





Abstract
Vascular‐disrupting agents (VDAs) represent a new class of chemotherapeutic agent that targets the existing vasculature in solid tumors. Preclinical and early‐phase trials have demonstrated the promising therapeutic benefits of VDAs but have also uncovered a distinctive toxicity profile highlighted by cardiovascular events.
We reviewed all preclinical and prospective phase I–III clinical trials published up to August 2010 in MEDLINE and the American Association of Cancer Research and American Society of Clinical Oncology meeting abstracts of small‐molecule VDAs, including combretastatin A4 phosphate (CA4P), combretastatin A1 phosphate (CA1P), MPC‐6827, ZD6126, AVE8062, and ASA404.
Phase I and II studies of CA1P, ASA404, MPC‐6827, and CA4P all reported cardiovascular toxicities, with the most common cardiac events being National Cancer Institute Common Toxicity Criteria (version 3) grade 1–3 hypertension, tachyarrhythmias and bradyarrhythmias, atrial fibrillation, and myocardial infarction. Cardiac events were dose‐limiting toxicities in phase I trials with VDA monotherapy and combination therapy.
Early‐phase trials of VDAs have revealed a cardiovascular toxicity profile similar to that of their vascular‐targeting counterparts, the angiogenesis inhibitors. As these agents are added to the mainstream chemotherapeutic arsenal, careful identification of baseline cardiovascular risk factors would seem to be a prudent strategy. Close collaboration with cardiology colleagues for early indicators of serious cardiac adverse events will likely minimize toxicity while optimizing the therapeutic potential of VDAs and ultimately enhancing patient outcomes.
摘要
背景血管阻断剂(VDAs)是一类以实体瘤现存血管结构为治疗靶点的新型化疗药物,临床前和早期临床研究已显示VDAs的治疗前景,但同时也发现了其特有的毒性谱,以心血管不良事件尤为明显。
方法综述2010年8月前发表并收录于MEDLINE,或是发表于美国癌症研究学会(AACR)及美国临床肿瘤学会(ASCO)的相关会议摘要中,所有小分子VDAs相关的临床前与前瞻性I~III期临床研究结果,VDAs包括考布他汀A4磷酸盐(CA4P)、考布他汀A1磷酸盐(CA1P)、MPC‐6827、ZD6126、AVE8062和ASA404。
结果CA1P、ASA404、MPC‐6827和CA4P的I、II期研究均报告了心血管毒性,最常见的心脏事件为国立癌症研究院常见毒性标准(NCIC‐CTC,第3版)界定的1~3级高血压、心动过速、心动过缓、房颤和心肌梗死。I期研究中,心脏事件是VDA单药治疗和联合治疗的剂量限制性毒性。
结论早期临床研究结果显示,VDAs的心血管毒性谱与其他靶向血管生成的抑制剂相似。由于这些药物多与主流化疗方案联合使用,因此仔细确认基线心血管危险因素是谨慎的策略。与心脏科同行密切合作,监控心脏严重不良事件的早期指标,有可能在获得VDAs最佳治疗作用的同时,使毒性最小化,并最终改善患者转归。
Introduction
Recently, there has been increasing interest in developing anticancer agents that target the tumor vasculature. Anti–vascular endothelial growth factor (anti‐VEGF) agents are being extensively investigated, and several have been approved by the U.S. Food and Drug Administration for the treatment of various tumor types. The next frontier in vasculature‐targeted drug development is the advent of tumor vascular‐disrupting agents (VDAs), which selectively disrupt existing tumor vasculature with minimal impact on the inhibition of angiogenesis or normal tissue vasculature [1].
The antitumor activity of VDAs is based on the concept that tumor vasculature and normal vessels have fundamental structural differences. In contrast to normal vessels, tumor‐related vasculature is characterized by a high proliferation rate, resulting in fragile, thin vessels with minimal innervation and smooth muscle reinforcement, as evidenced by the relative paucity of pericytes, making this type of vasculature a target for disruption [2–4]. As a result, VDAs target newly formed vasculature, resulting in small vessel destruction in the core of tumor tissue, but angiogenesis mechanisms remain intact on the border zones with the normal tissue. This rim of tumor tissue supplied by the latent vasculature of the surrounding noncancerous cells remains intact despite VDA therapy and leads to robust reactive angiogenesis and tumor regrowth after VDA exposure. This process underscores the shortcoming of VDA monotherapy and the necessity for combination regimens using VDAs [5].
Early‐phase clinical trials demonstrated that VDAs have significant antitumor activity in advanced and refractory solid tumors (Table 1). Treatment was generally well tolerated, but certain patients experienced cardiovascular events ranging from asymptomatic Common Terminology Criteria for Adverse Events (version 3) grade 1–2 hypertension to myocardial infarction (MI).
Vascular‐disrupting agents in clinical trials
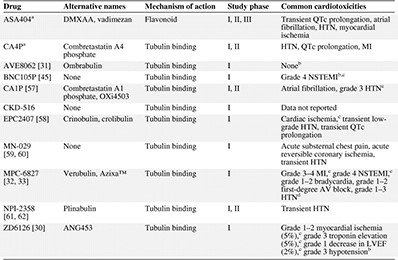
aASA404 and CA4P are the more extensively studied VDAs.
bGrade 1–2 events not reported.
cClassified as a dose‐limiting toxicity.
dGrade 5 toxicity resulting from grade 3 cerebral hemorrhage and grade 3 hypertension.
Abbreviations: AV, atrioventricular; HTN, hypertension; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NSTEMI, non‐ST‐elevation myocardial infarction.
Vascular‐disrupting agents in clinical trials
aASA404 and CA4P are the more extensively studied VDAs.
bGrade 1–2 events not reported.
cClassified as a dose‐limiting toxicity.
dGrade 5 toxicity resulting from grade 3 cerebral hemorrhage and grade 3 hypertension.
Abbreviations: AV, atrioventricular; HTN, hypertension; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NSTEMI, non‐ST‐elevation myocardial infarction.
Therefore, this review focuses on the cardiovascular adverse events noted in clinical trials of VDAs and their potential ramifications. The goal is to increase awareness of these cardiovascular toxicities associated with VDA therapy and to emphasize the importance of patient selection and the need for close cardiac monitoring of patients treated with VDAs. Strategies for the prevention and optimal therapeutic management of these complications are also discussed.
Methods
We searched MEDLINE and American Association for Cancer Research and American Society of Clinical Oncology meeting abstracts for preclinical and prospective phase I–III clinical trials of any small molecule VDA published before or during August 2010 using, as search terms, “vascular disrupting agents,” “VDA,” and each VDA separately.
Mechanisms of Action of VDAs
The small molecule tumor VDAs fall into two general classes: (a) tubulin‐binding molecules that target the highly dynamic tubulin heterodimers and (b) flavonoids, which purportedly increase cytokine activity in the tumor microenvironment [6–8]. Although these two classes have differing mechanisms of action, both ultimately lead to collapse of the vascular cytoskeleton, with subsequent cessation of blood flow, tumor hypoxia, and hemorrhagic necrosis (Fig. 1).
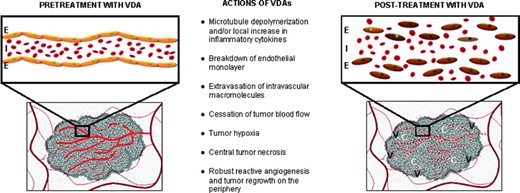
Purported mechanism of action of tumor VDAs: Disruption of the newly formed tumor vasculature renders them nonfunctional and results in a viable rim of tumor cells supplied by the nearby nontumor normal vasculature.
Abbreviations: C, central necrosis; E, endothelial monolayer; I, intravascular macromolecules; V, viable tumor rim with robust regrowth and angiogenesis; VDA, vascular disrupting agent.
Combretastatin A4 phosphate (CA4P, fosbretabulin tromethamine, Zybrestat™; OXiGENE, San Francisco, CA), the first VDA to be studied in clinical trials, induces microtubule disruption by targeting the colchicine‐binding sites on the β‐tubulin subunits of newly formed endothelial cells in the highly proliferative tumor environment [9]. These neovascular endothelial cells then separate from the vessel wall, disrupting tumor blood flow and producing central hemorrhagic necrosis in the tumor core. The composite in vivo findings most closely resemble an acute inflammatory condition; the number of visible blood vessels dramatically decreases and tumor blood flow drops precipitously to 5% of the baseline value within 1 hour of treatment with CA4P. Vascular permeability increases measurably, suggesting breakdown of the endothelial monolayer and a consequent leakage of macromolecules [10]. Because CA4P is the most tested small molecule VDA, its proposed mechanism can be credibly applied to the whole class of microtubule‐de‐polymerizing VDAs, including combretastatin A1 phosphate (CA1P, OXi4503; OXiGENE, San Francisco, CA), ZD6126 (AstraZeneca, Wilmington, DE), AVE8062 (ombrabulin, AC‐7700; Sanofi Aventis, Bridgewater Township, NJ), and MPC‐6827 (Azixa™, verubulin; Myrexis, Salt Lake City, UT) [10]. CA1P is a second‐generation small molecule VDA that attaches to the colchicine‐binding site, producing tubulin destabilization. CA1P is more potent than its earlier analog, CA4P, resulting not only from its vascular‐disrupting activity but also from its free radical–mediated tumor cell death [11].
In contrast to the microtubule‐destabilizing VDAs, the flavonoid VDAs, which are potent derivatives of flavone acetic acid, act by extensively increasing cytokine concentrations in the tumor microenvironment [12]. For instance, the flavonoid ASA404 (vadimezan, DMXAA, AS1404; Novartis, Cambridge, MA) promotes the production of proinflammatory vasoactive molecules, including tumor necrosis factor α, monocyte chemoattractant protein 1, macrophage inflammatory protein 1α, chemokine C‐C motif ligand 5, interleukin 6, interferon inducible protein 10, keratinocyte‐derived chemokine, and G‐CSF [13, 14]. Cytokine production from upstream signaling pathways and activation of nuclear factor κB (NF‐κB) in murine splenocyte cultures treated with DMXAA suggest that NF‐κB may be involved in a mechanism of post‐translational regulation [13, 15]. NF‐κB has been independently shown to alter the cytoskeletal structure of vascular endothelial cells, signifying yet another VDA‐induced cytotoxic event ultimately leading to endothelial cell apoptosis [16].
Overall, in vitro studies in endothelial cell lines using VDAs show widespread induction of caspase 3 activity upon exposure to a VDA, resulting in prompt termination of proliferation and widespread apoptosis of targeted cells [17]. Additional immunohistochemical markers, such as cytosolic release of cytochrome C and increased intracellular ceramide and carbonic anhydrase‐9 levels, all confirm mitochondrial compromise and tumor hypoxia as a result of VDA treatment, ultimately leading to tumor cell death [17].
Preclinical Models
Preclinical studies report a transient favorable response to VDAs when used as single agents but note robust tumor regrowth within days after VDA treatment. Xenograft models treated with a VDA in combination with chemotherapy or radiation show longer delays in disease progression. A significant delay in tumor regrowth was demonstrated in human ovarian OVCAR‐3 carcinoma xenografts in nude mice treated with MPC‐6827 and carboplatin in comparison with either agent alone, suggesting a synergistic therapeutic mechanism [18]. Similarly, mouse models of human non‐small cell lung carcinoma (NSCLC) given ASA404, paclitaxel, and carboplatin demonstrated better tumor growth inhibition than with paclitaxel and carboplatin alone as well as a significantly slower increase in tumor volume (63.5 days, versus 24.8 days in controls) [19]. Furthermore, an additional cycle of ASA404 alone after completion of the triple regimen of paclitaxel, carboplatin, and ASA404 resulted in further inhibition of tumor growth, with observations of tumor regression [19]. Additional studies in lung cancer xenografts treated with ASA404 with cisplatin/carboplatin or gemcitabine suggest that the combination of a VDA and classic chemotherapy is synergistic and necessary [12].
Tumor tissue extracted from mice treated with ASA404 showed significantly higher VEGF‐A mRNA expression, indicating the possibility that the tissue hypoxia produced by ASA404 activity acts as a positive feedback loop for greater VEGF production and activity [20]. The recruitment of circulating endothelial cells to VDA‐affected regions and the ensuing reactive angiogenesis plausibly account for treatment failure with VDA monotherapy and suggest that an angiogenesis inhibitor should be added to any VDA‐based treatment regimen [21]. Mouse models of primary human acute myelogenous leukemia and renal cell carcinoma exhibited significant tumor growth delay when CA1P was used in combination with the anti‐VEGF agent bevacizumab [22, 23]. Similarly, treatment with radiation and ZD6126 in a rodent KHT sarcoma model and human U87 glioblastoma mouse model showed a longer delay in the regrowth of the target lesions than with treatment with either modality alone [24]. Cardiotoxicities were not noted in early preclinical studies.
Clinical Trials
Data from early‐phase clinical trials of tubulin‐destabilizing VDAs and flavonoids indicate a toxicity profile marked by cardiovascular events (Table 2).
Reported vascular‐disrupting agent–associated cardiotoxicities

Reported vascular‐disrupting agent–associated cardiotoxicities
Cardiotoxicities in Phase I Clinical Trials
CA4P was the first VDA investigated in a phase I clinical trial in patients with advanced solid tumors [25]. Trials of CA4P monotherapy specified varying exclusion criteria for pre‐existing cardiac disease, ranging from no reported heart disease to a normal electrocardiogram (ECG) or exclusion of all patients with prior evidence of ischemic heart disease [25, 26]. Overall, the most commonly reported cardiac events were grade 1–2 atypical chest pain without corresponding evidence of ischemia on ECG and grade 1–2 tachycardia, hypertension, and QTc prolongation (Table 3). In a phase I study of 25 patients with advanced solid tumors and normal pre‐enrollment 12‐lead ECGs, two of the four dose‐limiting toxicities (DLTs) were cardiac events, specifically, acute nonfatal MIs requiring cardiac catheterization, both of which occurred in patients who received higher doses of CA4P (>60 mg/m2) [2, 25]. The first patient, who had no risk factors for cardiac disease, developed angina with ischemic changes on ECG and underwent a coronary angiogram that revealed minimal nonocclusive coronary artery disease (CAD). ECG changes resolved to baseline in the postprocedure period, and cardiac troponins were never elevated. The second cardiac DLT occurred in an asymptomatic patient with significant cardiac risk factors in whom a routine ECG noted acute ST‐T ischemic changes with concurrent cardiac troponin I elevation. Left heart catheterization confirmed the presence of occlusive CAD, which was treated with angioplasty [21].
Cardiotoxicities of CA4P
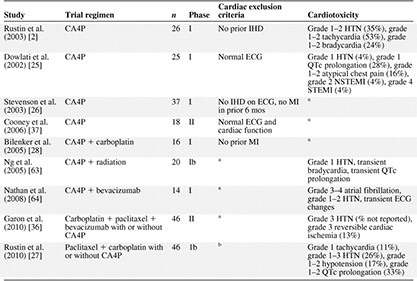
aNone reported.
bCardiac exclusion criteria not reported but HTN present in 22% and IHD in 2% of patients.
Abbreviations: CA4P, combretastatin A4 phosphate; ECG, electrocardiogram; HTN, hypertension; IHD, ischemic heart disease; MI, myocardial infarction; NSTEMI, non‐ST‐elevation myocardial infarction; STEMI, ST‐elevation myocardial infarction.
Cardiotoxicities of CA4P
aNone reported.
bCardiac exclusion criteria not reported but HTN present in 22% and IHD in 2% of patients.
Abbreviations: CA4P, combretastatin A4 phosphate; ECG, electrocardiogram; HTN, hypertension; IHD, ischemic heart disease; MI, myocardial infarction; NSTEMI, non‐ST‐elevation myocardial infarction; STEMI, ST‐elevation myocardial infarction.
Similar cardiovascular events were also reported when CA4P was administered in combination with cytotoxic agents. A phase 1b trial testing CA4P with paclitaxel and/or carboplatin for advanced refractory solid tumors assigned 46 patients into three treatment cohorts (arm 1, CA4P and carboplatin; arm 2, CA4P and paclitaxel; and arm 3, CA4P, carboplatin, and paclitaxel) [27]. Patients with prior MI, angina, QTc prolongation, arrhythmias, poorly controlled hypertension, peripheral vascular disease, or radiation‐induced vascular injury were excluded. This study demonstrated that 12 of 46 patients (26%) developed grade 1–3 hypertension. Of these 12 patients, eight had pre‐existing hypertension. Four of 34 patients (11%) without a history of hypertension had an episode of hypertension during the study [27]. Asymptomatic grade 3 hypertension in one patient treated at the highest dose of CA4P (72 mg/m2) was one of two DLTs in that trial and required the administration of antihypertensive medication [27]. The observed higher incidence of hypertension with dose escalation of CA4P prompted the addition of a prophylactic calcium channel blocker in an ongoing trial of CA4P's newer derivative CA1P [27]. Grade 1–2 QTc prolongation of >450 ms was noted in 33% of patients (15 of 46), grade 1–2 hypotension was noted in 17% of patients (eight of 46), and grade 1 tachycardia was noted in 11% of patients (five of 46). Grade 3 ataxia was the other DLT in that trial.
Like its predecessor CA4P, CA1P has completed early‐phase trials that have revealed a similar cardiotoxicity profile, with reports of dose‐limiting supraventricular arrhythmias and high‐grade hypertension [11, 28].
Another tubulin‐targeting VDA, ZD6126, completed phase I trials in 2008; findings revealed significant dose‐limiting cardiotoxicities occurring in five of 44 patients (11%), including myocardial ischemia, elevated troponins, and decreased left ventricular ejection fraction (LVEF), ultimately leading to the termination of this drug's development [29, 30].
Newer combretastatin derivatives may be less cardiotoxic, as evidenced by results of their early‐phase clinical trials. One phase I trial combined AVE8062, a synthetic CA4P derivative, with docetaxel in patients with advanced solid tumors [3, 31]. The DLTs were noncardiac (grade 3 asthenia and headache), with no reported high‐grade (grades 3–4) ischemic, arrhythmic, or hemodynamic events [31].
Another microtubule‐destabilizing VDA, MPC‐6827, was tested in a phase I dose‐escalation study of 48 patients with heavily pretreated advanced solid tumors. Grade 1–2 hypertension occurred in 21% of patients (n = 10), with two reports of nonfatal grade 3 MI and three patients with grade 1–2 first‐degree atrioventricular block and bradycardia [32]. These findings were echoed in another phase I trial of MPC‐6827 with temozolomide in patients with metastatic melanoma [33]. That study excluded patients with unstable angina, prior MI, or an abnormal troponin I level. Of 12 patients treated at the highest dose level (3.3 mg/m2), one experienced grade 3 hypertension and grade 3 cerebral hemorrhage, which were subsequently fatal (grade 5). One patient in the low‐dose treatment arm (2.1 mg/m2) experienced a grade 4 nonfatal MI. These results suggest that patients with underlying cerebrovascular disease should be excluded from treatment with MPC‐6827 in future clinical trials [33].
Flavonoid VDAs share a cardiovascular toxicity profile with their tubulin‐depolymerizing counterparts. ASA404, the most tested flavonoid VDA, disrupts tumor vascular endothelial cells by activating inflammatory cytokines within the tumor microenvironment, resulting in extensive necrosis of the tumor core (Table 4). A phase I trial in 63 patients receiving ASA404 monotherapy demonstrated asymptomatic QTc prolongation in 21% of patients, and a second phase I study reported low‐grade hemodynamic toxicities, including grade I hypertension and hypotension in nine (20%) and six (13%) patients, respectively [34, 35].
Cardiotoxicity of vadimezan (ASA404; DMXAA)
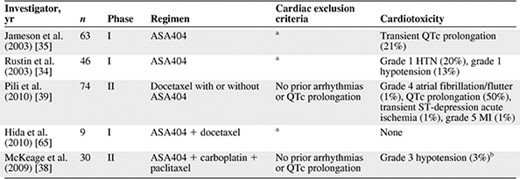
aCardiac history not reported but patients with “serious medical conditions” were excluded.
bGrade 1–2 toxicities not reported; no reported grade 3–4 arrhythmias or cardiac ischemia.
Abbreviations: HTN, hypertension; MI, myocardial infarction.
Cardiotoxicity of vadimezan (ASA404; DMXAA)
aCardiac history not reported but patients with “serious medical conditions” were excluded.
bGrade 1–2 toxicities not reported; no reported grade 3–4 arrhythmias or cardiac ischemia.
Abbreviations: HTN, hypertension; MI, myocardial infarction.
Cardiotoxicities in Phase II Clinical Trials
Recent trials combine a VDA with a cytotoxic (e.g., taxanes) or cytostatic (e.g., VEGF inhibitors) agent to reproduce the therapeutic synergy reported in preclinical models. Trials combining a VDA with an angiogenesis inhibitor have progressed with caution, given the projected aggregative cardiotoxicity. A randomized phase II study of carboplatin, paclitaxel, and bevacizumab with CA4P (treatment cohort) and without CA4P (control) in previously untreated stage IIIb/IV NSCLC patients reported no significant toxicities attributable to the addition of CA4P. Patients receiving CA4P had comparable progression‐free survival and response rates but had a higher overall survival rate [36]. The reported adverse events included grade 3 hypertension (also noted at a comparable rate in the control cohort) and three cases of grade 3 reversible cardiac ischemia not requiring hospitalization [36].
Contrasting findings were reported in a phase IIb study of CA4P monotherapy for metastatic anaplastic thyroid carcinoma [37]. That study administered a modest dose of CA4P (45 mg/m2), shown to cause limited adverse events in earlier trials, and excluded patients with cardiovascular disease (as evidenced by an abnormal ECG or cardiac function) and those who had received prior systemic chemotherapy for metastatic disease. Preliminary data from that trial reveal no high‐grade cardiovascular events, suggesting that careful patient selection may limit VDA‐induced cardiovascular toxicity [37].
In a phase II study of ASA404 in patients with NSCLC, all 23 patients in the ASA404 treatment arm experienced transient QTc prolongation in the peri‐infusional period [38]. This short‐lived ECG finding was also noted in a second phase II trial in castration‐refractory metastatic prostate cancer (CRMPC) patients, but it returned to baseline within 4–6 hours postinfusion [39]. Despite the predilection of QTc prolongations to advance to more malignant arrhythmias, there are no reports of ventricular tachyarrhythmias in published early‐phase trials of VDAs. However, the CRMPC trial, which did exclude patients with significant arrhythmias or heart rate–corrected QTc prolongation, reported four cases of supraventricular tachycardia and myocardial ischemia and MI in the ASA404 treatment arm, leading to three patient withdrawals and one fatality [39].
Cardiotoxicities in Phase III Clinical Trials
The first randomized, double‐blind, placebo‐controlled phase III trial of VDAs combined carboplatin and paclitaxel with or without ASA404 as first‐line treatment for patients with locally advanced or metastatic NSCLC (the Antivascular Targeted Therapy: Researching ASA404 in Cancer Treatment [ATTRACT] 1 study). An interim analysis revealed no survival benefit conferred to patients in the investigational arm in comparison with the standard therapy arm, leading to the termination of the study [40].
In that trial's successor, ATTRACT‐2 (ClinicalTrials.gov Identifier, NCT00738387), an ongoing randomized, double‐blind phase III trial, patients with locally advanced or metastatic (stage IIIb/IV) NSCLC are being randomized to receive docetaxel with or without ASA404 as second‐line therapy. Patients with uncontrolled hypertension or baseline prolonged QTc are excluded, highlighting the importance of careful patient selection for potentially cardiotoxic therapy. The adverse event profile from this ongoing trial will be essential in establishing an acceptable therapeutic index for this class of antineoplastics.
Pathophysiology of VDA‐Associated Cardiovascular Complications
Although the exact mechanisms of VDA‐associated cardiotoxicity have yet to be unequivocally clarified, certain overarching principles account for the adverse cardiac events reported.
Pre‐existing Hypertension and Ischemic Heart Disease
In clinical studies of VDAs, the presence of a pre‐existing cardiac diagnosis (defined as hypertension or undiagnosed CAD) predisposed patients to a study drug–related exacerbation, a finding further supported by the absence of high‐grade cardiovascular events in studies in which patients with abnormal ECGs and cardiac function were excluded [27, 37]. Furthermore, the presence of previously undiagnosed occlusive CAD appears to be unmasked by systemic exposure to VDAs [25].
Increased Systemic Vascular Resistance
VDA‐induced adverse events are partly attributable to postinfusion alterations to systemic vascular resistance, with compensatory systemic responses of cardiac variables (particularly blood pressure and heart rate) [25]. The observed hypertension with escalating doses produces transient bradycardia with a concomitant drop in cardiac output, accounting for the commonly noted grade 1–2 toxicities associated with the hemodynamic parameters of pulse and blood pressure [2]. These clinical findings confirm the preclinical studies, which show a systemic increase in vascular resistance after CA4P administration, not only in the tumor microenvironment but also in nontumor vascular beds such as the heart and kidneys, with subsequent compensatory hypertension and bradycardia [41]. These changes in pulse and blood pressure frequently resolve to pretreatment levels in the postinfusional period.
Transient Vasospasm
A parallel mechanism of cardiotoxicity suggests that exposure to a systemic VDA produces transient vasospastic alterations in small vessel vascular beds, including coronaries and the gastrointestinal tract (leading to the oft‐reported diffuse abdominal cramping) [25]. This proposed mechanism is further supported by the case of a patient without cardiac risk factors or known structural heart disease who experienced myocardial ischemia; subsequent cardiac catheterization showed no evidence of occlusive coronary disease, suggesting drug‐induced vasospasm [25]. With regard to coronary syndromes, vasospasm has not been implicated as the only causative mechanism. The most plausible explanation for an acute coronary syndrome in patients undergoing VDA therapy is a ruptured atherosclerotic plaque that results in transient ischemia (as is common in the general population).
Drug‐Induced Effect on Cardiac Repolarization
The mechanism of VDA‐induced alteration in duration of ventricular depolarization and repolarization, which manifests as a prolongation of the QTc interval, remains to be defined. Similar effects on the cardiac conduction system have been reported with other potent vascular‐targeting agents such as the tyrosine kinase inhibitors sunitinib, dasatinib, and nilotinib, for which the incidence of QTc prolongation correlates with dose dependency, greater frequency of occurrence at night time, electrolyte imbalances, female gender, and concurrent medications [42–44]. Dose reduction and avoidance of concurrent therapy with cytochrome P450 3A4 inhibitors that prolong the QTc interval, including macrolides and azole antifungals, may be prudent.
Strategies for Prevention of Cardiotoxicity
Systemic VDAs with Selective Activation by the Tumor
Preclinical studies in tumor‐bearing mice have investigated one unique approach to minimizing the systemic toxicities of VDAs—creating a novel VDA, ICT2588 (Incanthera Ltd, West Yorkshire, U.K.), that is selectively cleaved to its active metabolite by a matrix metalloproteinase expressed only in tumor tissue [21]. In mouse xenografts with fibrosarcoma model HT1080 tumor implants, combination therapy with ICT2588 and doxorubicin produced a significantly longer delay in tumor growth than with either agent alone. Additionally, the cardiac tissue of the treated mice demonstrated no detectable levels of the active metabolite of ICT2588, suggesting a lower likelihood of cardiovascular complications [21]. Such targeted activation of VDA selectively in the tumor environment mitigates the cardiotoxicity of combination therapies coupling a VDA and another cardiotoxic agent.
Increasing VDA Specificity for Activated Endothelial Cells
BNC105P (Bionomics, Ltd., Thebarton, Australia) and its dephosphorylated active metabolite BNC105 are novel tubulin‐depolymerizing small molecule VDAs. In vitro studies show an 80‐fold higher specificity for rapidly proliferating stimulated endothelial cells [45–47]. In the first phase I trial of BNC105P monotherapy in patients with advanced solid tumors, dynamic contrast‐enhanced magnetic resonance imaging evaluation of pharmacodynamic activity demonstrated less tumor blood flow and greater microtubule depolymerization, confirming drug activity at the targeted site. Of 21 enrolled patients, one in the highest dosing cohort had a grade 4 nonfatal MI, a DLT [45].
A Single Agent with Dual Roles
Given that the fundamental flaw of VDA monotherapy is the remnant rim of viable tumor tissue in the periphery of the mass, the development of novel VDAs has veered toward molecules that not only demonstrate vascular‐disrupting activity but also possess antiangiogenic properties.
One such agent with dual mechanisms of action undergoing preclinical studies is BPR0L075, a structural analogue of CA4P. In vitro evaluations in rat aorta endothelial cells show the disruption of capillary formation with BPR0L075 treatment, and in vivo inhibition of VEGF‐associated angiogenesis was seen in mice models [48]. Murine xenografts of human solid tumors from the lower intestine, lung, and cervix have demonstrated significant growth suppression with BPR0L075 monotherapy [49]. Additionally, a more robust tumor response to BPR0L075 was demonstrated when it was used in combination with cisplatin, suggesting a synergistic effect [48].
Another novel VDA in preclinical studies, VB‐111 (VBL Therapeutics, Or Yehuda, Israel), also has demonstrated vascular‐disrupting activity with antiangiogenic properties [50]. VB‐111 is constructed with a nonreplicating adenovirus vector together with a modified murine endothelial angiogenic‐specific promoter, a proprietary vascular targeting system (VTS™), and a proapoptotic Fas and tumor necrosis factor Chimera c transgene [50, 51]. Preclinical testing in a highly vascular malignancy such as glioblastoma showed a significant decrease in tumor size and longer survival. A phase I study of 27 patients with advanced refractory solid malignancies showed good patient tolerance to VB‐111 monotherapy, without any DLTs [50, 51]. Constructing condition‐specific viral vector–based molecules for vascular targeting holds significant promise for minimizing systemic VDA‐induced toxicities to nontumor vascular beds.
Close Surveillance for Cardiotoxicity
Careful selection of patients for treatment with VDAs and close surveillance for the earliest signs of cardiotoxicity during the post‐treatment period are the hallmarks of patient safety (Table 5). Optimization of surveillance for cardiac adverse events has been explored in trials with other classes of chemotherapeutic agents with significant cardiotoxicity, such as human epidermal growth factor receptor 2–positive breast cancer treated with the monoclonal antibody trastuzumab [52]. In searching for biomarkers to identify the earliest signs of cardiotoxicity, the study investigators measured cardiac troponin I levels prior to and after every infusion of trastuzumab, with concurrent measurement of left ventricular (LV) function, including the LVEF. The composite findings showed that 47% of patients with a trastuzumab‐induced decrease in LV function had elevated troponin I levels; furthermore, this subset of patients with elevated troponin I was less likely to have reversal of this cardiotoxicity postchemotherapy. Such studies suggest that frequent monitoring of troponin I or other cardiac biomarkers as an early marker of cardiac injury and sign of impending and potentially irreversible cardiotoxicities should play a role in future trials of VDAs [52].
Strategies for the prevention of vascular‐disrupting agent (VDA)‐associated cardiotoxicity

Strategies for the prevention of vascular‐disrupting agent (VDA)‐associated cardiotoxicity
Current Challenges and Treatment Considerations
Elucidation of the pathophysiology and mechanisms of VDA‐induced cardiac events will ultimately guide their management. Given the prevalence of hypertension, diabetes, obesity, and other cardiac risk factors leading to atherosclerosis and CAD in the general population, it may be challenging to exclude all patients with these comorbidities. A more practical strategy may be to exclude patients with severe heart disease and aggressively treat and monitor other pre‐existing conditions for patients who may benefit from VDA‐containing therapy.
The management of cardiovascular complications in patients treated with VDAs has not been standardized. However, a management strategy has been explored for patients with hypertension treated with anti‐VEGR agents [53]. Recommendations made by the interdisciplinary panel convened by the Investigational Drug Steering Committee of the National Cancer Institute included (a) a thorough in‐office pretreatment risk assessment, including a patient history, physical examination, blood pressure measurements, and targeted blood and urine analyses to establish baseline levels and to assess subclinical hypertension‐induced complications (e.g., microalbuminuria, retinopathy); (b) stabilization of hypertension through optimization of antihypertensives prior to initiation of antineoplastic therapy; (c) frequent blood pressure monitoring during treatment, especially during the first cycle; and (d) control of anti‐VEGF–induced hypertension to 140/90 mm Hg (or less, depending on comorbidities) in order to continue use of these agents and limit long‐term effects of the drug [53]. Such strategies developed for hypertension induced by anti‐VEGF therapy could be adapted to VDA‐induced hypertension and continually modified as the mechanisms of VDA‐induced cardiotoxicities are further elucidated.
Additionally, patients with important cardiac risk factors and comorbidities, including diabetes and hyperlipidemia, will benefit from agents such as statins and aspirin for the treatment and prevention of cardiac ischemic events. Because thromboembolism is the most likely mechanism of chest pain (caused by ischemia resulting from VDA therapy), the potential benefit of aspirin (most importantly, a lower mortality rate) outweighs the potential risk for bleeding; if the platelet count decreases to <50,000, aspirin should be withheld [54].
Recent evidence suggests that the neuroendocrine response also plays a role in tumor biology, specifically invasion and metastasis. Activation of the tumor cell protein kinase signaling cascade through β2‐adrenergic activation was found to lead to greater tumor vascularization, suggesting that selective β‐blockade for patients receiving therapy with a VDA may be worth exploration because dosing levels at which β‐blockers inhibit angiogenesis are not defined [55, 56].
Conclusions
Although VDAs show promise as a therapeutic option to decrease the tumor burden and lengthen the progression‐free survival interval, cardiovascular adverse events secondary to VDA exposure have been DLTs in several early‐phase human trials. The data unequivocally confirm the shortcomings of VDAs as monotherapy and have led to the use of VDAs in combination with other cytotoxic chemotherapies. Although early‐phase clinical trials have revealed promising antineoplastic activity, the concurrent emerging cardiovascular toxicities of VDAs hamper their therapeutic index. Some of these toxicities, like grade 1–2 hypertension, are easily manageable, but others such as grade 3–4 non‐ST‐elevation MI and atrial fibrillation, can significantly impact clinical outcomes and quality of life in later‐phase clinical trials. Additionally, regimens that combine VDAs with other cardiotoxic chemotherapy raise concerns about aggregate toxicities. As randomized phase II and III trials of VDAs in combination with other chemotherapies mature and as more data become available on toxicity profiles, evidence‐based guidelines for identifying baseline cardiovascular risk factors and early signs of serious cardiac adverse events are imperative if we are to accomplish the goal of minimizing toxicity while optimizing the therapeutic index and patient outcomes.
Author Contributions
Conception/Design: Apostolia M. Tsimberidou
Provision of study material or patients: Apostolia M. Tsimberidou
Collection and/or assembly of data: Apostolia M. Tsimberidou, Ishwaria M. Subbiah
Data analysis and interpretation: Apostolia M. Tsimberidou, Daniel J. Lenihan
Manuscript writing: Apostolia M. Tsimberidou, Ishwaria M. Subbiah, Daniel J. Lenihan
Final approval of manuscript: Apostolia M. Tsimberidou, Ishwaria M. Subbiah, Daniel J. Lenihan
References
Author notes
Disclosures: Ishwaria M. Subbiah: None; Daniel J. Lenihan: Consultant/advisory role: AstraZeneca; Apostolia M. Tsimberidou: None.
The content of this article has been reviewed by independent peer reviewers to ensure that it is balanced, objective, and free from commercial bias. No financial relationships relevant to the content of this article have been disclosed by the independent peer reviewers.

{kind=link}