primary pancreatic cancer samples from the adjuvant trial RTOG-9704 [116]. It fell short of having a validated hENT1 cutoff value for metastatic cases, thus making the interpretation of results controversial. In fact, the current definition of hENT1 expression level is arbitrary and without standardization. In the original biomarker study of RTOG-9704 cohort, a murine monoclonal anti-hENT antibody and a different scoring system was used [116]. High hENT1 expression was defined as strong reactivity in >50% of neoplastic cells, no hENT1 expression meant no staining in >50% of cells, and low hENT1 expression were cases in between. In Poplin's study [115], a rabbit monoclonal anti-hENT1 antibody was used. High hENT1 expression was defined as unequivocal membranous staining in >50% of tumor cells, and all other cases were defined as hENT1 low expression. Furthermore, another biomarker study exploring the role of hENT1 levels in predicting survival in pancreatic patients after adjuvant chemotherapy used a mouse monoclonal anti-hENT1 antibody but classified high and low hENT1 expression by the median H score of the same study [117]. A standardized immunohistochemical study protocol and a validated scoring system using a metastatic cohort are required to further develop the role of hENT1 in predicting response to GEM.
A few other potential biomarkers have been suggested by correlative studies. In a crossover phase III study, APC patients with K-ras wild-type tumors apparently derived better survival benefits from erlotinib than patients with K-ras mutant tumors [118]. In the phase I/II clinical trial of nab-paclitaxel plus GEM, patients with SPARC staining-positive tumors were reported to have significantly better ORR and PFS than patients with negative staining. In the phase I/II study evaluating IGF-1R antibody MK-0646 with GEM and erlotinib in APC, patients with IGF-1R expressed tumor were associated with more disease control [119]. Although preliminary, these findings well illustrate the possibility of a biomarker-driven approach in the systemic management of APC.
Conclusion
The development of molecular targeted therapy in treating APC patients is rather slow when compared with other solid cancers. Many agents failed the test during phase III trials despite scientifically plausible mechanisms of action and encouraging results in early phase clinical trials. We now understand that pancreatic cancer is a heterogeneous disease. Drug resistance is likely to be due to a multitude of compensatory pathways and complex crosstalk within the signaling network. Future treatment direction might encompass combinations of multiple targeted agents to achieve parallel or vertical blockade. Novel approaches using the knowledge in microenvironment and using immunotherapy may bypass the limitation of agents with single-site activity within particular pathways.
Chemotherapy remains the mainstream of treatment in APC. Most clinical trials comprise a study drug plus GEM as backbone. This approach is logical but fails to take into account the complex biology of this cancer. Many new agents are in development. As experience in the use of these agents in combination grows, more flexible trial design is anticipated. The traditional receipt of GEM-based study also leads to another perplexing question about the choice of second-line treatment. Most current second-line studies use a 5FU-based regimen. After the publication of PRODIGE 4/ACCORD11 trial, FOLFIRINOX has become the first-line treatment when feasible. The efficacy of GEM after failure of FOLFIRINOX is not known. The question is further complicated with the release of MPACT study. Although numerically FOLFIRNOX appears to give longer survival than the GEM/nab-paclitaxel combination, they have different toxicity profile and consist of different cytotoxic agents, and there probably will never be a head-to-head comparison between the two regimens. Because some patients might respond to one regimen but not the other, and most pancreatic cancer patients deteriorate quickly after first-line treatment, it is important to choose an effective and appropriate treatment in the first place. Recently an ongoing prospective pharmacogenomics (PG) study at Memorial Sloan-Kettering Cancer Center might help tackle this problem [120]. In this innovative trial, total RNA is extracted from circulating tumor cells taken from patients’ blood. The RNA is subjected to a PG model that predicts sensitivity to cytotoxic agents commonly used in APC. Preliminary results showed that patients who received treatment predicted to be effective had a longer time to progression than patients who received treatment predicted to be ineffective. Furthermore, Hedgehog pathway overexpression appeared to associate with resistance to GEM but clinical response to 5FU. This innovative approach opens the door to a new paradigm of personalized medicine. This approach might be applicable to targeted therapy in the future.
In light of research in biomarker and genetic profiling, future personalized medicine in APC might comprise a repertoire of treatment strategies. Comprehensive genomic assessment of tumors may involve DNA profiling for pathway defects, study of protein expression, or even PG signature. Upcoming research in epigenetic and RNA sequencing will further unravel the mechanism behind treatment resistance. Research in pancreatic cancer is challenging, but we have seen major progress and many breakthroughs in the last few years. Promising new strategies are on the way.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Author Contributions
Conception/Design: Thomas Yau, Joanne W. Chiu, Hilda Wong, Tan-To Cheung
Provision of study material or patients: Thomas Yau, Tan-To Cheung, Sheung-Tat Fan, Ronnie Poon
Collection and/or assembly of data: Thomas Yau, Joanne W. Chiu, Hilda Wong, Roland Leung, Roberta Pang
Data analysis and interpretation: Thomas Yau, Joanne W. Chiu, Hilda Wong, Sheung-Tat Fan, Ronnie Poon
Manuscript writing: Thomas Yau, Joanne W. Chiu, Hilda Wong, Roland Leung, Roberta Pang, Tan-To Cheung, Sheung-Tat Fan, Ronnie Poon
Final approval of manuscript: Thomas Yau, Joanne W. Chiu, Hilda Wong, Roland Leung, Roberta Pang, Tan-To Cheung, Sheung-Tat Fan, Ronnie Poon
Disclosures
The authors indicated no financial relationships.
For Further Reading:Kian-Huat Lim, Eugene Chung, Adeel Khan, Dengfeng Cao et al. Neoadjuvant Therapy of Pancreatic Cancer: The Emerging Paradigm? The Oncologist 2012;17:192–200.
Abstract:Pancreatic cancer remains one of the deadliest cancers due to difficulty in early diagnosis and its high resistance to chemotherapy and radiation. It is now clear that even patients with potentially resectable disease require multimodality treatment including chemotherapy and/or radiation to improve resectability and reduce recurrence. Tremendous efforts are currently being invested in refining preoperative staging to identify optimal surgical candidates, and also in developing various neoadjuvant or adjuvant regimens to improve surgical outcome. Although at present no studies have been done to directly compare the benefit of neoadjuvant versus adjuvant approaches, accumulating evidence suggests that the neoadjuvant approach is probably beneficial for a subset of the patient population, particularly those with borderline resectable disease in which complete surgical resection is almost certainly unachievable. In this article, we review the literature and rationales of neoadjuvant chemotherapy and chemoradiation, as well as their potential limitations and caveats. We also review the pathological findings following neoadjuvant therapies, and potential surgical complications that may be associated with neoadjuvant therapies.
References
Author notes
Disclosures of potential conflicts of interest may be found at the end of this article.

{kind=link}